Introduction
Sepsis is caused by polymicrobial infections
associated with severe systemic inflammatory response syndrome that
leads to multiple organ failure, including acute lung injury and
renal and hepatic failure, as well as septic shock (1–3).
Bacterial endotoxin lipopolysaccharide (LPS) is a major component
of the outer membrane of gram-negative bacteria and is important
for stimulating mononuclear phagocytes (macrophages and monocytes)
to secrete various inflammatory mediators (e.g. cytokines, reactive
oxygen species, prostanoids/leukotrienes, proteases and nitric
oxide), including alarmins (1,3,4).
Alarmins are identified as endogenous mediators that
have potent immune-activating abilities. To date, distinct
molecules of alarmins have been identified, including antibacterial
peptide (defensin and cathelicidin), cytokines (IL-1β and IL-33),
heat shock proteins, adenosine triphosphate, uric acid and nuclear
components (HMG proteins, nucleosome and histone) (4–9).
Generally, alarmins are extracellularly released by the following
mechanisms: i) Passive release by necrotic (unprogrammed) cell
death, which is caused by infective or noninfective tissue damage
or ii) active release by certain immune cells without cell death by
a specialized secretion pathway, in which alarmins are transported
from the nucleus to the cytoplasm, then into the lysosome and
eventually released into the extracellular space through exocytosis
(9,10). Additionally, previous studies have
revealed that pyroptosis, a form of programmed necrotic cell death
accompanied by the formation of inflammasomes, caspase-1 activation
and IL-1β production, induces the secretion of certain alarmins
(11,12). Alarmins augment the innate and
adaptive immune responses by distinct mechanisms and are capable of
promoting the recruitment and activation of immune cells, including
antigen-presenting cells (13,14).
Under septic conditions, excessive release of several alarmins,
including HMGB1 and IL-1β, leads to uncontrolled inflammation in
the host (15,16). By contrast, alarmins also exhibit
beneficial effects on the host by promoting the mobilization and
activation of immune cells to eliminate potential pathogens
(13,14). Thus, alarmins are crucial in the
pathological process of sepsis/septic shock.
High mobility group nucleosomal binding protein-1
(HMGN1), a highly conserved, non-histone chromosomal protein, which
binds to the inner side of the nucleosomal DNA, regulates chromatin
dynamics and transcription in cells (17,18).
By contrast, HMGN1 acts as a cytokine in the extracellular milieu.
HMGN1 induces the recruitment and maturation of antigen-presenting
cells (dendritic cells) to enhance Th1-type antigen-specific immune
responses (13). In addition,
HMGN1 promotes the secretion of inflammatory cytokines from
dendritic cells via direct interaction with Toll-like receptor 4
(TLR4), followed by the activation of myeloid differentiation
primary response gene 88/nuclear factor κB (NF-κB) and the mitogen
activated protein kinase (MAPK) pathway (14). Thus, HMGN1 is expected to act as an
alarmin, when released into the extracellular milieu, and to be
important in the pathogenesis of sepsis/septic shock. However, the
mechanism of HMGN1 release from immune cells remains to be
elucidated. The present study therefore investigated the release
mechanism of HMGN1 from macrophages, which is important in
sepsis/septic shock by producing inflammatory mediators in response
to bacterial pathogen-associated molecular patterns (PAMPs), using
mouse macrophage-like RAW264.7 cells.
Materials and methods
Reagents
LPS (from E. coli serotype O111:B4) was
purchased from Sigma-Aldrich (St. Louis, MO, USA).
Ac-Tyr-Val-Ala-Asp-H (Ac-YVAD-CHO), a specific caspase-1 inhibitor
was purchased from Peptide Institute, Inc. (Osaka, Japan).
Necrostatin-1 (Nec-1), a selective inhibitor of necroptosis and its
inactive analog necrostatin-1 (Nec-1i) were purchased from Merck
Millipore (Darmstadt, Germany).
Quantification of HMGN1 released from
LPS-stimulated RAW264.7 cells
A murine macrophage cell line RAW264.7 was purchased
from the European Collection of Cell Cultures (Wiltshire, UK) and
maintained in Dulbecco's modified Eagle's medium (DMEM;
Sigma-Aldrich) supplemented with 10% fetal calf serum (endotoxin
level <10 EU/ml; Cell Culture Technologies, Herndon, VA, USA)
and 100 U/ml penicillin/100 µg/ml streptomycin (Nacalai
Tesque, Inc., Kyoto, Japan) at 37°C in a 5% CO2
incubator. RAW264.7 cells (2×105 cells/well) were seeded
into a 12-well culture plate and incubated overnight at 37°C in 5%
CO2. Subsequently, cells were washed with fresh media
and incubated with or without LPS (100 ng/ml) in the absence or
presence of Ac-YVAD-CHO, Nec-1 or Nec-1i at a final concentration
of 10 µM (each) in the media. Culture supernatants were
recovered at 20 h after the incubation and HMGN1 in the culture
supernatants was quantitated by western blotting. Prior to
electrophoresis, supernatants were concentrated by trichloroacetic
acid (TCA) precipitation. Briefly, 900 µl of supernatants
were added with ice-cold 1/9 volume of 100% TCA (100 µl),
vortexed and incubated on ice for 30 min. The mixtures were
centrifuged at 8,000 × g at 4°C for 15 min and the supernatants
were aspirated. Pellets were washed with 1 ml of 100% ice-cold
acetone and centrifuged at 8,000 × g at 4°C for 5 min, and
dissolved in 1X Laemmli sample buffer. Dissolved samples were
subjected to 15% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (Nacalai Tesque, Inc.), and the resolved proteins
were electrophoretically transferred onto polyvinylidine difluoride
nitrocellulose membranes (Immobilon-P; Merck Millipore). The
membranes were blocked in Blocking One (Nacalai Tesque, Inc.) and
sequentially probed with rabbit anti-HMGN1 polyclonal antibody (2
µg/ml; cat. no. ab5212; Abcam, Cambridge, MA, USA), and
horseradish peroxidase-conjugated goat anti-rabbit IgG polyclonal
antibody (1:2,000 dilution; cat. no. AP187P; Chemicon
International, Temecula, CA, USA). HMGN1 was finally visualized
using Super Signal West Dura Chemiluminescent Substrate (Pierce
Biotechnology, Inc., Rockford, IL, USA) and the detected bands were
analyzed using Multi Gauge software (version 3.0) and an LAS-3000
Image Analyzer (Fujifilm, Tokyo, Japan).
Quantification of lactate dehydrogenase
(LDH) activities
LDH activity in the supernatants of LPS-stimulated
RAW264.7 cells was determined for evaluating cell death. LDH
activity in the supernatants and 1% Triton X-100-lysed cells (as a
total activity of 100%) were measured using a commercially
available LDH assay kit (Takara Bio, Inc., Shiga, Japan), according
to the manufacturer's instructions.
Quantification of IL-1β
The quantity of IL-1β in the culture supernatants of
LPS-stimulated RAW264.7 cells was measured using a commercially
available mouse IL-1β ELISA kit (detection limits of 8 pg/ml;
eBioscience, San Diego, CA, USA), according to the manufacturer's
instructions.
Quantification of apoptosis
Apoptotic and necrotic cell death was assessed by
staining cells with annexin V-fluorescein isothiocyanate and
propidium iodide (PI). RAW264.7 cells (2×105 cells/well)
were seeded into a 12-well culture plate and incubated overnight at
37°C in 5% CO2. Subsequently, cells were washed with
fresh media and incubated with or without LPS (100 ng/ml) in the
absence or presence of Ac-YVAD-CHO, Nec-1 or Nec-1i (10 µM
each). After 8 h, cells were washed with 0.05%
ethylenediaminetetraacetic acid (EDTA)/phosphate-buffered saline
(PBS) and detached with 0.25% trypsin-EDTA (Life Technologies;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). Subsequently,
annexin V/PI staining was performed using a MEBCYTO Apoptosis kit
(Medical & Biological Laboratories Co., Ltd., Nagoya, Japan),
according to the manufacturer's instructions. Briefly, cells were
centrifuged in a 1.5 ml tube (200 × g, 5 min) and supernatants were
discarded. Cells were sequentially rinsed with DMEM and ice-cold
PBS, and resuspended in 85 ml of binding buffer. A total of 10
µl of annexin V stock solution and 5 µl of PI stock
solution was then added to the cells, and incubated for 15 min in
the dark at room temperature. Thereafter, cells were immediately
analyzed by flow cytometry (FACSCalibur cell analyzer;
Becton-Dickinson, San Jose, CA, USA).
Statistical analysis
Data are presented as the mean ± standard deviation.
Statistical significance was determined by Student's t-test or
one-way analysis of variance (GraphPad Prism Software Inc., La
Jolla, CA, USA) with Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Release of HMGN1 from LPS-stimulated
RAW264.7 cells
Initially, the present study aimed to determine
whether LPS stimulation induces the extracellular release of HMGN1
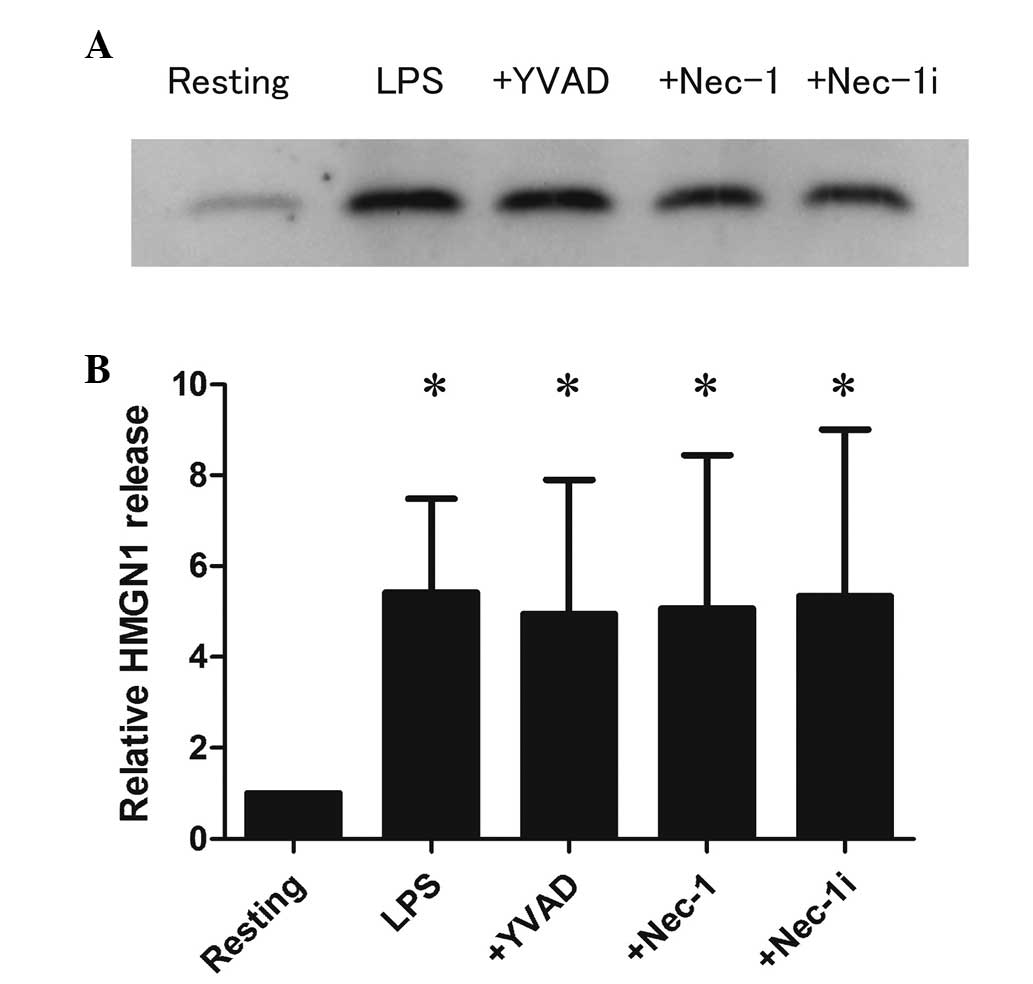
from RAW264.7 cells. As shown in Fig.
1, the level of HMGN1 in the supernatants increased by 5.4-fold
following LPS (100 ng/ml) stimulation compared with resting
(P<0.05). The percentage of HMGN1 release was calculated from
the total quantity of HMGN1 in the cell lysates. HMGN1 release was
<1% in resting cells; however, increased up to 2.7–5.9% in
LPS-stimulated cells (data not shown).
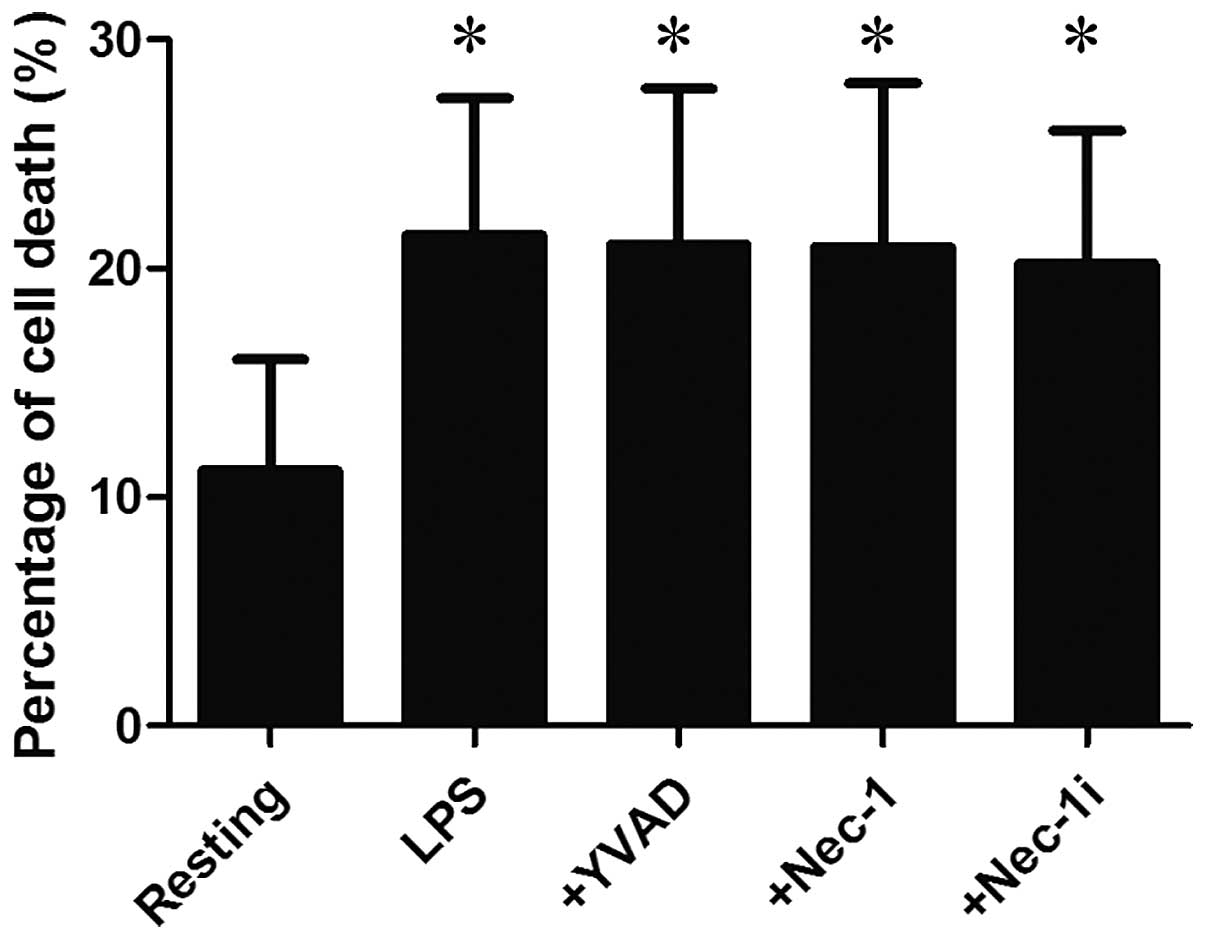
Furthermore, LDH assay indicated that the LDH
activity in the supernatants increased from 11.2±4.9% in resting
cells to 21.5±6.0% in LPS-stimulated cells (P<0.05; Fig. 2), suggesting that HMGN1 release is
accompanied by cell death.
Effects of Ac-YVAD-CHO and Nec-1 on HMGN1
release from LPS-stimulated RAW264.7 cells
Subsequently, the patterns of cell death involved in
HMGN1 release from LPS-stimulated RAW264.7 cells were determined
using Ac-YVAD-CHO (YVAD) and Nec-1. YVAD, a selective inhibitor of
caspase 1, suppresses pyroptosis, whereas Nec-1, an allosteric
inhibitor of the death domain receptor-associated adaptor kinase
RIP1, inhibits necroptosis (19).
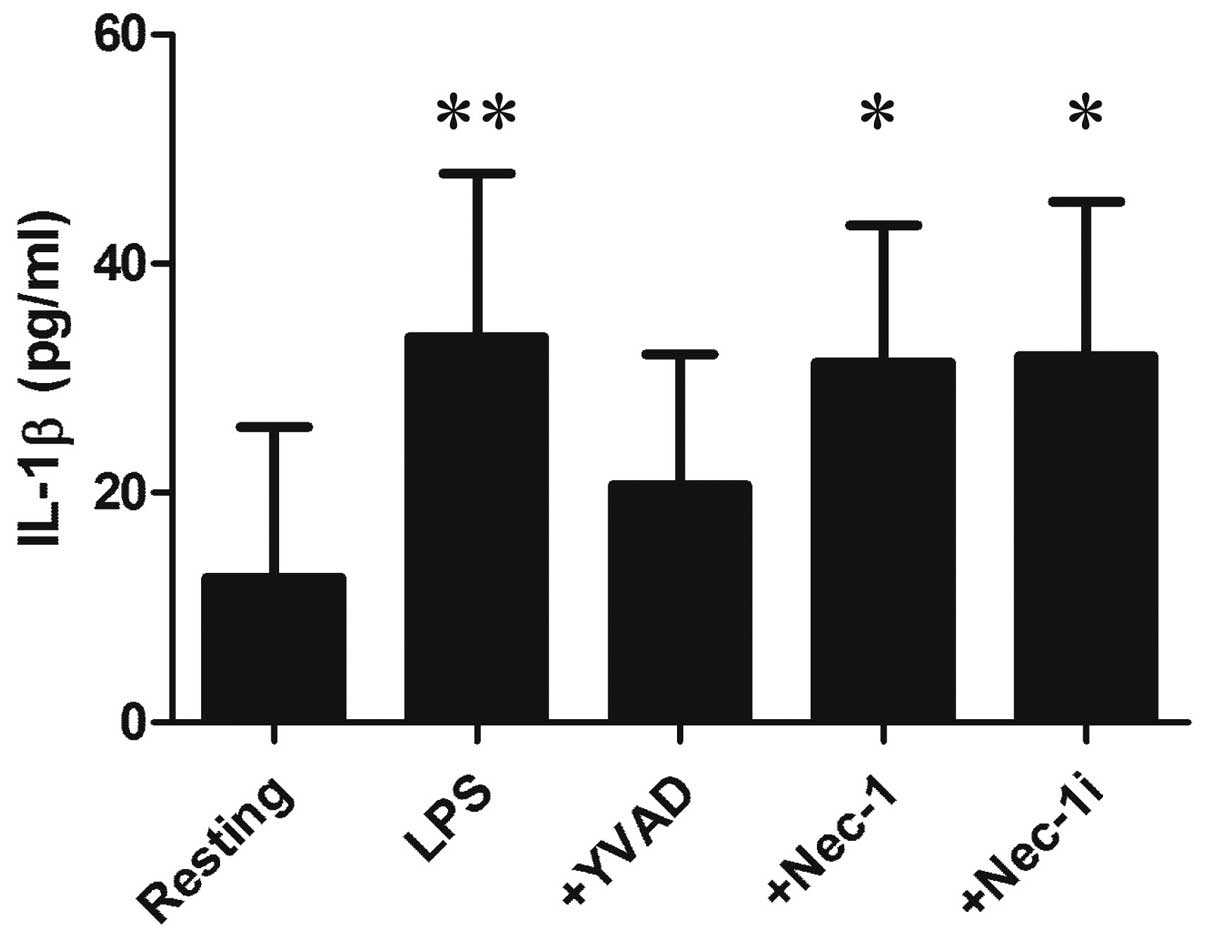
YVAD did not affect the release of HMGN1 or LDH from LPS-stimulated
RAW264.7 cells (Figs. 1 and
2), although it inhibited the
release of IL-1β as a marker of caspase-1 activation (Fig. 3). Similarly, Nec-1 did not affect
HMGN1 release or LDH release from LPS-stimulated RAW264.7 cells
(Figs. 1 and 2). A previous study confirmed that Nec-1
suppressed the release of HMGN1 from RAW264.7 cells treated with
LPS plus zVAD (a pan-caspase inhibitor), which induces necroptosis
of macrophages (20,21).
These observations suggest that HMGN1 release from
LPS-stimulated RAW264.7 cells is not associated with pyroptosis
(caspase-1-acivated cell death) or necroptosis (RIP1-dependent
programmed cell death).
Involvement of necrosis in HMGN1 release
from LPS-stimulated RAW264.7 cells
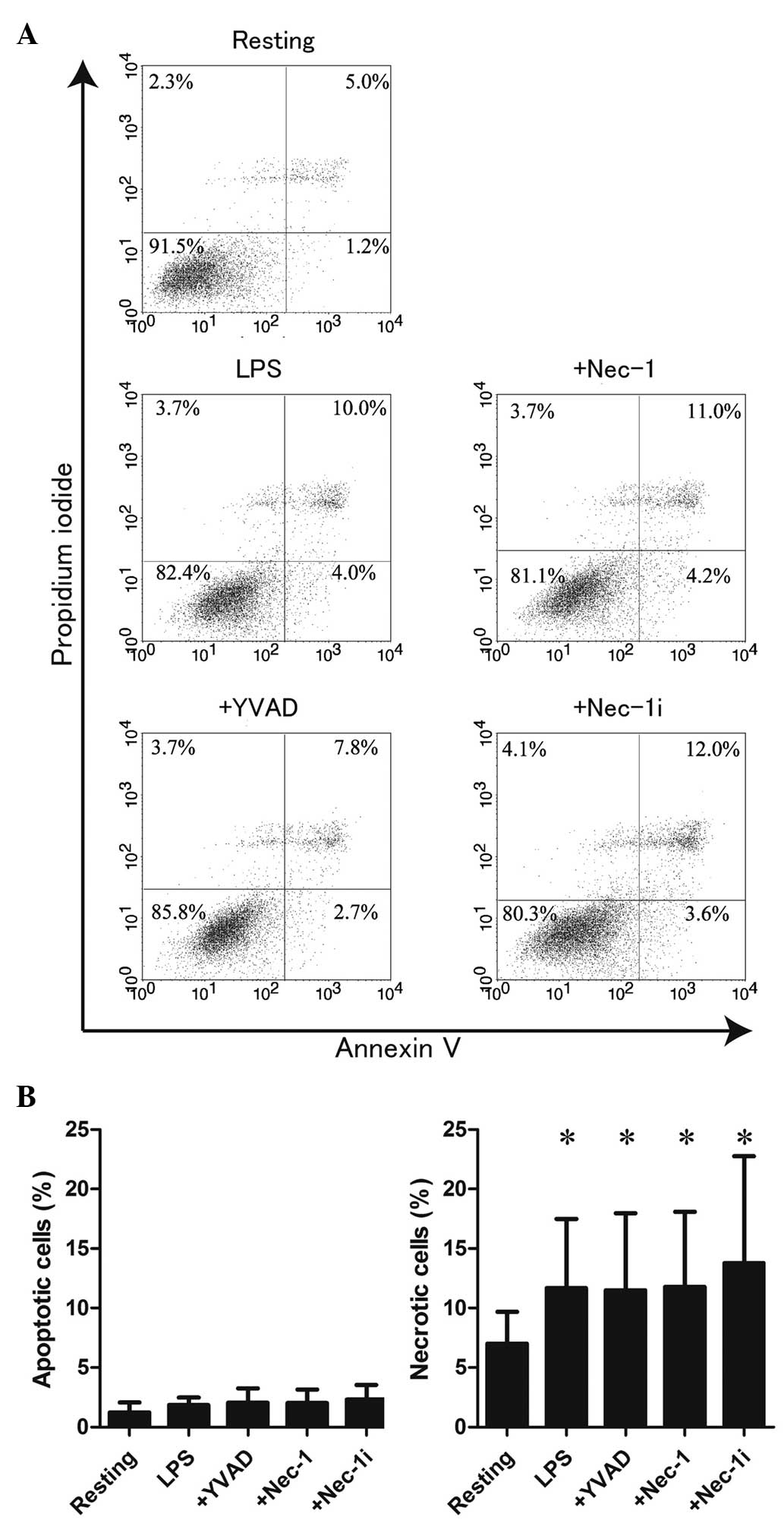
Finally, the involvement of necrotic cell death in
HMGN1 release was determined by staining LPS-stimulated RAW264.7
cells with annexin V and PI. As shown in Fig. 4A and B, LPS stimulation did not
significantly increase apoptotic cells (lower right division) but
significantly increased necrotic cells (P<0.05, upper right and
left divisions) compared with resting. Of note, it has been
reported that PI-positive cells are increased by pyroptosis and
necroptosis (22,23). However, YVAD and Nec-1 did not
affect these patterns of cell death. Together these findings
suggest that LPS stimulation is not likely to induce apoptosis,
pyroptosis or necroptosis of RAW264.7 cells, and HMGN1 is released
from PI-positive necrotic RAW264.7 cells stimulated with LPS.
Discussion
Endotoxin/septic shock is a severe and abnormal
condition that is induced during infections with gram-negative
bacteria. It is characterized by systemic inflammatory responses of
the host to invading microorganisms, PAMPs and damage-associated
molecular patterns, including endogenous danger signal molecules
termed alarmins (4). A previous
study revealed that HMGN1, an alarmin, acts as a cyto-kine in the
extracellular milieu, which enhances Th1-type antigen-specific
immune responses (13). In
addition, HMGN1 stimulates immune cells, including monocytes and
dendritic cells, to secrete inflammatory cytokines via the
TLR4/NF-κB and MAPK pathways (14). Since the activation of NF-κB and
excessive production of inflammatory cytokines from
mono-cytes/macrophages are key events in sepsis, the extracellular
release of HMGN1 from monocytes/macrophages is expected to be
important in the pathogenesis of sepsis/septic shock (14). However, the release mechanism of
HMGN1 from monocytes/macrophages remains to be elucidated. The
present study revealed that HMGN1 is extracellularly released from
LPS-stimulated RAW264.7 macrophage-like cells, accompanied by
unprogrammed necrotic cell death but not pyroptosis, necroptosis or
apoptosis.
Cell death has been discussed dichotomously as
either apoptosis or necrosis (24). Apoptosis is typically described as
a programmed process of self-destruction that avoids release of
inflammatory cellular contents (25). The execution of apoptosis minimizes
the leakage of cellular constituents by forming an apoptotic body,
a result of non-lytic cellular shrinkage (26). By contrast, necrosis is the most
common form of cell death, which is characterized by membrane
rupture and leakage of prophlogistic cellular constituents
(24). Thus, necrosis is
considered to be the potent source of immunoactivating danger
signal molecules, including alarmins. In addition, a previous study
expanded categorization of the two types of novel cell deaths i.e.
pyroptosis and necroptosis (26).
Pyroptosis is identified as caspase-1-dependent cell death of
macrophages and dendritic cells found in bacterial infection
(27). Pyroptosis is induced by
the formation of the inflammasome that facilitates the activation
of caspase-1, and the generation of cytokines, including IL-1β,
based on the cleavage of their precursors by activated caspase-1
(12). During pyroptosis, cellular
contents are rapidly released into the extracellular space by pore
formation and plasma membrane loss (28). Another form of cell death,
necroptosis was originally defined as a potent immunogenic
programmed cell death induced by the presence of zVAD-fmk (a
pan-caspase inhibitor) and tumor necrosis factor receptor signaling
that involves the activation of RIP1 and mitochondrial instability
(20,29). During necroptosis, swelling of
cellular organelles and plasma membrane disruption are induced, and
consequently inflammatory cellular contents are extracellularly
released (25).
In the present study, it was revealed that HMGN1 is
released from LPS-stimulated RAW264.7 cells, accompanied by cell
death as assessed by the release of LDH. Subsequently, the patterns
of cell death involved in HMGN1 release from LPS-stimulated
RAW264.7 cells were determined using YVAD (a caspase-1 inhibitor)
and Nec-1 (a RIP1 inhibitor). YVAD and Nec-1 did not alter
LPS-induced HMGN1 and LDH release, although YVAD and Nec-1
inhibited LPS-stimulated IL-1β release and LPS/zVAD-stimulated
HMGN1 release, respectively. These observations suggest that
pyroptosis (caspase-1-activated cell death) and necroptosis
(RIP1-dependent programmed cell death) are not involved in the
release of HMGN1 from LPS-stimulated RAW264.7 cells. In addition,
flow cytometric analysis indicated that LPS stimulation did not
induce apoptosis but substantially augmented necrosis, as evidenced
by annexin V/PI-staining. Together these findings suggest that
HMGN1 is released from necrotic (unprogrammed cell death) but not
apoptotic, pyroptotic or necroptotic (a programmed form of
necrosis) RAW264.7 cells following stimulation with LPS. Supporting
this theory, pyroptosis and necroptosis were not induced in
RAW264.7 cells by LPS stimulation under our experimental condition,
since YVAD (a pyroptosis inhibitor) and Nec-1 (a necroptosis
inhibitor) did not inhibit LPS-induced LDH release and PI staining,
although these inhibitors were capable of suppressing LDH release
and PI staining associated with pyroptosis and necroptosis
(27,30–32).
In addition, IL-1β was extracellularly released from RAW264.7 cells
by LPS stimulation, but its maximum level was 30 pg/ml (Fig. 3). Notably, pyroptosis-associated
IL-1β release is reported to reach 1–2 ng/ml (11,33,34).
These observations suggest that in our experiment, caspase-1 is
activated by LPS stimulation to process and release IL-1β; however,
it is unlikely that the activation is sufficient to induce
pyroptosis.
In conclusion, the release mechanism of HMGN1 from
macrophages was evaluated using RAW264.7 cells and it was revealed
that its extracellular release from LPS-stimulated RAW264.7 cells
is predominantly dependent on necrosis (unprogrammed cell death)
but not apoptosis, pyroptosis or necroptosis. Under conditions of
sepsis/septic shock, alarmins are hypothesized to be released from
immune cells due to cell death or cell activation at the site of
infection and inflammation, and have a fundamental role in the
regulation of host defense and tissue repair (35,36).
Therefore, HMGN1 released from macrophages and other immune cells
by stimulation with PAMPs, including LPS, is likely to be important
in sepsis/septic shock as an alarmin. Anti-cytokine based therapies
for sepsis-associated systemic inflammation have not been
successful (37). Therefore, the
modulation of cell death/cell activation and subsequent release of
alarmins, including HMGN1 is expected to be a promising target for
a novel therapeutic strategy against sepsis/septic shock.
Acknowledgments
The present study was supported in part by a
Grant-in-Aid for Young Scientists B and for Scientific Research C
(grant no. 2290416) from the Japan Society for the Promotion of
Science, and a Grants-in-Aid (grant no. S1201013) from the Ministry
of Education, Culture, Sports, Science and Technology (Japan).
Abbreviations:
|
HMGN1
|
high mobility group nucleosome binding
domain 1
|
|
LPS
|
lipopolysaccharide
|
|
HMGB1
|
high mobility group box 1
|
|
HMG
|
high mobility group
|
|
NF-κB
|
nuclear factor κB
|
|
MAPK
|
mitogen activated protein kinase
|
References
|
1
|
Cohen J: The immunopathogenesis of sepsis.
Nature. 420:885–891. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
O'Brien JM Jr, Ali NA, Aberegg SK and
Abraham E: Sepsis. Am J Med. 120:1012–1022. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Van Amersfoort ES, Van Berkel TJ and
Kuiper J: Receptors, mediators, and mechanisms involved in
bacterial sepsis and septic shock. Clin Microbiol Rev. 16:379–414.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Manson J, Thiemermann C and Brohi K:
Trauma alarmins as activators of damage-induced inflammation. Br J
Surg. 99(Suppl 1): S12–S20. 2012. View
Article : Google Scholar
|
|
5
|
Tewary P, de la Rosa G, Sharma N,
Rodriguez LG, Tarasov SG, Howard OM, Shirota H, Steinhagen F,
Klinman DM, Yang D and Oppenheim JJ: β-Defensin 2 and 3 promote the
uptake of self or CpG DNA, enhance IFN-α production by human
plasmacytoid dendritic cells, and promote inflammation. J Immunol.
191:865–874. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nagaoka I, Tamura H and Hirata M: An
antimicrobial cathelicidin peptide, human CAP18/LL-37, suppresses
neutrophil apoptosis via the activation of formyl-peptide
receptor-like 1 and P2×7. J Immunol. 176:3044–3052. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Suzuki K, Murakami T, Kuwahara-Arai K,
Tamura H, Hiramatsu K and Nagaoka I: Human anti-microbial
cathelicidin peptide LL-37 suppresses the LPS-induced apoptosis of
endo-thelial cells. Int Immunol. 23:185–193. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moussion C, Ortega N and Girard JP: The
IL-1-like cytokine IL-33 is constitutively expressed in the nucleus
of endothelial cells and epithelial cells in vivo: A novel
'alarmin'? PLoS One. 3:e33312008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bianchi ME: DAMPs, PAMPs and alarmins: All
we need to know about danger. J Leukoc Biol. 81:1–5. 2007.
View Article : Google Scholar
|
|
10
|
Yang Z, Li L, Chen L, Yuan W, Dong L,
Zhang Y, Wu H and Wang C: PARP-1 mediates LPS-induced HMGB1 release
by macrophages through regulation of HMGB1 acetylation. J Immunol.
193:6114–6123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hu Z, Murakami T, Suzuki K, Tamura H,
Kuwahara-Arai K, Iba T and Nagaoka I: Antimicrobial cathelicidin
peptide LL-37 inhibits the LPS/ATP-induced pyroptosis of
macrophages by dual mechanism. PLoS One. 9:e857652014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lamkanfi M and Dixit VM: Modulation of
inflammasome pathways by bacterial and viral pathogens. J Immunol.
187:597–602. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wei F, Yang D, Tewary P, Li Y, Li S, Chen
X, Howard OM, Bustin M and Oppenheim JJ: The Alarmin HMGN1
contributes to antitumor immunity and is a potent immunoadjuvant.
Cancer Res. 74:5989–5998. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang D, Postnikov YV, Li Y, Tewary P, de
la Rosa G, Wei F, Klinman D, Gioannini T, Weiss JP, Furusawa T, et
al: High-mobility group nucleosome-binding protein 1 acts as an
alarmin and is critical for lipopolysaccharide-induced immune
responses. J Exp Med. 209:157–171. 2012. View Article : Google Scholar :
|
|
15
|
Magna M and Pisetsky DS: The role of HMGB1
in the pathogenesis of inflammatory and autoimmune diseases. Mol
Med. 20:138–146. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Murakami T, Suzuki K, Tamura H and Nagaoka
I: Suppressive action of resolvin D1 on the production and release
of septic mediators in D-galactosamine-sensitized endotoxin shock
mice. Exp Ther Med. 2:57–61. 2011.PubMed/NCBI
|
|
17
|
Kugler JE, Horsch M, Huang D, Furusawa T,
Rochman M, Garrett L, Becker L, Bohla A, Hölter SM, Prehn C, et al:
High mobility group N proteins modulate the fidelity of the
cellular transcriptional profile in a tissue- and variant-specific
manner. J Biol Chem. 288:16690–16703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gerlitz G: HMGNs, DNA repair and cancer.
Biochim Biophys Acta. 1799:80–85. 2010. View Article : Google Scholar :
|
|
19
|
Cho Y, McQuade T, Zhang H, Zhang J and
Chan FK: RIP1-dependent and independent effects of necrostatin-1 in
necrosis and T cell activation. PLoS One. 6:e232092011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kaczmarek A, Vandenabeele P and Krysko DV:
Necroptosis: The release of damage-associated molecular patterns
and its physiological relevance. Immunity. 38:209–223. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moquin DM, McQuade T and Chan FK: CYLD
deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate
kinase activation and programmed necrosis. PLoS One. 8:e768412013.
View Article : Google Scholar
|
|
22
|
Wree A, Eguchi A, McGeough MD, Pena CA,
Johnson CD, Canbay A, Hoffman HM and Feldstein AE: NLRP3
inflammasome activation results in hepatocyte pyroptosis, liver
inflammation and fibrosis in mice. Hepatology. 59:898–910. 2014.
View Article : Google Scholar :
|
|
23
|
Khan MJ, Rizwan Alam M, Waldeck-Weiermair
M, Karsten F, Groschner L, Riederer M, Hallström S, Rockenfeller P,
Konya V, Heinemann A, et al: Inhibition of autophagy rescues
palmitic acid-induced necroptosis of endothelial cells. J Biol
Chem. 287:21110–21120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zong WX and Thompson CB: Necrotic death as
a cell fate. Genes Dev. 20:1–15. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Davidovich P, Kearney CJ and Martin SJ:
Inflammatory outcomes of apoptosis, necrosis and necroptosis. Biol
Chem. 395:1163–1171. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Inoue H and Tani K: Multimodal immunogenic
cancer cell death as a consequence of anticancer cytotoxic
treatments. Cell Death Differ. 21:39–49. 2014. View Article : Google Scholar
|
|
27
|
Cervantes J, Nagata T, Uchijima M, Shibata
K and Koide Y: Intracytosolic listeria monocytogenes induces cell
death through caspase-1 activation in murine macrophages. Cell
Microbiol. 10:41–52. 2008.
|
|
28
|
Lamkanfi M and Dixit VM: Mechanisms and
functions of inflammasomes. Cell. 157:1013–1022. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar
|
|
30
|
Yamanaka K, Urano Y, Takabe W, Saito Y and
Noguchi N: Induction of apoptosis and necroptosis by
24(S)-hydroxycholesterol is dependent on activity of acyl-CoA:
Cholesterol acyltransferase 1. Cell Death Dis. 5:e9902014.
View Article : Google Scholar
|
|
31
|
Steinhart L, Belz K and Fulda S: Smac
mimetic and demeth-ylating agents synergistically trigger cell
death in acute myeloid leukemia cells and overcome apoptosis
resistance by inducing necroptosis. Cell Death Dis. 4:e8022013.
View Article : Google Scholar
|
|
32
|
Tseng WA, Thein T, Kinnunen K, Lashkari K,
Gregory MS, D'Amore PA and Ksander BR: NLRP3 inflammasome
activation in retinal pigment epithelial cells by lysosomal
destabilization: Implications for age-related macular degeneration.
Invest Ophthalmol Vis Sci. 54:110–120. 2013. View Article : Google Scholar :
|
|
33
|
Sauer JD, Witte CE, Zemansky J, Hanson B,
Lauer P and Portnoy DA: Listeria monocytogenes triggers
AIM2-mediated pyroptosis upon infrequent bacteriolysis in the
macrophage cytosol. Cell Host Microbe. 7:412–419. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hua KF, Chou JC, Ka SM, Tasi YL, Chen A,
Wu SH, Chiu HW, Wong WT, Wang YF, Tsai CL, et al: Cyclooxygenase-2
regulates NLRP3 inflammasome-derived IL-1β production. J Cell
Physiol. 230:863–874. 2015. View Article : Google Scholar
|
|
35
|
Diener KR, Al-Dasooqi N, Lousberg EL and
Hayball JD: The multifunctional alarmin HMGB1 with roles in the
pathophysiology of sepsis and cancer. Immunol Cell Biol.
91:443–450. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang H, Ward MF and Sama AE: Targeting
HMGB1 in the treatment of sepsis. Expert Opin Ther Targets.
18:257–268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Webster NR and Galley HF: Immunomodulation
in the critically ill. Br J Anaesth. 103:70–81. 2009. View Article : Google Scholar : PubMed/NCBI
|