Introduction
Hepatitis is an inflammation of the liver without a
specific cause and presents a major threat to human health
worldwide (1). Hepatitis is a
medical condition defined by inflammation of the liver and
characterized by the presence of inflammatory cells in the organ
tissue. Previous studies have indicated the main causes as follows:
i) Various disorders, including a viral or bacterial infection of
the liver (1,2); ii) the intake of toxic substances
(1,3); iii) interruption of the normal blood
supply to the liver; and iv) an autoimmune disorder (4). Notably, the hepatitis viruses,
designated hepatitis A, B and C, and autoimmune hepatitis (AIH),
result in the greatest liver injury. During investigations of AIH,
concanavalin A (Con A)-induced hepatitis, a well-established mouse
model of immune-mediated liver injury, has been recognized as the
best experimental model for AIH research (5–7). In
addition, previous studies suggest that pro-inflammatory cytokines
and T lymphocytes are important in the pathogenesis of AIH
(6,8). Furthermore, a single intravenous
(i.v.) injection of Con A has been demonstrated to induce
fulminant, T cell-dependent hepatitis and major inflammatory
cytokine production, including tumor necrosis factor (TNF)-α,
interferon (IFN)-γ and interleukin (IL)-6 (8). Overexpression of pro-inflammatory
cytokines is associated with recruitment and activation of immune
cell infiltration, which results in aberrant expression of
inflammatory genes, ultimately resulting in hepatitis or other
diseases (such as lung injury, lung cancer, gastritis and
neurogenic inflammation) (9).
Fractalkine (FKN), also termed chemokine (C-X3-C
motif) ligand 1 (CX3CL1) is important in the recruitment of
intraepithelial lymphocytes and the adhesion of inflammatory cells
(10,11). Chemokine (C-X3-C motif) receptor 1
(CX3CR1) is a specific CX3CL1 receptor, which has been implicated
in the pathogenesis of liver injury in humans (11). Wollberg et al (12) and Pirvulescu et al (13) demonstrated the underlying
mechanisms of CX3CR1 in cellular adhesion, migration, metastasis,
and CX3CL1/CX3CR1-induced migration and recruitment of inflammatory
cells. Furthermore, CX3CL1 expression in endothelial and vascular
smooth muscle cells, and CX3CL1-mediated leukocyte adhesion
markedly promote the development of inflammatory diseases (14). However, the role of CX3CL1 in Con
A-induced liver injury remains to be elucidated. Previous studies
have demonstrated that the CX3CL1/CX3CR1 axis was markedly
upregulated via activation of the nuclear factor
κ-light-chain-enhancer of activated B cells (NF-κB) signaling
pathway (14,15). Notably, the generation of
inflammatory cytokines, resulting from CX3CL1/CX3CR1 acceleration,
promotes increased production of inflammatory cytokines and
exacerbation of the inflammatory response (16).
Protectin D1 (PD1) is a bioactive product generated
from docosahexaenoic acid (DHA), which has been reported to exert
anti-inflammatory effects in various disorders, including acute
kidney injury, neurodegenerative diseases and acute lung injury
(17–20). Previous studies suggest PD1
attenuates inflammatory action by inhibiting inflammatory signaling
pathways, such as NF-κB and p38 mitogen-activated protein kinases
(20). Furthermore, Yan et
al (20) demonstrated that ω-3
fatty acids (including DHA and eicosapentaenoic acid) potentially
have the ability to inhibit caspase-1 expression, IL-1β secretion
and NLR family, pyrin domain containing 3 (NLRP3) inflammasome
formation. Hence, the present study hypothesized that PD1, as a
bioactive product generated from DHA, may suppress NLRP3
expression. Furthermore, Tsutsui et al (21) indicated that Toll-like receptor
(TLR) 4, NF-κB and NLRP3-mediated pro-inflammatory cytokine and
chemokine expression is a major factor in the development of liver
injury. Therefore, the current study hypothesizes that PD1 may be
key in the alleviation of Con A-induced hepatitis. The present
study identified that PD1 pretreatment suppressed systemic
inflammation, in part, via inhibition of NF-κB activation, CX3CL1
expression and NLRP3 inflammasome formation. These findings suggest
that PD1 may be considered a promising agent for the treatment of
liver-related diseases.
Materials and methods
Animals and administration of Con A and
PD1
A total of 60 male C57BL/6 mice (age, 8 weeks;
weight, 20–25 g) were purchased from the Animal Experimentation
Center of the Second Military Medical University (Shanghai, China).
Mice were acclimatized to their environment for one week. They were
housed in a pathogen-free, temperature and humidity-controlled
environment (25±2°C; 50±5% humidity) with a standard 12-h
light/dark cycle, and allowed free access to food and water. All
procedures were performed in accordance with the Regulations of
Experimental Animal Administration issued by the Ministry of
Science and Technology of the People's Republic of China
(http://www.most.gov.cn). The Institutional Animal
Care and Use Committee at Jilin University (Changchun, China)
approved the animal study protocols. PD1 was obtained from the
Cayman Chemical Company (Ann Arbor, MI, USA) and prepared in Hank's
buffer (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
The PD1 solution was prepared 10 min prior to use and placed in
cold storage upon reaching a peak concentration of 200
µg/ml. Con A (cat. no. 11028-71-0; purity≥98%) was purchased
from Santa Cruz Biotechnology, Inc (Dallas, TX, USA) and dissolved
in Hank's balanced salt solution buffer (Gibco; Thermo Fisher
Scientific, Inc.) at a concentration of 25 mg/ml. The mice were
randomly divided into four groups as follows: i) Control group (a
tail vein injection with 0.5 ml Hank's buffer); ii) Con A group (30
mg/kg); iii) 20 µg/kg PD1 pretreatment group (HPD1; high
dose of PD1); iv) 10 µg/kg PD1 pretreatment group (LPD1; low
dose of PD1). Mice were deprived of food for 24 h, but given free
access to water. The mice were pretreated with 20 or 10
µg/kg PD1 via i.v. injection for 4 h. They were subsequently
injected (i.v.) with Con A (30 mg/kg) for 24 h, at different
time-points (4, 12 and 24 h), blood samples (1.5 ml) were collected
by cardiac puncture following sevoflurane anesthesia.
Serum transaminase activity assays and
pro-inflammatory cytokine levels
Serum was obtained following centrifugation at 650 ×
g for 15 min. Alanine transaminase (ALT) and aspartate transaminase
(AST) serum levels were analyzed spectrophotometrically using an
Olympus AU1000 automated chemistry analyzer (Olympus Corporation,
Tokyo, Japan). The TNF-α (DY410), IFN-γ (DY485), IL-6 (DY406) and
CX3CL1 (DY472) levels in the serum (markers of Con A-induced liver
injury) were measured in the mice using ELISA kits (R&D
Systems, Inc., Minneapolis, MN, USA) according to the
manufacturer's protocols. The high mobility group protein B1
(HMGB1) was quantified using sandwich immunoassays from Bio-Gene
Technology, Ltd. (Shanghai, China). The remaining serum was stored
at −80°C.
Histopathological examination of the
liver
Liver tissue samples were obtained 24 h after Con A
administration, fixed in 10% neutral buffered formalin and embedded
in paraffin (both purchased from Nanjing Jiancheng Biology
Engineering Institute, Nanjing, China). For morphometric analysis,
the sections were stained with hematoxylin and eosin (Thermo Fisher
Scientific, Inc.) to monitor histological changes. Following
dehydration, thin sections (4–6 µm) were evaluated by light
microscopy (OLYMPUS CX23; Olympus Corporation) according to the
previously described method (22).
Preparation of liver mononuclear cells
(MNCs)
Liver samples were collected 24 h after Con A
administration. The liver MNCs were prepared according to the
method by Tsutsui et al (21) with certain modifications. To avoid
CD3+ interference, blood was collected from the eyeball
rather than by conducting liver perfusion. The liver tissue samples
were infiltrated with collagenase type II (Invitrogen; Thermo
Fisher Scientific, Inc.) for 40 min. Liver samples were washed with
cold Hank's buffer three times, the tissues were compressed and
passed through a stainless steel mesh (pores, size 60;
Sigma-Aldrich, St. Louis, MO, USA), and suspended in RPMI-1640
medium (Gibco; Thermo Fisher Scientific, Inc.). The cell
suspensions were centrifuged at 75 × g for 5 min to remove debris
and impurities prior to being filtered through a nylon mesh (Jilin
Futian Bioproduct Corporation), which had been presoaked in Hank's
buffer. The supernatants containing hepatic MNCs were harvested and
washed once with Hank's buffer, and the cells were re-suspended in
40% Percoll® (Sigma-Aldrich). All cell suspensions were
placed over 70% Percoll® and centrifuged for 30 min at
350 × g. MNCs were enriched at interphase, and washed twice in
Hank's buffer.
Flow cytometric analysis
Single-cell suspensions of liver tissue were
collected 24 h following Con A injection (30 mg/kg). Cells were
immediately stained with fluorescent-labeled antibodies, anti-CD4
allophycocyanin (APC) (dilution, 1:1,000; cat. no. 17-0041-82),
anti-CD8 phycoerythrin (PE) (dilution, 1:1,000; cat. no.
11-0081-85), anti-killer cell lectin-like receptor subfamily B,
member 1 (NKT1.1) PE (dilution, 1:1,000; cat. no. 12-5941) and
anti-CD3 APC (dilution, 1:1,000; cat. no. 17-0031), which were
obtained from eBioscience, Inc., San Diego, CA, USA). The number of
CD4+, CD8+ and NKT cells infiltrating the
mouse livers was analyzed by flow cytometry (Miltenyi Biotec GmbH,
Bergisch Gladbach, Germany).
Cell viability and cell cycle
analysis
A rapid method for isolating mice lymphocytes was
used to evaluate the inhibitory effect of PD1 on T cell
proliferation and pro-inflammatory responses. T lymphocytes of
mouse livers (that had not been administered with therapeutic
agents) were separated and prepared in Hank's buffer. The isolation
was based on the preparation of liver MNCs. All the cells were
washed using Hank's buffer (containing 0.1% bovine serum albumin;
BSA; Sigma-Aldrich China, Inc., Shanghai, China) three times and
resuspended in the RPMI-1640 culture medium. The cells
(1×107 cells/ml) were seeded in 24- or 96-well plates
containing RPMI-1640 with 10% fetal bovine serum, 100 U/ml
penicillin and 100 µg/ml streptomycin (Sigma-Aldrich China,
Inc.). The cells were either untreated or treated with 20
µg/ml Con A in the presence of 0, 2.5, 5, 10 or 20 nM PD1
for 48 h. The cells were maintained in an atmosphere of 5%
CO2 at 37°C. The MTT assay was used to analyze cell
viability. All of the medium was removed and 5 µl MTT
solution (10 mg/ml; Sigma-Aldrich China, Inc.) was added to 100
µl phenol red-free growth medium (Sigma-Aldrich China,
Inc.), and the plates were incubated at 37°C in 5% CO2
for 4 h. Subsequently, a microplate reader (Model 550; Bio-Rad
Laboratories, Inc., Hercules, CA, USA) was used to measure the
absorbance of each well at a wavelength of 540 nm. For cell cycle
analysis, cells were plated at 1×107 cells/well in
24-well plates and left untreated or treated with 20 µg/ml
Con A in the presence of 20 nM PD1 for 18 h. Subsequently, the
cells were incubated with 20 µg/ml RNase A, followed by 25
µg/ml propidium iodide (PI; Sigma-Aldrich China, Inc.). A
flow cytometer (MACSQuant; Miltenyi Biotec GmbH,) with an argon
laser and 570-nm Bandpass filter (Sigma-Aldrich China, Inc.) was
used to measure the intensity of PI fluorescence to determine the
phase of the cell cycle.
Western blot analysis
The liver tissues and cells were homogenized in 10%
(w/v) hypotonic buffer [25 mM Tris-HCl (pH 8.0), 1 mM EDTA, 5
µg/ml leupeptin, 1 mM Pefabloc® SC, 50
µg/ml aprotinin, 5 µg/ml soybean trypsin inhibitor
and 4 mM benzamidine (Shanghai Bogoo Biotech, Co., Ltd., Shanghai,
China)] to yield a homogenate. The final supernatants were obtained
by centrifugation at 12,000 rpm for 20 min. Protein concentration
was determined using an ASSAYSMicro BCA protein assay kit (Thermo
Fisher Scientific, Inc.) with BSA serving as a standard. The total
protein extract (5 µg) was used for western blot analysis.
Equal quantities of total protein from the liver tissues samples
were subjected to 10 or 12% SDS-PAGE (Sigma-Aldrich China, Inc.;
150 V for 1 h). Immunoblotting was conducted using the following
primary polyclonal rabbitantibodies: Rabbit anti-GAPDH (dilution,
1:1,000; cat. no. 5174), NF-κB (dilution, 1:1,000; cat. no. 8242),
phosphorylated-NF-κB (dilution, 1:1,000; cat. no. 4887), TLR4
(dilution, 1:1,000; cat. no. 14358), nuclear factor of κ light
polypeptide gene enhancer in B-cells inhibitor, α (IκBα) (dilution,
1:1,000; cat. no. 4814), inhibitor of nuclear factor κ-B kinase
subunit beta (IKKβ) (dilution, 1:1,000; cat. no. 8943), myeloid
differentiation primary response gene 88 (MyD88) (dilution,
1:2,000; cat. no. 4283), NLRP3 (dilution, 1:1,000; cat. no. 13158),
IL-1β (dilution, 1:1,000; cat. no. 12426), caspase-1 (dilution,
1:1,000; cat. no. 3866) (Cell Signaling Technology, Inc., Danvers,
MA, USA), CX3CR1 (cat. no. ab8021) and CX3CL1 (dilution, 1:1,000;
cat. no. ab25088) (Abcam, Cambridge, MA, USA). Immunoreactive bands
were visualized by a Pierce enhanced chemiluminescence immunoblot
detection system (Thermo Fisher Scientific, Inc.) and exposed to
Kodak X-ray film (Kodak, Rochester, NY, USA). Expression levels of
each protein were defined as grey values (ImageJ software, version
1.4.2b; ImageJ, National Institutes of Health, Bethesda, MD, USA),
standardized to the housekeeping gene, GAPDH and expressed as a
fold of the control.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from tissue and cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.) and
1 µg total RNA was reverse transcribed using the M-MLV-RT
system (Promega Corporation, Madison, WI, USA). This was performed
at 42°C for 1 h and terminated by deactivation of the enzyme at
70°C for 10 min. qPCR was conducted using SYBR® Green
(Bio-Rad Laboratories, Inc.) in an ABI PRISM 7900HT detection
system (Applied Biosystems; Thermo Fisher Scientific, Inc.). All
the primers for GAPDH, TNF-α, NF-κB p65, IL-6, IL-1β, IFN-γ, IκBα,
IKKβ, caspase-1, NLRP3, HMGB1, CX3CL1 and CX3CR1 were produced by
Thermo Fisher Scientific, Inc. and the sequences are presented in
Table I. Amplification of
pre-denatured products was conducted at 94°C for 60 sec; followed
by 45 cycles at 95°C for 30 sec, 58°C for 30 sec and 72°C for 30
sec; followed by 95°C for 10 sec, 65°C for 45 sec, and 40°C for 60
sec. Fold induction values were calculated according to
2−ΔΔCq expression (15), where ΔCq represents the differences
in cycle threshold number between the target gene and GAPDH, and
ΔΔCq represents the relative change in the differences between the
control and treatment groups.
 | Table IPrimer sequences used in the present
study. |
Table I
Primer sequences used in the present
study.
| Gene | Primer sequence
(5′-3′) |
|---|
| TNF-α | F:
CAGGCGGTGCCTATGTCTC |
| R:
CGATCACCCCGAAGTTCAGTAG |
| IL-2 | F:
TGAGCAGGATGGAGAATTACAGG |
| R:
GTCCAAGTTCATCTTCTAGGCAC |
| IL-6 | F:
CTGCAAGAGACTTCCATCCAG |
| R:
AGTGGTATAGACAGGTCTGTTGG |
| HMGB1 | F:
GCATCCTGGCTTATCCATTGG |
| R:
GGCTGCTTGTCATCTGCTG |
| NF-κB | F:
ATGGCAGACGATGATCCCTAC |
| R:
CGGATCGAAATCCCCTCTGTT |
| IκBα | F:
AAGATGTCGTTCAAGGAGGTGCG |
| R:
ATCCTCTGAGATTTGACGCTTTG |
| IKKβ | F:
GGTGTGAAATTGAGACAATTGAAAAC |
| R:
GTTTCCTGTCAGTACCAAGGTTGA |
| Caspase-1 | F:
ACGCCTTGCCCTCATAAT |
| R:
TCTAATACATCTGGGACTTCTT |
| CX3CL1 | F:
GCTAGCATGGCTCCCTCGCCGCTCGCG |
| R:
ATGCAGGACCACGGTCACACTTGCGCA |
| IL-1β | F:
CCAGGATGAGGACCCAAGCA |
| R:
TCCCGACCATTGCTGTTTCC |
| IFN-γ | F:
CCTCAAACTTGGCAATACTCA |
| R:
CTCAAGTGGCATAGATGTGGA |
| CX3CR1 | F:
TCATCCAGACGCTGTTTTCC |
| R:
AGGCATTTCCCATACAGGTG |
| NLRP3 | F:
CTTCTCTGATGAGGCCCAAG |
| R:
GCAGCAAACTGGAAAGGAAG |
| GAPDH | F:
TTCACCACCATGGAGAAGGC |
| R:
GGCATGGACTGTGGTCATGA |
Immunoprecipitation (IP)
IP was performed using a Pierce Classic IP kit
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. Lysates from cell cultures were obtained by
homogenizing the cells in 10% (w/v) hypotonic lysis buffer
(Shanghai Bogoo Biotech, Co., Ltd., Shanghai, China). Then, 100
µl bead slurry (Thermo Fisher Scientific, Inc.) was added to
the lysate and incubated for 10 to 30 min at 4°C with gentle
agitation. To increase the yield, the beads were washed 1 or 2
additional times in lysis buffer, and the supernatants were
collected. The cell lysate was put on ice and, in a microcentrifuge
tube, 100 µl culture supernatant (Thermo Fisher Scientific,
Inc.) was added to 10–50 µg cell lysate. The samples were
then incubated with the following primary polyclonal antibodies
overnight at 4°C: rabbit anti-NF-kB (dilution, 1:1000, cat. no.
8242), TLR4 (dilution, 1:1000; cat. no. 14358), IκB (dilution,
1:1000; cat. no. 4814), MyD88 (dilution, 1:2000; cat. no. 4283)
(all purchased from Cell Signaling Technologies, Inc.), CX3CL1
(dilution, 1:1000; cat. no. ab25088) and CX3CR1 (dilution, 1:1000;
cat. no. ab8021) (both purchased from Abcam). The samples were then
incubated with horseradish peroxidase-conjugated rabbit anti-biotin
(dilution, 1:15,000; cat. no. 5571; Cell Signaling Technologies,
Inc.) for 1.5 h at 37°C. The beads were then washed with washing
buffer or lysis buffer three times to remove non-specific binding.
Samples were then centrifuged at 1,000 × g and western blot
analysis was performed using 30 µg protein samples. Protein
expression levels were defined as grey value and analyzed using
Image J software (version 1.4.2b, National Institutes of Health,
Bethesda, MA, USA), and standardized to GAPDH and expressed as a
fold of the control.
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. Treated tissue samples and the corresponding controls
were compared using GraphPad PRISM software (version 6.0; GraphPad
Software, Inc., La Jolla, CA, USA), and one-way analysis of
variance with Dunn's least significant difference tests or
Student's t-tests. P<0.05 was considered to indicate a
statistically significant difference.
Results
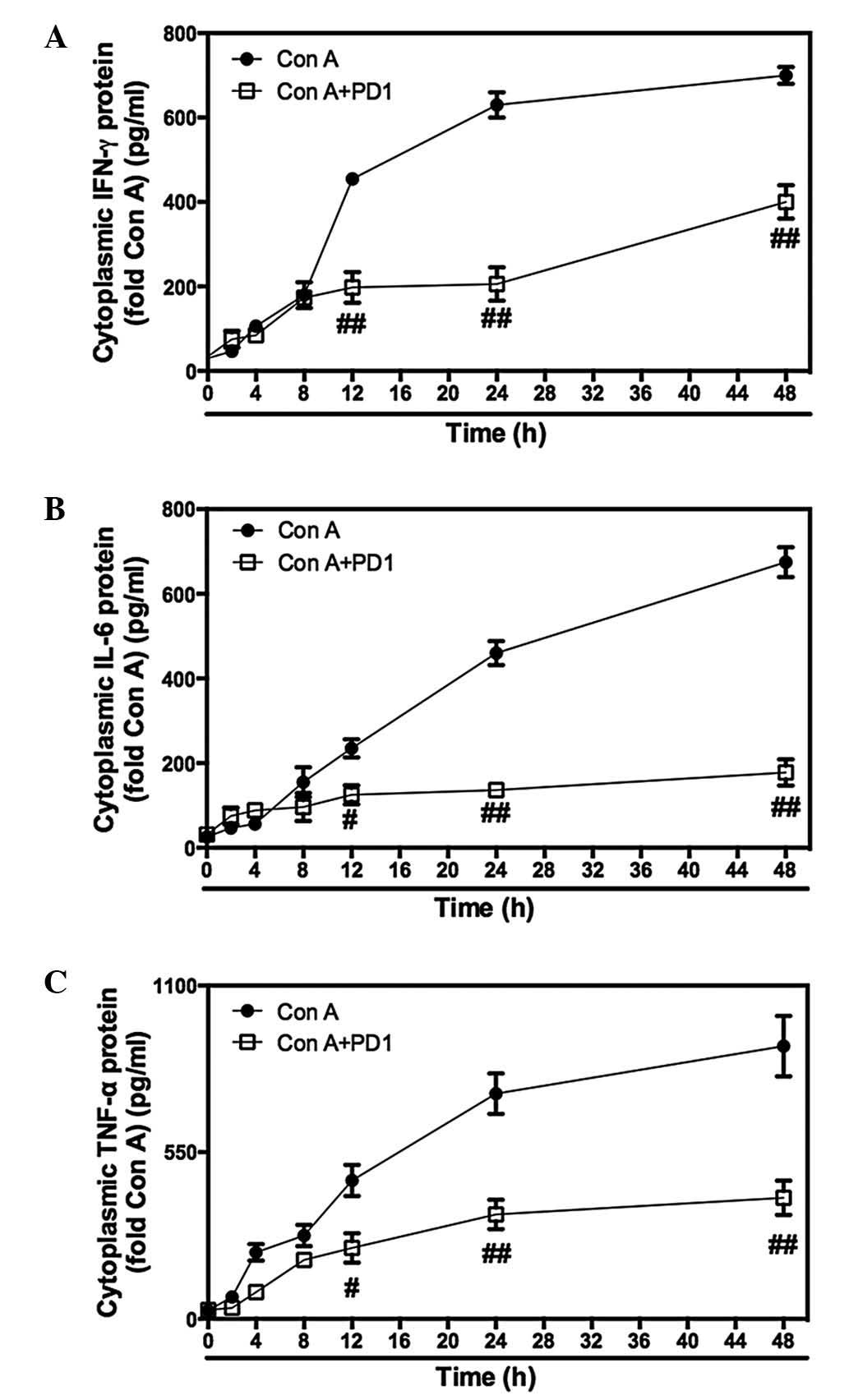
Effect of PD1 on serum transaminase
activity and pro-inflammatory cytokine secretion in Con A-induced
hepatitis
Serum ALT and AST levels significantly increased
following intravenous Con A administration. By contrast,
pretreatment with HPD1 and LPD1 markedly suppressed the serum
concentration of AST and ALT when compared with that of the Con A
group (Fig. 1A and B), indicating
that PD1 has a protective effect on Con A-induced hepatitis. The
association between pro-inflammatory cytokine secretion and the
inhibitory effect of PD1 was also investigated. Following Con A
injection, ELISA kits were used to measure the key pro-inflammatory
cytokines and analyze the inhibitory effect of PD1 on IL-1β, TNF-α,
IFN-γ, IL-6, CX3CL1 and HMGB1 expression levels. As expected, the
results demonstrated that PD1 downregulated the release and
production of inflammatory cytokines in a dose-dependent manner
(Fig. 1C–H). These results
indicate that PD1 inhibits the hepatitis induced by Con A i.v.
injection.
 | Figure 1PD1 inhibits Con A-induced liver
injury in mice. Following Con A administration (30 mg/kg) for 24 h,
the serum samples were obtained by cardiac puncture at different
time-points. (A) ALT and (B) AST serum levels were analyzed using
an Olympus AU1000 automated chemistry analyzer. (C) HMGB1 was
quantified via sandwich immunoassay. ELISA kits were used to
examine levels of (D) CX3CL1, (E) IL-1β, (F) IFN-γ, (G) IL-6 and
(H) TNF-α in the serum. The bars indicate the mean ± standard error
of the mean (n=9). *P<0.05, **P<0.01,
***P<0.001 vs. Con A (30 mg/kg). PD1, protectin D1;
Con A, con-canavalin A; ALT, alanine transaminase; AST, aspartate
transaminase; HMGB1, high mobility group B1; CX3CL1, chemokine
(C-X3-C motif) ligand 1; IL, interleukin; IFN-γ, interferon-γ;
TNF-α, tumor necrosis factor-α; HPD1, 20 µg/kg PD1
pretreatment group; LPD1, 10 µg/kg PD1 pretreatment
group. |
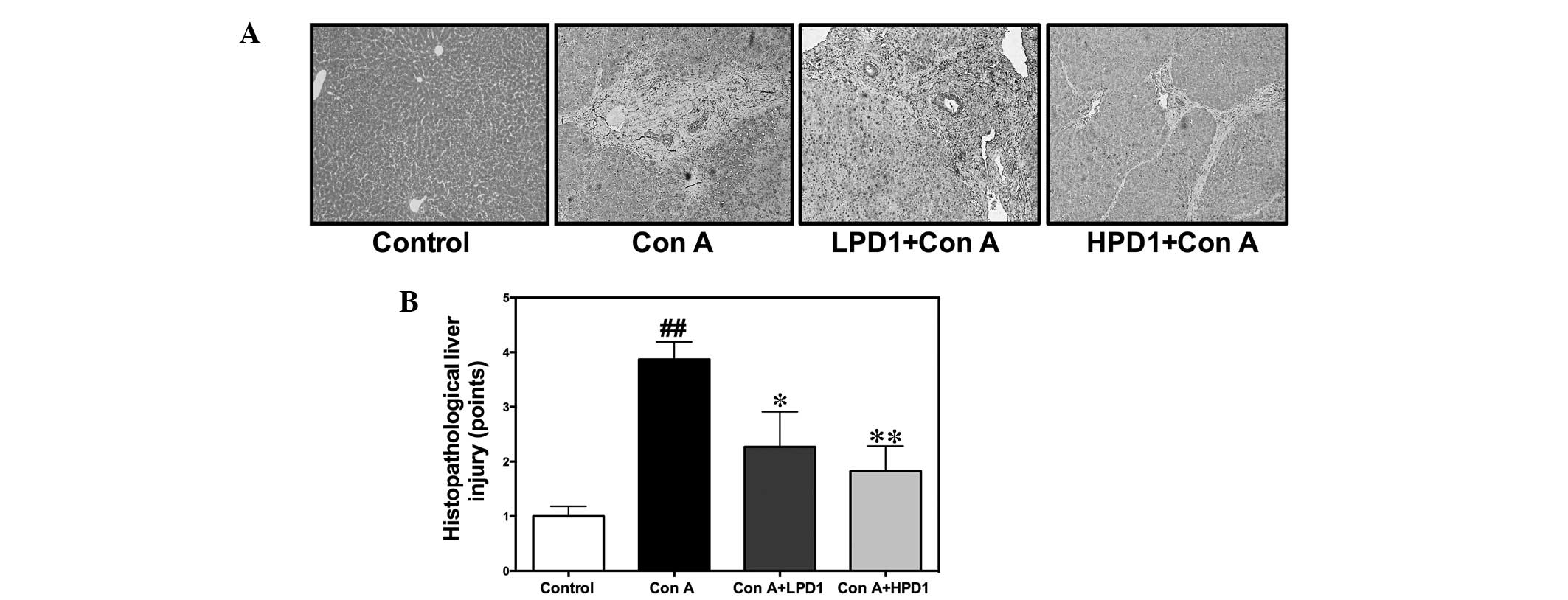
PD1 attenuates Con A-induced liver injury
in mice
To further confirm the protective effect of PD1 on
liver damage, hematoxylin and eosin staining of the liver was
performed to evaluate whether PD1 inhibits pathological
development. Following i.v. Con A administration, marked hepatocyte
necrosis was observed in the Con A group. Four pathologists blinded
to the experimental conditions evaluated the sections, indicating
that the Con A group induced marked bridging necrosis, which was
observed under a light microscope (Fig. 2A). Furthermore, the pathological
scores in the PD1 pretreatment groups were lower than that of the
Con A group (Fig. 2B).
Effect of PD1 on inflammation-associated
gene expression in Con A-induced hepatitis
Pro-inflammatory cytokines and inflammatory
signaling pathways are important in Con A-induced hepatitis.
Results from the present study demonstrated that in PD1
pretreatment groups, including LPD1 and HPD1, the upregulated
messenger (m)RNA expression levels of TNF-α, NF-κB p65, IL-6,
IL-1β, IFN-γ, IκBα, IKKβ, HMGB1, CX3CL1 and CX3CR1 in response to
Con A were attenuated by pretreatment with PD1 (Fig. 3). Hence, these results suggest that
the NF-κB-mediated CX3CL1/CX3CR1 pathway was associated (directly
or indirectly) with the development of inflammation in Con
A-induced liver injury.
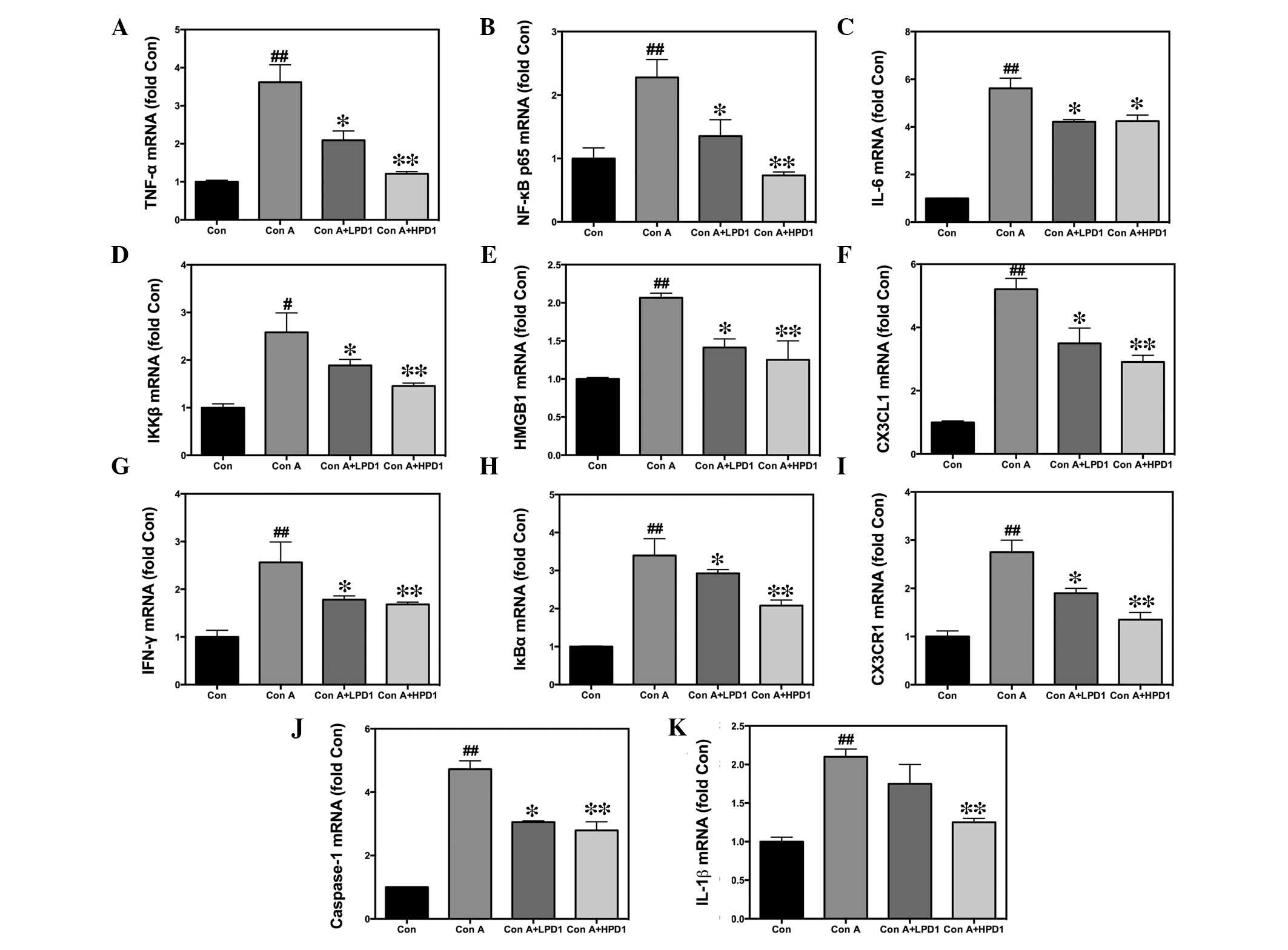 | Figure 3Effects of PD1 on inflammatory gene
expression in Con A-induced hepatitis. Livers were removed and RNA
was extracted using TRIzol. Quantification of mRNA expression
levels of (A) TNF-α, (B) NF-κB p65, (C) IL-6, (D) IKKβ, (E) HMGB1,
(F) CX3CL1, (G) IFN-γ, (H) IκBα, (I) CX3CR1, (J) caspase-1 and (K)
IL-1β in Con A-induced liver tissue. The bars indicate the mean ±
standard error of the mean (n=10). #P<0.05,
##P<0.01 vs. control group; *P<0.05,
**P<0.01 vs. Con A (30 mg/kg). PD1, protectin D1; Con
A, concanavalin A; HPD1, 20 µg/kg PD1 pretreatment group;
LPD1, 10 µg/kg PD1 pretreatment group; TNF-α, tumor necrosis
factor-α; NF-κB p65, nuclear factor κ-light-chain-enhancer of
activated B cells p65 subunit; IL, interleukin; IKKβ, inhibitor of
nuclear factor κ-B kinase subunit β; HMGB1, high mobility group B1;
CX3CL1, chemokine (C-X3-C motif) ligand 1; IFN-γ, interferon-γ;
IκBα, nuclear factor of κ light polypeptide gene enhancer in
B-cells inhibitor, α; CX3CR1, chemokine (C-X3-C motif) receptor 1;
mRNA, messenger RNA. |
Pretreatment with PD1 inhibited
CD4+, CD8+ and NKT cell infiltration in mouse
livers
In the present study, the percentage of
CD4+ and CD8+ T cells in the liver was
assessed by flow cytometry. As expected, compared with the Con A
group, the percentage of infiltrating CD4+ and
CD8+ T cells was significantly decreased in the liver
tissue samples (Fig. 4A and B).
The percentage of NKT cells in the liver tissue samples was also
analyzed and the data demonstrated that the absolute quantities of
infiltrating NKT cells in the liver were significantly upregulated
in the Con A group, compared with mice in the PD1 treatment groups
(Fig. 4C). These results suggest
that PD1 markedly suppressed the recruitment of CD4+,
CD8+ and NKT cells into the liver in Con A-induced liver
injury.
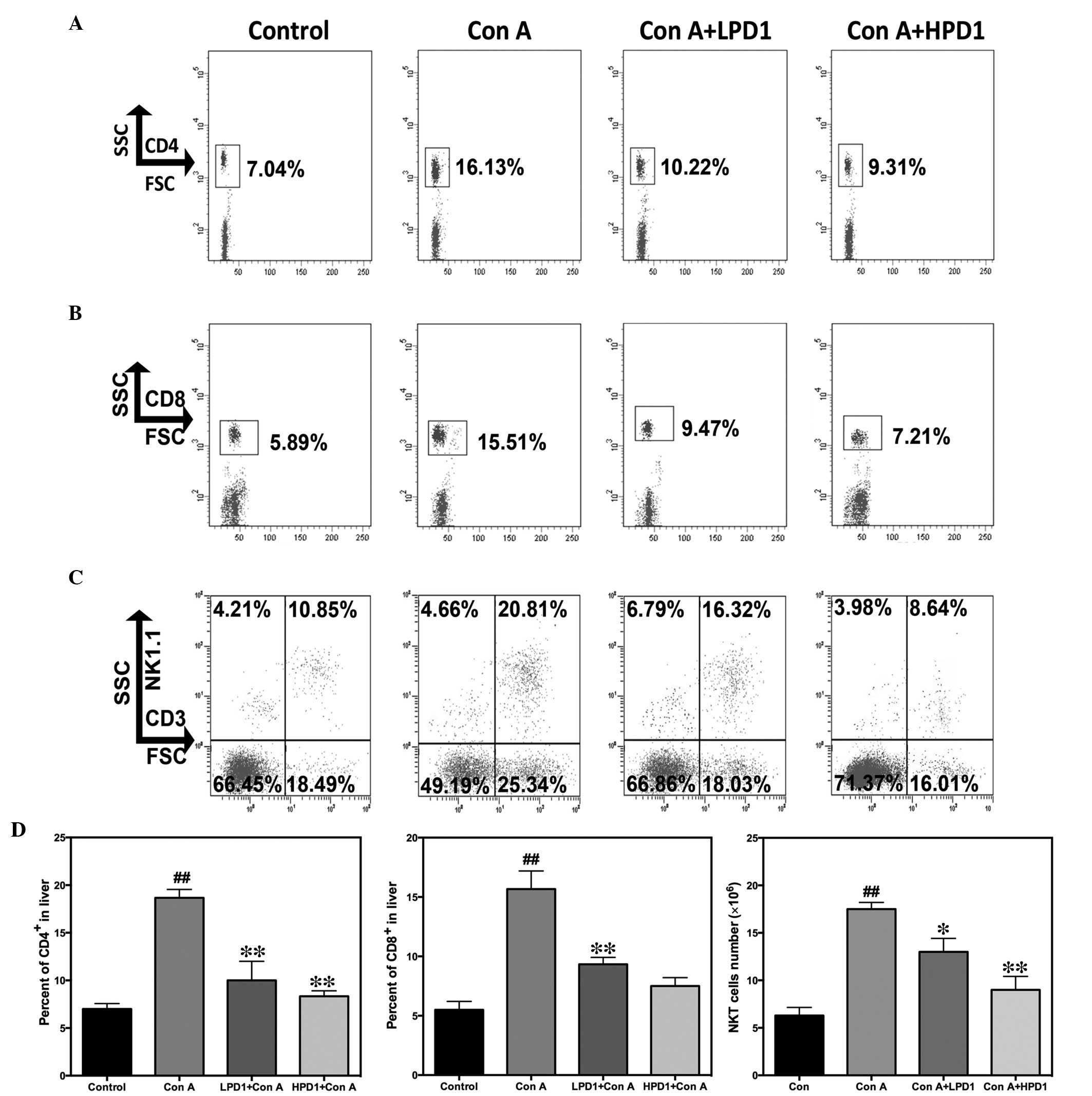 | Figure 4Pretreatment of PD1 inhibited
CD4+, CD8+ and NKT cells infiltrating the
liver, as demonstrated by flow cytometric analysis of hepatic
lymphocytes following 24 h Con A (30 mg/kg) administration. The T
cells were stained with allophycocyanin- or
phycoerythrin-conjugated monoclonal antibodies. Flow cytometric
analysis of (A) CD4+ T cells, (B) CD8+ T
cells and (C) NKT cells. (D) The percentages of CD4+ and
CD8+ T cells were counted and the absolute number of
infiltrated NKT cells was calculated by multiplying the total
number of hepatic mononuclear cells with the percentage of NKT
cells. The bars indicate the mean ± standard error of the mean
(n=10). ##P<0.01 vs. control group;
*P<0.05, **P<0.01 vs. Con A. CD,
cluster of differentiation; NKT, natural killer T cells; Con A,
concanavalin A; PD1, protectin D1; HPD1, 20 µg/kg PD1
pretreatment group; LPD1, 10 µg/kg PD1 pretreatment group;
SSC, side scatter; FSC, forward scatter; NK1.1, killer cell
lectin-like receptor subfamily B, member 1. |
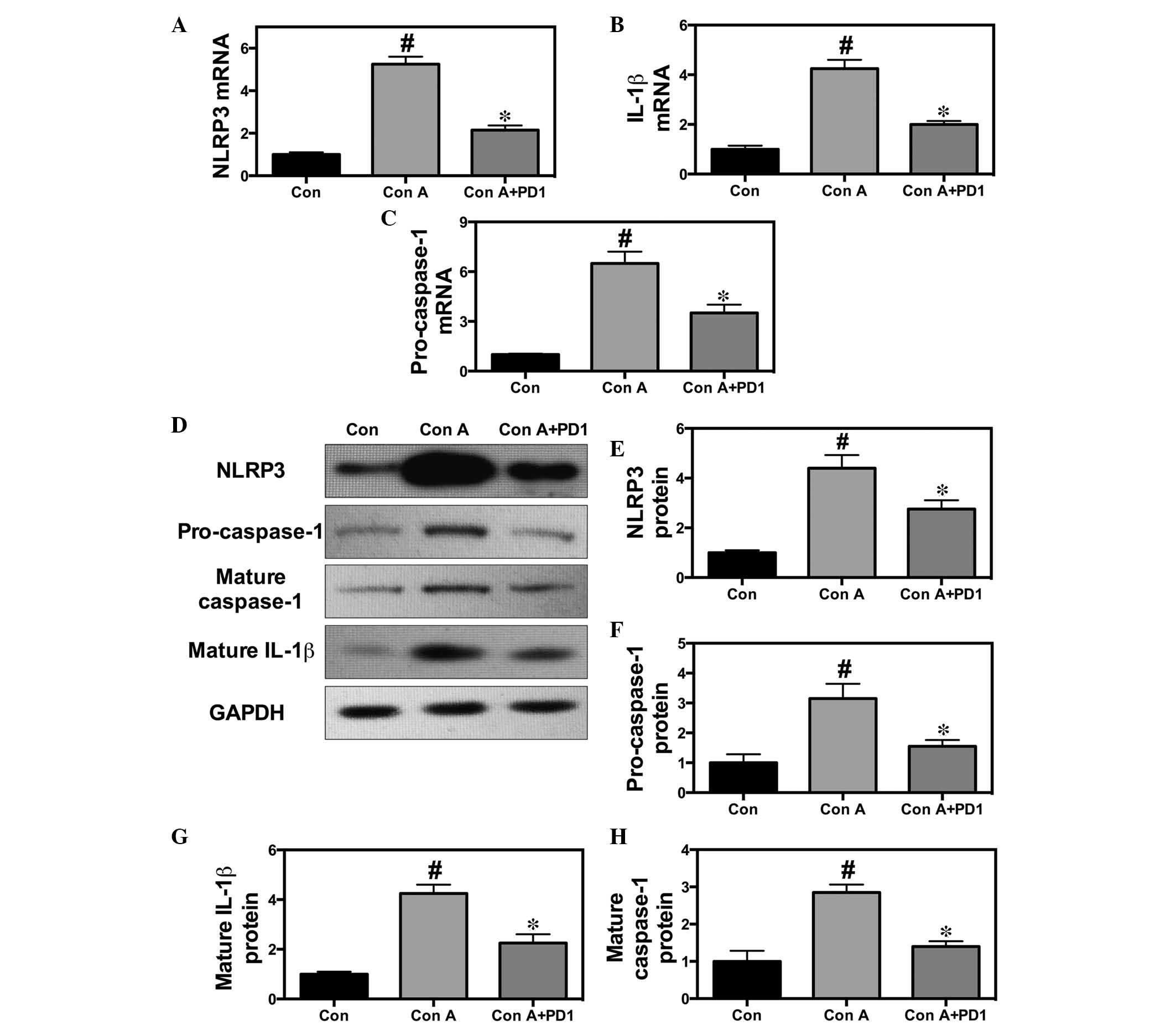
PD1 suppresses inflammatory signaling
pathways
The NF-κB-mediated CX3CL1/CX3CR1 signaling pathway
has been reported to be important in hepatitis and associated with
inflammatory responses. Hence, the present study investigated
whether PD1 inhibits the CX3CL1/CX3CR1 and the TLR4/MyD88/NF-κB
signaling pathways using RT-qPCR. PD1 significantly downregulated
the expression of CX3CL1 and CX3CR1 proteins, and NF-κB
phosphorylation. In addition, the IL-1β, NLRP3 and HMGB1 protein
expression levels were suppressed by PD1 treatment, which indicates
that NLRP3 and HMGB1 are involved in the development of liver
injury (Fig. 5).
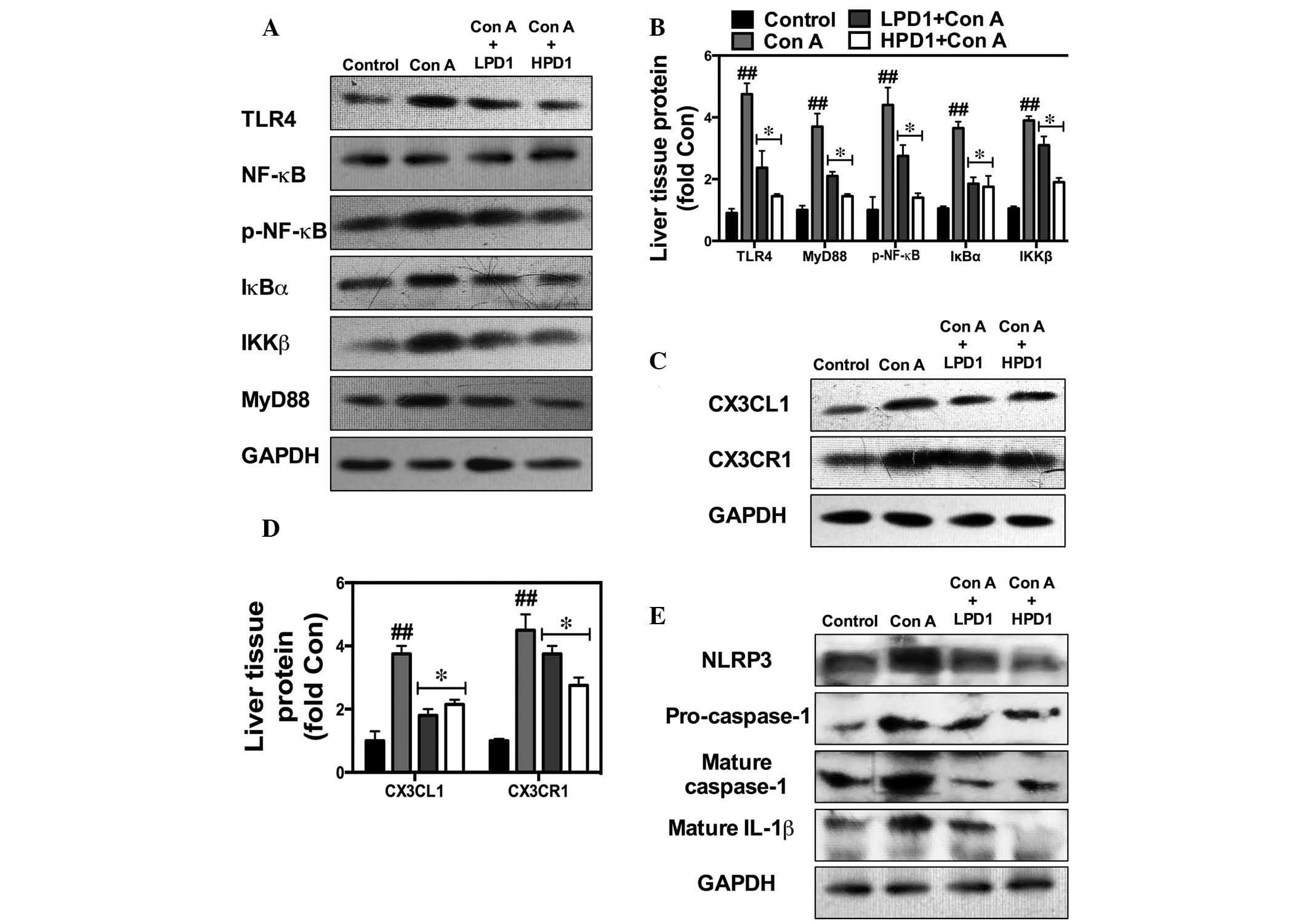 | Figure 5PD1 inhibits NF-κB-stimulated
CX3CL1/CX3CR1-dependent inflammatory pathway activation and
generation of the inflammasome. Western blot analysis for NF-κB and
CX3CL1/CX3CR1 activity was used to demonstrate the effect of PD1 on
Con A-induced liver injury and the underlying mechanisms. (A and B)
Bands and quantification of TLR4, p-NF-κB, IκBα, IKKβ and MyD88
protein. (C and D) Bands and quantification of CX3CL1 and CX3CR1
protein. (E) Bands and quantification of NLRP3 and caspase-1
protein. The bars indicate means ± standard error of the mean
(n=10). ##P<0.01 vs. control group;
*P<0.05 vs. Con A (30 mg/kg). PD1, protectin D1; Con
A, concanavalin A; HPD1, 20 µg/kg PD1 pretreatment group;
LPD1, 10 µg/kg PD1 pretreatment group; TLR4, Toll-like
receptor 4; NF-κB, nuclear factor κ-light-chain-enhancer of
activated B cells; p-NF-κB, phosphorylated NF-κB; IκBα, nuclear
factor of κ light polypeptide gene enhancer in B-cells inhibitor,
α; IKKβ, inhibitor of nuclear factor κ-B kinase subunit β; MyD88,
myeloid differentiation primary response gene 88; CX3CL1, chemokine
(C-X3-C motif) ligand 1; CX3CR1, chemokine (C-X3-C motif) receptor
1; NLRP3, NLR family, pyrin domain containing 3; IL-1β,
interleukin-1β. |
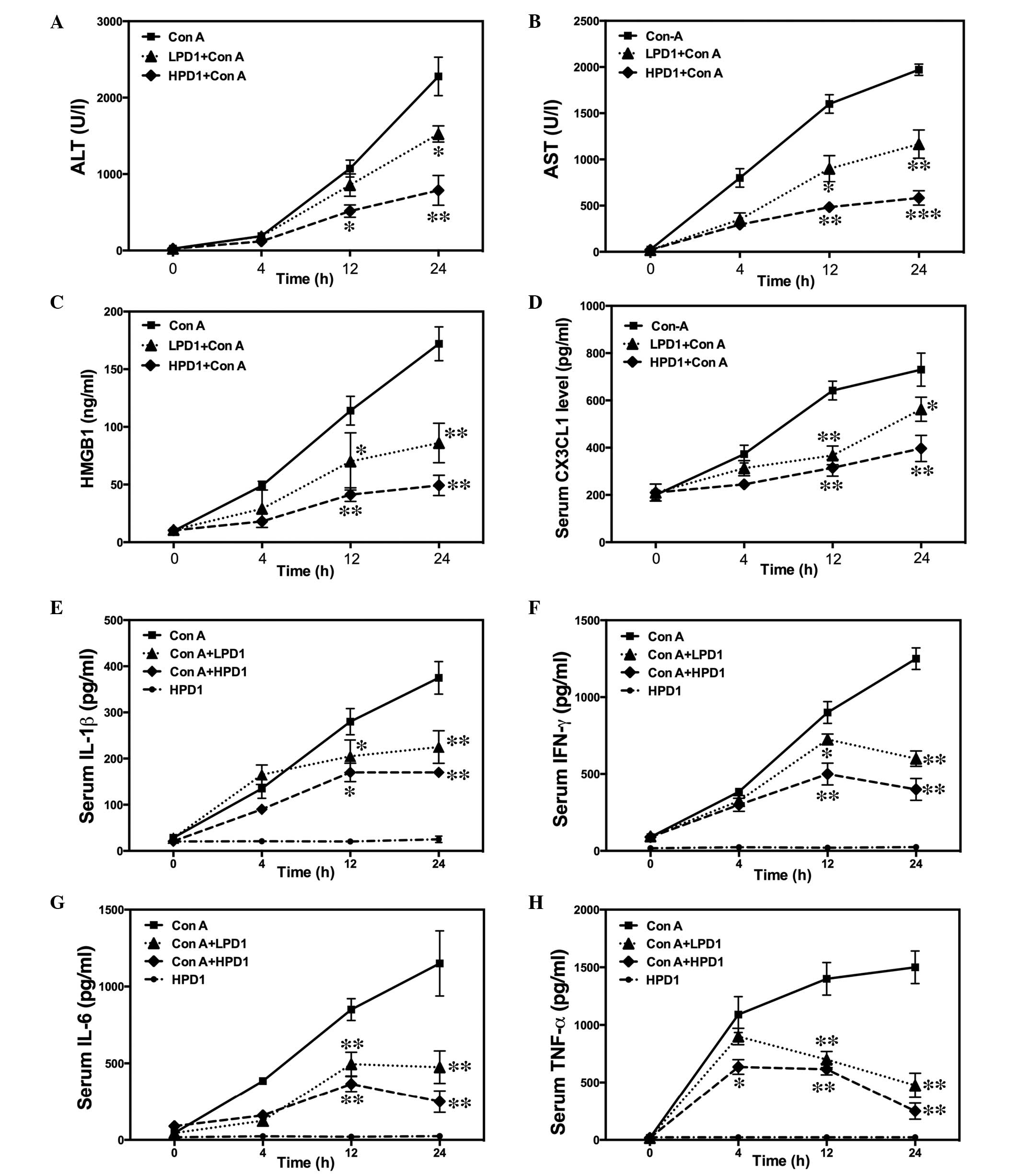
PD1 inhibited T lymphocyte proliferation
and inflammatory signaling pathways
Various concentrations of PD1 (0, 2.5, 5, 10 and 20
nM) were investigated to assess the inhibition of proliferation in
Con A-stimulated T lymphocytes. As presented in Fig. 6A, PD1 pretreatment markedly
inhibited cell proliferation compared with the Con A group.
Furthermore, it was observed that 20 nM PD1 affects cell viability
in a time-dependent manner (Fig.
6B). Evaluation of cell cycle progression indicated that Con A
improved cell cycle progression in T cells, as fewer cells were
observed in the G0/G1 phase and increasing
cells were arrested in the S and G2/M phase; this effect
was partially suppressed by pretreatment with PD1 (Fig. 6C). Subsequently, the inhibitory
effect of PD1 on inflammatory cytokines was investigated and
RT-qPCR indicated that treatment of PD1 significantly downregulated
cytokine expression levels, including TNF-α, IFN-γ and IL-6
(Fig. 6D–F and Fig. 7). Furthermore, PD1 treatment was
observed to inhibit NF-κB/CX3CL1/CX3CR1 activation; IP results
indicated that PD1 decreased the formation of NF-κB/IκBα,
CX3CL1/CX3CR1 and TLR4/MyD88 complexes (Fig. 8). Finally, the inhibitory effect of
PD1 on the NLRP3 signaling pathway was investigated. The data
indicated that treatment with PD1 significantly suppressed IL-1β,
caspase-1 and NLRP3 mRNA and protein expression levels (Fig. 9).
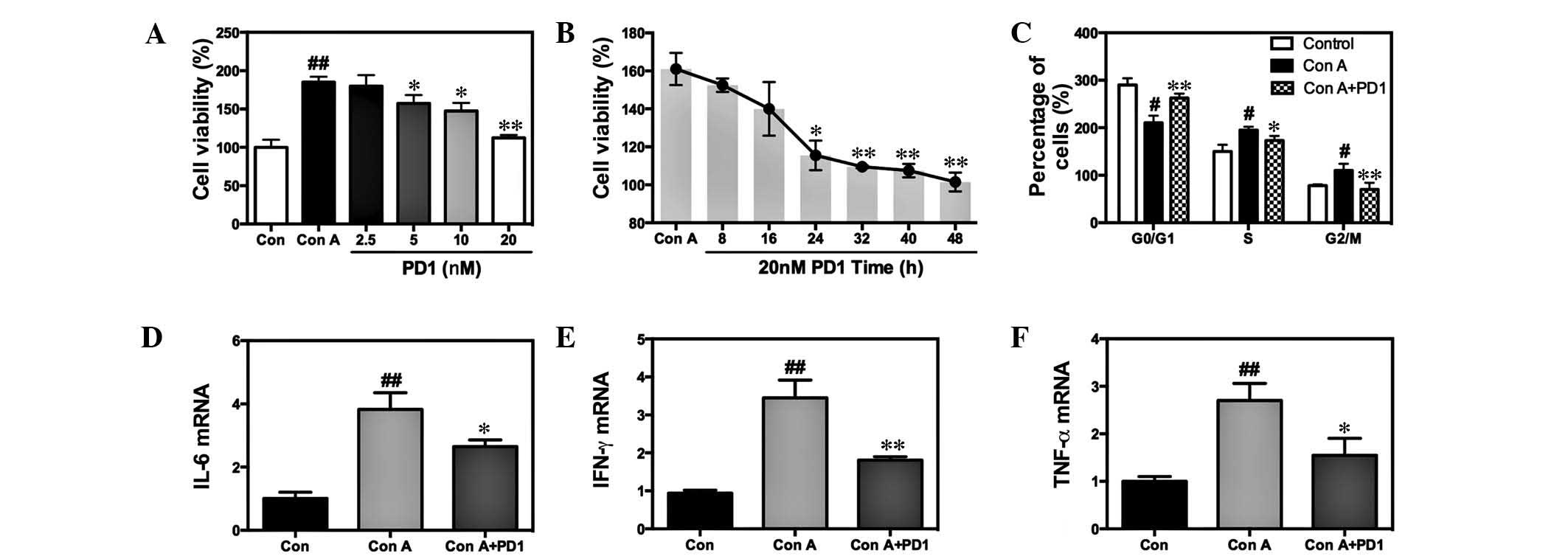 | Figure 6Effects of PD1 on the proliferation
of Con A-induced T lymphocytes. (A) T lymphocytes were treated with
or without 20 µg/ml Con A in the presence of 0, 2.5, 5, 10
and 20 nM PD1 for 48 h. After the PD1 administration, cell
viability was investigated using MTT assay to evaluate the
influence of PD1 on pro-liferation. (B) MTT assay examining cell
viability in the condition of Con A+20 nM PD1 treatment. (C) Cell
cycles were examined by flow cytometry. Reverse quantitative
transcription-polymerase chain reaction demonstrated the mRNA
expression levels of (D) IL-6, (E) IFN-γ and (F) TNF-α. The bars
indicate the mean ± standard error of the mean (n=10).
#P<0.05, ##P< 0.01 vs. control group.
*P< 0.05, **P< 0.01 vs. Con A. PD1,
protectin D1; Con A, concanavalin A; IL-6, interleukin-6; TNF-α,
tumor necrosis factor-α; IFN-γ, interferon-γ; mRNA, messenger
RNA. |
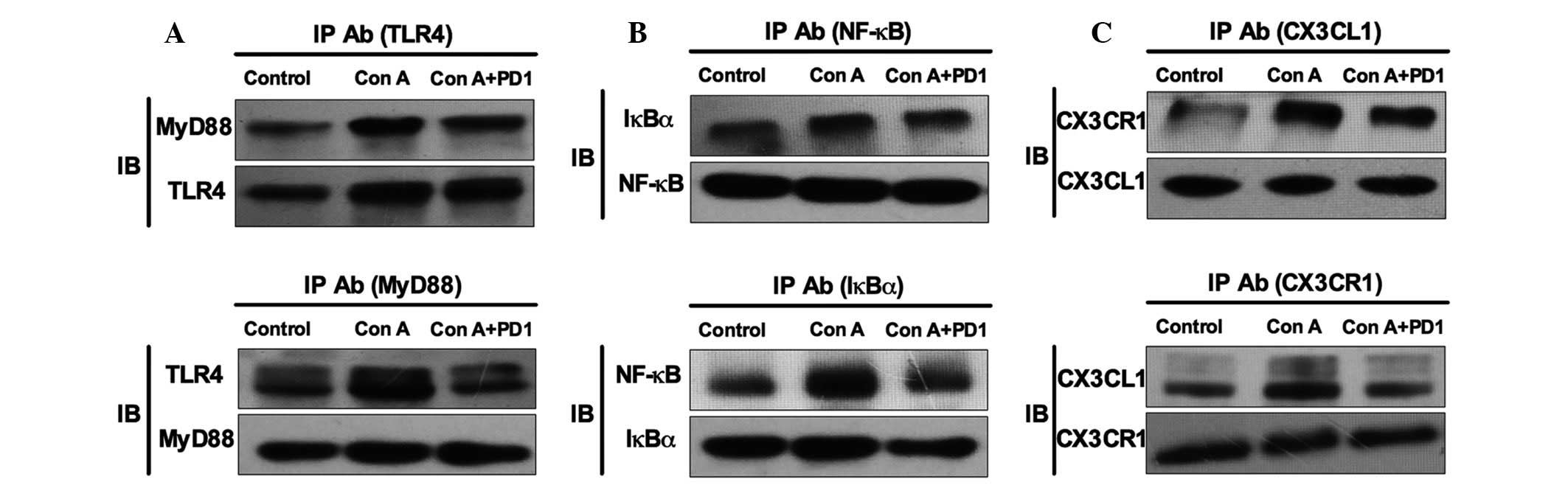 | Figure 8Effects of Con A (20 µg/ml)
and PD1 (20 nM) on NF-κB activated CX3CL1/CX3CR1 signaling pathway.
IP was used to investigate (A) TLR4/MyD88, (B) NF-κB/IκBα and (C)
CX3CL1/CX3CR1 complex formation. The bars indicate the mean ±
standard error of the mean. (n=10). IP, immunoprecipitation; Ab,
antibody; PD1, protectin D1; Con A, concanavalin A; TLR4, Toll-like
receptor 4; MyD88, myeloid differentiation primary response gene
88; NF-κB, nuclear factor κ-light-chain-enhancer of activated B
cells; IκBα, nuclear factor of κ light polypeptide gene enhancer in
B-cells inhibitor; CX3CL1, che-mokine (C-X3-C motif) ligand 1;
CX3CR1, chemokine (C-X3-C motif) receptor 1. |
Discussion
Hepatitis is a serious global health problem
associated with high mortality and infection rates (1,2).
Therapeutic strategies that target the underlying mechanisms of
liver injury are required, particularly in viral and autoimmune
hepatitis (2,3). Despite advances in medical science
and research regarding the pathogenesis of hepatitis, the search
for effective therapeutic strategies remains a major challenge.
Hence, elucidation of the physiological and genetic mechanisms of
liver damage may aid with the development of effective therapeutic
agents. Thus, in the current study, the protective effect of PD1 on
Con A-induced hepatitis was investigated. Previous studies have
indicated that PD1 exerts a marked anti-inflammatory effect in
various disorders, such as acute kidney injury, neurodegenerative
diseases and acute lung injury (18,19).
In the present study, pretreatment with PD1 demonstrated a
potential protective effect against Con A-induced hepatitis as
levels of ALT and AST were downregulated, as was the severity of
hepatic necrosis. Notably, the underlying mechanisms of PD1 may be
associated with the suppression of inflammatory signaling pathways
and lymphocyte infiltration by regulating the cell cycle and
cycle-related protein expression. Previous studies in a mouse model
of Con A-induced hepatitis demonstrated that TNF-α, IL-6 and IFN-γ
were significant. Kato et al (23) demonstrated that TNF-α, IFN-γ and
IL-6 are, directly or indirectly, involved in the regulation of
inflammatory cytokine release, as IFN−/− and
TNF−/− mice were free from liver injury
following administration of Con A. Therefore, the current study
investigated the expression of major inflammatory cytokines, such
as TNF-α, IFN-γ, IL-6 and HMGB1 in Con A-induced hepatitis. PD1
pretreatment suppressed the release of pro-inflammatory cytokines,
including TNF-α, IFN-γ, IL-6 and HMGB1 production, as well as their
mRNA expression in the liver and T cells. In addition,
CD4+ and CD8+ infiltrating T lymphocytes are
involved in the development of Con A-induced hepatitis (18–20).
In the present study, PD1 treatment markedly inhibited
CD4+, CD8+ and NKT cell infiltration in the
liver as compared with the Con A group. These results indicated
that PD1 exerted a protective effect, partly dependent on the
inhibition of CD4+, CD8+ and NKT cells,
against liver injury. Furthermore, NK cells are a class of
lymphocyte distinct from T and B cells, and are the predominant
cells involved in autoimmune disorders or viral infections and may
result in liver damage by destroying foreign cells (for example,
neoplastic cells) and contribute to the progression of inflammation
(20,21,24).
In the present study, in vitro studies with isolates of T
lymphocytes were performed to assess whether treatment with PD1
inhibits Con A-induced T lymphocyte proliferation in inflammation
and to determine the underlying mechanisms by which it may restore
inflammatory responses. The results indicated that different
concentrations of PD1 (particularly 20 nM) significantly suppressed
lymphocyte proliferation in a time-dependent manner (0–48 h).
Notably, PD1 may be important in cell cycle regulation.
CX3CL1 (also referred to as FKN) is a major CX3C
chemokine, which binds to its receptor, CX3CR1 and has been
associated with the development of inflammation (15). There are two different forms of
CX3CL1, the membrane-anchored form and the soluble form, and CX3CL1
may be induced by pro-inflammatory cytokines, TNF-α and IFN-γ.
TNF-α and IFN-γ upregulate the expression of CX3CL1 and CX3CR1 in
various different types of cells via NF-κB activation. In addition,
the soluble form of CX3CL1, which is released by TNF-α or IFN-γ
changing enzyme, is a potent chemoattractant for recruiting
monocytes/macrophages, T cells or NK cells. The present study
demonstrated that CX3CL1 is involved (directly or indirectly) in
the development of inflammation and liver injury in Con A-induced
hepatitis. Furthermore, CX3CL1 is involved in the embryonic
development of the central nervous system and is key in T cell
development and activation (15).
Previous studies have demonstrated that the NF-κB signaling
pathway, and TNF-α and IFN-γ cytokines are involved in the
development of Con A-induced liver injury (20,21).
Thus, the current study investigated whether NF-κB induced CX3CL1
expression via TNF-α- or IFN-γ-stimulated inflammatory responses.
To determine the role of the CX3CL1/CX3CR1 axis in Con A-induced
hepatitis, Con A was used to simulate AIH in a mouse model. The
results indicated that CX3CL1 expression and NF-κB activation were
significantly inhibited at high and low doses of PD1. Furthermore,
the ELISA results demonstrated that soluble CX3CL1 in the serum was
downregulated by PD1 administration. In addition, the present study
investigated the inhibitory effect of PD1 on Con A-induced T
lymphocyte inflammatory responses in vitro. Experimental
data demonstrated that PD1 markedly inhibited Con A-induced
inflammatory cytokine expression via suppression of NF-κB
activation and CX3CL1/CX3CR1 pathways at different time-points. The
results of the IP analysis indicated that the CX3CL1/CX3CR1 axis
was involved in the progression of Con A-induced inflammation, and
that the formation of CX3CL1/CX3CR1 complexes was markedly
inhibited by PD1. These results were consistent with previous
studies, which demonstrated that PD1 blocks the Con A-induced liver
damage by inhibiting NF-κB-stimulated CX3CL1/CX3CR1-dependent
inflammatory signals (10–13).
NLRP3 is a major component of the macromolecular
complex that triggers caspase-1-dependent maturation of IL-1β and
IL-18 cytokine precursors. It has been indicated that the NLRP3
inflammasome is activated by various cellular signals and directly
controls collagen synthesis, resulting in increased deposition of
collagens in the tissues, such as the lungs, liver, heart and skin
(22). Notably, Con A-activated
NF-κB promotes synthesis of pro-IL-1β, IL-18 and NLRP3 proteins to
further promote the inflammatory response (24). Therefore, the role of NLRP3 in the
pathological process of Con A-induced liver damage was
investigated. Data from the present study demonstrated that PD1
downregulated NLRP3 expression in the Con A-induced damaged liver.
In addition, the protein and mRNA expression levels of caspase-1
increased. Thus, pretreatment with PD1 suppresses Con A-induced
hepatitis and may indirectly contribute to the inhibition of NLRP3
expression, as well as trigger caspase-1.
In conclusion, the current study demonstrates that
PD1 suppressed hepatitis by directly or indirectly inhibiting
TLR4/NF-κB-stimulated CX3CL1/CX3CR1-dependent inflammatory signals
and the NLRP3 inflammasome. TLR4 was implicated in the inflammatory
response to Con A exposure, in which CX3CL1/CX3CR1-activation is
important in the sustained production of pro-inflammatory mediators
by regulating nuclear NF-κB-dependent transcription. These results
provide novel insight into the molecular mechanisms that link viral
and autoimmune hepatitis to inflammation in liver injury that is
caused by various factors. PD1 is a bioactive product generated
from DHA and has been demonstrated to suppress Con A-induced
hepatitis and inflammatory responses by inhibiting, in part, the
NF-κB-stimulated CX3CL1/CX3CR1 pathway, the NLRP3 inflammasome,
lymphocyte proliferation and infiltration of CD4+,
CD8+ and NKT cells to the liver. Therefore, inhibition
of inflammation by PD1 may provide a potential therapeutic strategy
to recover Con A-induced liver injury.
Acknowledgments
The present study was supported by research grants
from the National Natural Science Foundation of China (grant no.
813000142) and the Health Research Programs of Jilin Province
(grant no. 2013Z051).
References
|
1
|
American Gastroenterological: AGA
Institute guidelines on hepatitis B reactivation (HBVr): Clinical
decision support tool. Gastroenterology. 148:2202015. View Article : Google Scholar
|
|
2
|
Lee H, Ainechi S, Dresser K and Kurian EM:
Central portalization correlates with fibrosis but not with risk
factors for nonalcoholic steatohepatitis in steatotic chronic
hepatitis C. Int J Hepatol. 2014:3292972014.PubMed/NCBI
|
|
3
|
Gerlich WH: Prophylactic vaccination
against hepatitis B: Achievements, challenges and perspectives. Med
Microbiol Immunol. 204:39–55. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hardtke-Wolenski M, Taubert R, Noyan F,
Sievers M, Dywicki J, Schlue J, Falk CS, Ardesjö Lundgren B, Scott
HS, Pich A, et al: Autoimmune hepatitis in a murine autoimmune
polyendocrine syndrome type 1 model is directed against multiple
autoantigens. Hepatology. 61:1295–1305. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hart J, Ehlken H, Weiler-Normann C, Sebode
M, Kreuels B, Pannicke N, Zenouzi R, Glaubke C, Lohse AW and
Schramm C: Patient selection based on treatment duration and liver
biochemistry increases success rates after treatment withdrawal in
autoimmune hepatitis. J Hepatol. 62:642–646. 2015. View Article : Google Scholar
|
|
6
|
Lee JH, Won JH, Choi JM, Cha HH, Jang YJ,
Park S, Kim HG, Kim HC and Kim DK: Protective effect of ellagic
acid on concanavalin A-induced hepatitis via toll-like receptor and
mitogen-activated protein kinase/nuclear factor κB signaling
pathways. J Agric Food Chem. 62:10110–10117. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tu CT, Han B, Yao QY, Zhang YA, Liu HC and
Zhang SC: Curcumin attenuates concanavalin A-induced liver injury
in mice by inhibition of Toll-like receptor (TLR) 2, TLR4 and TLR9
expression. Int Immunopharmacol. 12:151–157. 2012. View Article : Google Scholar
|
|
8
|
Chen YF, Wang SH, Chang SJ, Shiau AL, Her
LS, Shieh GS, Chen CF, Chang CC, Su YC, Wu CL and Wu TS: Zhankuic
acid A as a novel JAK2 inhibitor for the treatment of concanavalin
A-induced hepatitis. Biochem Pharmacol. 91:217–230. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Crispe IN: Hepatic T cells and liver
tolerance. Nat Rev Immunol. 3:51–62. 3002. View Article : Google Scholar
|
|
10
|
Panek CA, Ramos MV, Mejias MP,
Abrey-Recalde MJ, Fernandez-Brando RJ, Gori MS, Salamone GV and
Palermo MS: Differential expression of the fractalkine chemokine
receptor (CXCR1) in human monocytes during differentiation. Cell
Mol Immunol. 12:669–680. 2015. View Article : Google Scholar
|
|
11
|
Isse K, Harada K, Zen Y, Kamihira T,
Shimoda S, Harada M and Nakanuma Y: Fractalkine and CX3CR1 are
involved in the recruitment of intraepithelial lymphocytes of
intrahepatic bile ducts. Hepatology. 41:506–516. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ridderstad Wollberg A, Ericsson-Dahlstrand
A, Juréus A, Ekerot P, Simon S, Nilsson M, Wiklund SJ, Berg AL,
Ferm M, Sunnemark D and Johansson R: Pharmacological inhibition of
the chemokine receptor CX3CR1 attenuates disease in a
chronic-relapsing rat model for multiple sclerosis. Proc Natl Acad
Sci USA. 111:5409–5414. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pirvulescu MM, Gan AM, Stan D, Simion V,
Calin M, Butoi E and Manduteanu I: Subendothelial resistin enhances
monocyte transmigration in a co-culture of human endothelial and
smooth muscle cells by mechanisms involving fractalkine, MCP-1 and
activation of TLR4 and Gi/o proteins signaling. Int J Biochem Cell
Biol. 50:29–37. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bourd-Boittin K, Basset L, Bonnier D,
L'helgoualc'h A, Samson M and Théret N: CX3CL1/fractalkine shedding
by human hepatic stellate cells: contribution to chronic
inflammation in the liver. J Cell Mol Med. 13:1526–1535. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Efsen E, Grappone C, DeFranco RM, Milani
S, Romanelli RG, Bonacchi A, Caliguri A, Failli P, Annunziato F,
Pagliai G, et al: Up-regulated expression of fractalkine and its
receptor CX3CR1 during liver injury in humans. J Hepatol. 37:39–47.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aoyama T, Inokuchi S, Brenner DA and Seki
E: CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced
liver inflammation and fibrosis in mice. Hepatology. 52:1390–1400.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu ZZ, Liu XJ, Berta T, Park CK, Lü N,
Serhan CN and Ji RR: Neuroprotectin/protectin D1 protects against
neuropathic pain in mice after nerve trauma. Ann Neurol.
74:490–495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li X, Li C, Liang W, Bi Y, Chen M and Dong
S: Protectin D1 promotes resolution of inflammation in a murine
model of lipopolysaccharide-induced acute lung injury via enhancing
neutrophil apoptosis. Chin Med J (Engl). 127:810–814. 2014.
|
|
19
|
Duffield JS, Hong S, Vaidya VS, Lu Y,
Fredman G, Serhan CN and Bonventre JV: Resolvin D series and
protectin D1 mitigate acute kidney injury. J Immunol.
177:5902–5911. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yan Y, Jiang W, Spinetti T, Tardivel A,
Castillo R, Bourquin C, Guarda G, Tian Z, Tschopp J and Zhou R:
Omega-3 fatty acids prevent inflammation and metabolic disorder
through inhibition of NLRP3 inflammasome activation. Immunity.
38:1154–1163. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tsutsui H, Imamura M, Fujimoto J and
Nakanishi K: The TLR4/TRIF-mediated activation of NLRP3
inflammasome underlies endotoxin-induced liver injury in mice.
Gastroenterol Res Pract. 2010:6418652010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu J, Wang J, Sheng Y, Zou Y, Bo L, Wang
F, Lou J, Fan X, Bao R, Wu Y, et al: Baicalin improves survival in
a murine model of polymicrobial sepsis via suppressing inflammatory
response and lymphocyte apoptosis. PLoS One. 7:e355232012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kato N, Matsumoto M, Kogawa M, Atkins GJ,
Findlay DM, Fujikawa T, Oda H and Ogata M: Critical role of p38
MAPK for regeneration of the sciatic nerve following crush injury
in vivo. J Neuroinflam. 10:Jan 3–2013. View Article : Google Scholar
|
|
24
|
DeSantis DA, Ko CW, Liu Y, Liu X, Hise AG,
Nunez G and Croniger CM: Alcohol-induced liver injury is modulated
by Nlrp3 and Nlrc4 inflammasomes in mice. Mediators Inflamm.
2013:7513742013. View Article : Google Scholar
|