Introduction
The mammalian target of rapamycin (mTOR) is a large
protein kinase of the phosphatidylinositol 3-kinase (PI3K)-related
kinase family (1). There are two
conserved TOR cell complexes: TOR complex (TORC)1 and TORC2
(2). Upstream and downstream
effectors of TOR have been identified using cultured mammalian
cells. Upstream, multiple signals exist, including insulin
signaling through PI3K and AKT, and energy signaling [via the
adenosine triphosphate:adenosine monophosphate (AMP) ratio] through
AMP-activated protein kinase. These signals converge in the
tuberous sclerosis (TSC)1-TSC2 complex, which serves as a GTPase
exchange factor for Rheb, whereas Rheb in GTP-bound form activates
mTOR through direct binding (3).
Downstream of mTOR, S6 kinase 1 (S6K1) and eukaryotic initiation
factor 4E-binding protein (4EBP1) are the two most studied
effectors, which are phosphorylated by mTORC1 but not mTORC2.
Phosphorylation of these two effectors, particularly S6K1
phosphorylation at threonine 389, has been widely used to indicate
mTORC1 activity. These effectors regulate translational initiation
and control protein synthesis (4).
The present study hypothesized that inhibiting mTOR
signaling may induce cellular stability, in which energy
consumption and cellular metabolism decrease. In this state,
damaged nuclear DNA is primarily repaired and mitochondrial
dysfunction is controlled. Subsequently, the cell death cascade is
suppressed. Therefore, mTOR inhibition-induced cell stability may
be considered promising protection against neurodegenerative
insults.
Neurotrophins exert diverse effects in the
development and regeneration of neural circuits in the vertebrate
visual system (5). In the retina,
neurotrophins influence proliferation, neurite outgrowth and cell
survival in the visual system in vitro and in vivo
(6). Neurotrophin availability is
critical for controlling normal cell death, since the majority of
retinal neurons depend on growth factors for their survival, and
cells may die when they lack adequate survival factors (6). In addition, neurotrophins rescue
photoreceptors from degeneration (7). The present study used serum
deprivation to mimic neurotrophin loss in retinal neurons, and
explored the neuroprotective mechanisms following suppression of
the mTOR pathway.
The 661W cell line was cloned from the retinal
tumors of a transgenic mouse line, and expresses simian virus 40T
antigen under the control of the human interphotoreceptor
retinol-binding protein promoter. These cells usually grow as a
monolayer and behave as photoreceptor cells, which express blue and
green cone pigments, transducin and cone arrestin, but not retinal
pigment epithelial cell-specific proteins. Furthermore, 661W cells
are sensitive to photooxidative stress, similar to normal retinal
photoreceptor cells (8).
The present study used the 661W cell line to
investigate the molecular mechanisms underlying serum
deprivation-induced cell death. In addition, the mTOR pathway was
blocked using a specific inhibitor, rapamycin. The results
demonstrated that inhibiting mTOR resulted in increased stability
of photoreceptor cells and cell cycle arrest at G2/M
stage. Furthermore, intracellular levels of reactive oxygen species
(ROS) and apoptotic markers were markedly decreased. Therefore,
inhibiting the mTOR pathway may have a neuroprotective effect
against serum deprivation-induced cell death.
Materials and methods
Chemicals and reagents
Cell culture media and additives were purchased from
Hyclone (GE Healthcare Life Sciences, Logan, UT, USA). Plastic
cultureware was obtained from Greiner Bio-One GmbH (Frickenhausen,
Germany). Rabbit antibodies against phosphorylated (p)-P70S6 kinase
(P70S6K) (cat. no. 11284), p-4EBP1 (cat. no. 11223) and mouse
β-actin (cat. no. 21800-1) were purchased from Signalway Antibody
LLC (College Park, MD, USA). Rabbit antibodies against p-mTOR (cat.
no. BS4706), heme oxygenase-1 (HO-1) (cat. no. BS6626), cyclin D1
(cat. no. BS6532) and cyclin D3 (cat. no. BS6139) were purchased
from Bioworld Technology, Inc. (St. Louis Park, MO, USA). Rabbit
antibodies against poly (ADP-ribose) polymerase 1 (PARP-1) (cat.
no. 9542), cleaved caspase-3 (cat. no. 9662) and cyclin D2 (cat.
no. 3741) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Goat anti-apoptosis inducing factor (AIF) (cat.
no. sc-9416) was obtained from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Rapamycin, dichloro-dihydro-fluorescein
diacetate (DCFH-DA), JC-1, MitoTracker Green and other reagents
were purchased from Sigma-Aldrich Shanghai Trading Co., Ltd.
(Shanghai, China).
Cell culture
The 661W photoreceptor cell line was generously
provided by Dr. Muayyad Al-Ubaidi (Department of Cell Biology,
University of Oklahoma Health Sciences Center, Oklahoma City, OK,
USA). Cells were cultured in Dulbecco's modified Eagle's medium
supplemented with 10% heat-inactivated fetal calf serum (Hyclone;
GE Healthcare Life Sciences) and 1% penicillin/streptomycin, at
37°C in a humidified atmosphere containing 5% CO2. Cells
have a doubling time of ~20 h under these conditions, and were
passaged by trypsinization at a ratio of 1:6 every 3–4 days. For
the serum deprivation experiments, the 661W cells were cultured in
96- or 24-well plates for 24 h with normal medium, washed with PBS
three times and then cultured with serum-free medium for 1, 2, 4 or
6 days. For the rapamycin experiments, the 661W cells were
additionally treated with 100 nM rapamycin during serum deprivation
for 2, 4 or 6 days.
Intracellular ROS measurement
Intracellular ROS were measured using the
oxidation-sensitive fluorescent probe DCFH-DA (9). Cells were cultured in 6-well plates
for 2 days, were washed twice with fresh medium, and were then
incubated with 10 µM DCFH-DA at 37°C for 20 min. Oxidized
2,7-dichlorofluorescein fluorescence was visualized under the
IX-ULWCD fluorescent microscope (Olympus Corporation, Tokyo,
Japan). Fluorescent intensities were measured using ImageJ
software, version 1.46 (National Institutes of Health, Bethesda,
MD, USA). Relative fluorescence intensities of the cells were
assessed using the following formula (10): Biomarker relative intensity =
[Foreground intensity (cell staining) / surface area] / [background
intensity / surface area].
Propidium iodide (PI) staining
The 661W cells were cultured in 6-well plates for 24
h. After serum deprivation at the indicated time points, cells were
stained with PI solution (2 µg/ml) and incubated in the dark
for 10 min at room temperature. PI-positive cells were visualized
under the IX-ULWCD fluorescent microscope (Olympus Corporation).
For quantitative analysis, cells were harvested by trypsinization,
rinsed with phosphate-buffered saline (PBS), and analyzed by flow
cytometry (FACSCanto II; BD Biosciences, San Diego, CA, USA).
Cell cycle analysis
Cell cycle distribution was analyzed by flow
cytometry (11). Six groups of
cells (~1×106 cells/group) were harvested by
trypsinization, rinsed with PBS and fixed with cold 70% ethanol at
4°C overnight. Cells were then washed twice with PBS and
re-suspended in 1 ml staining solution (50 µg/ml PI, 50
µg/ml RNaseA, 0.1% Triton X-100 in citrate buffer; pH 7.8)
at room temperature for 30 min. Cells were acquired and the
percentage of cells in each cell cycle stage was analyzed by flow
cytometry (FACSCanto II; BD Biosciences).
Mitochondrial-membrane potential
assay
JC-1 selectively accumulates within intact
mitochondria to form multimeric J-aggregates, which emit
fluorescent light at 590 nm (red) at a higher membrane potential
(12). In the present study, the
cells were seeded in 6-well plates, and cultured with serum-free
medium for 2 days. Subsequently, the medium was removed, and the
cells were washed with Ca2+/Mg2+-free PBS.
Cells were stained with JC-1 (10 µg/ml) for 30 min at 37°C
and were examined under the IX-ULWCD fluorescent microscope
(Olympus Corporation) at 590 nm.
Mitochondrial staining
Mitochondria of the 661W cells were stained using
MitoTracker Green, as described previously by Stojkovic et
al(13). Briefly, 661W cells
were incubated in serum-free medium containing 0.2 mM MitoTracker
Green for 10 min at 37°C. Subsequently, the 661W cells were washed
several times in fresh medium, mounted on glass slides, and were
observed under the IX-ULWCD fluorescent microscope (Olympus
Corporation) at 490 nm. Images were captured and analyzed using
ImageJ software, version 1.46 (National Institutes of Health).
Western blotting
Immunoblot assay was performed as described
previously by Li et al(14). The 661W cells were sonicated in
protein lysis buffer, and a bicinchoninic acid assay was used to
determine protein concentration. An equal amount of cell lysate (20
µg) was dissolved in sample buffer, and samples were boiled
for 3 min. Electrophoresis was performed using 10% polyacrylamide
gels containing 0.1% sodium dodecyl sulfate. Proteins were
transferred to nitrocellulose membranes and were subsequently
blocked for 1 h at 25°C in 5% (w/v) non-fat dried milk. The blots
were incubated for 3 h at room temperature with primary antibodies
(1:1,000), followed by an incubation with goat anti-rabbit, rabbit
anti-mouse or anti-goat biotinylated secondary antibodies (1:1,000;
Sigma-Aldrich; cat. nos. A0545, A9044 and A5420, respectively) for
1.5 h. Signals were developed using enhanced chemiluminescence and
images were captured using a charge-coupled device camera (Tanon
Science & Technology Co., Ltd., Shanghai, China). Densitometry
analysis was performed using Quantity One software, version V4.62
(Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Each experiment was repeated at least three times.
Data are presented as the mean ± standard error of the mean.
Differences between the means were evaluated using one-way analysis
of variance followed by a Bonferroni test with the SPSS software,
version 17 (SPSS Inc., Chicago, IL, USA). Significance for all
cases was P<0.001.
Results
Inhibition of mTOR protects 661W cells
from serum deprivation-induced injury
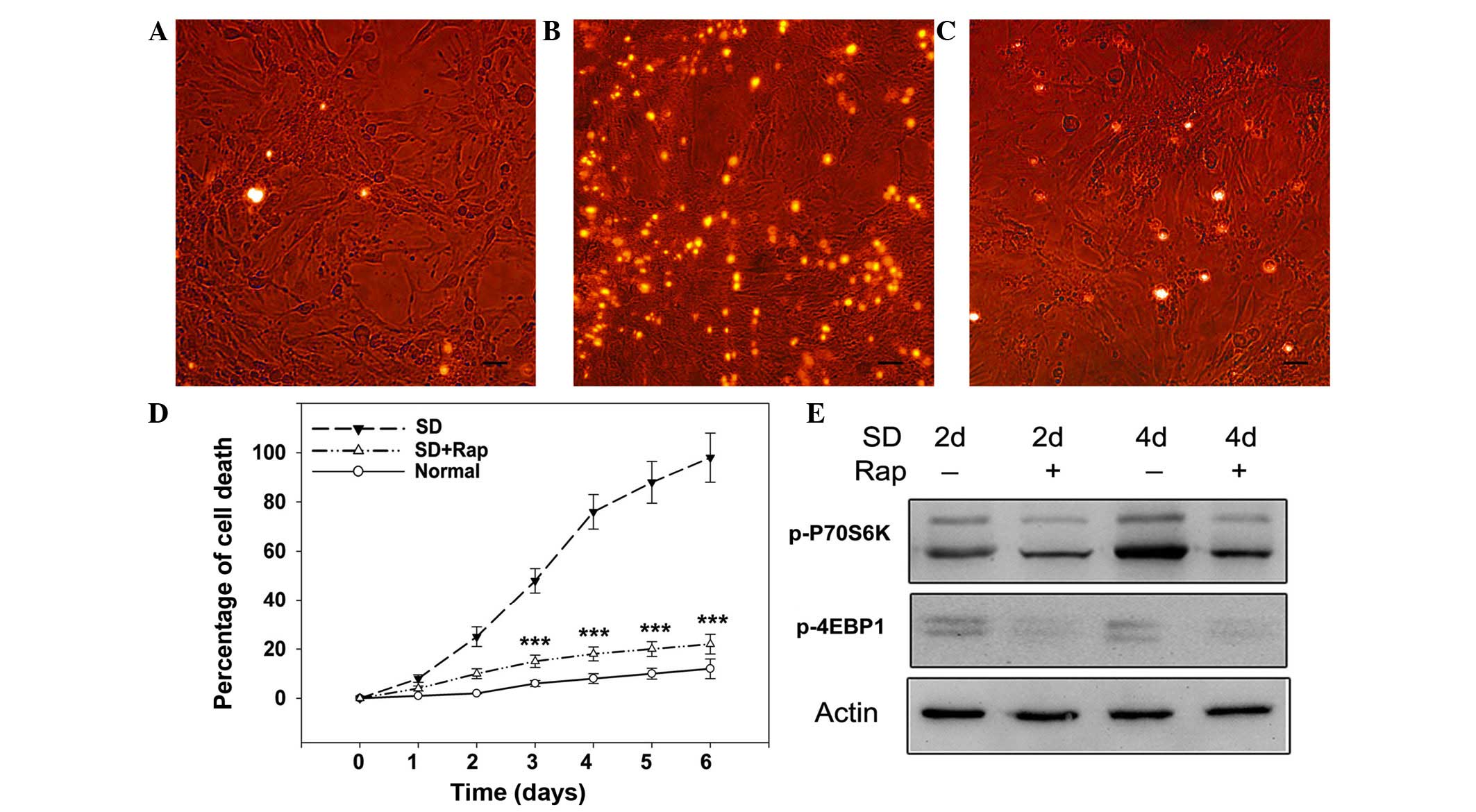
The present study investigated the effects of serum
deprivation on 661W cells by measuring cell death using PI.
Compared with the cells cultured in normal medium (Fig. 1A), on day 3 of serum deprivation
photoreceptor cells underwent programmed cell death, and were
positively stained for PI (Fig.
1B). Conversely, rapamycin (100 nM) attenuated serum
starvation-induced cell death, as evidenced by fewer PI-positive
cells (Fig. 1C). Cell death was
monitored with flow cytometry up to 6 days, by which time nearly
all cells had died. As shown in Fig.
1D, cell death in the serum-starved group increased over time
compared with in the control cells cultured in normal medium. On
day 3, serum deprivation killed ~50% of the cells, and by day 4 80%
of the cells were dead. By days 5 and 6, >90% of the cells were
dead, as measured by PI staining and flow cytometry. The percentage
of dead control cells at this time point was ~15%.
Subsequently, the effects of mTOR inhibition on
serum-deprived photoreceptor cells were assessed. Initially, it was
confirmed that rapamycin inhibited mTOR signaling by western blot
analysis. As shown in Fig. 1E,
rapamycin (100 nM) markedly suppressed the expression levels of
downstream factors of mTOR, as evidenced by reduced protein
expression of p-P70S6 and p-4EBP1 on days 2 and 4 of serum
deprivation. Consistent with inhibition of mTOR, cell death
analysis indicated that rapamycin exerted neuroprotective effects
against serum deprivation in photoreceptor cells, with cell death
maintained at <20% up to 6 days (Fig. 1D; P<0.001 vs. the serum-deprived
group). These results suggest that mTOR inhibition may protect
photoreceptor cells from neurotrophin withdrawal.
Inhibition of mTOR suppresses serum
starvation-induced oxidative stress
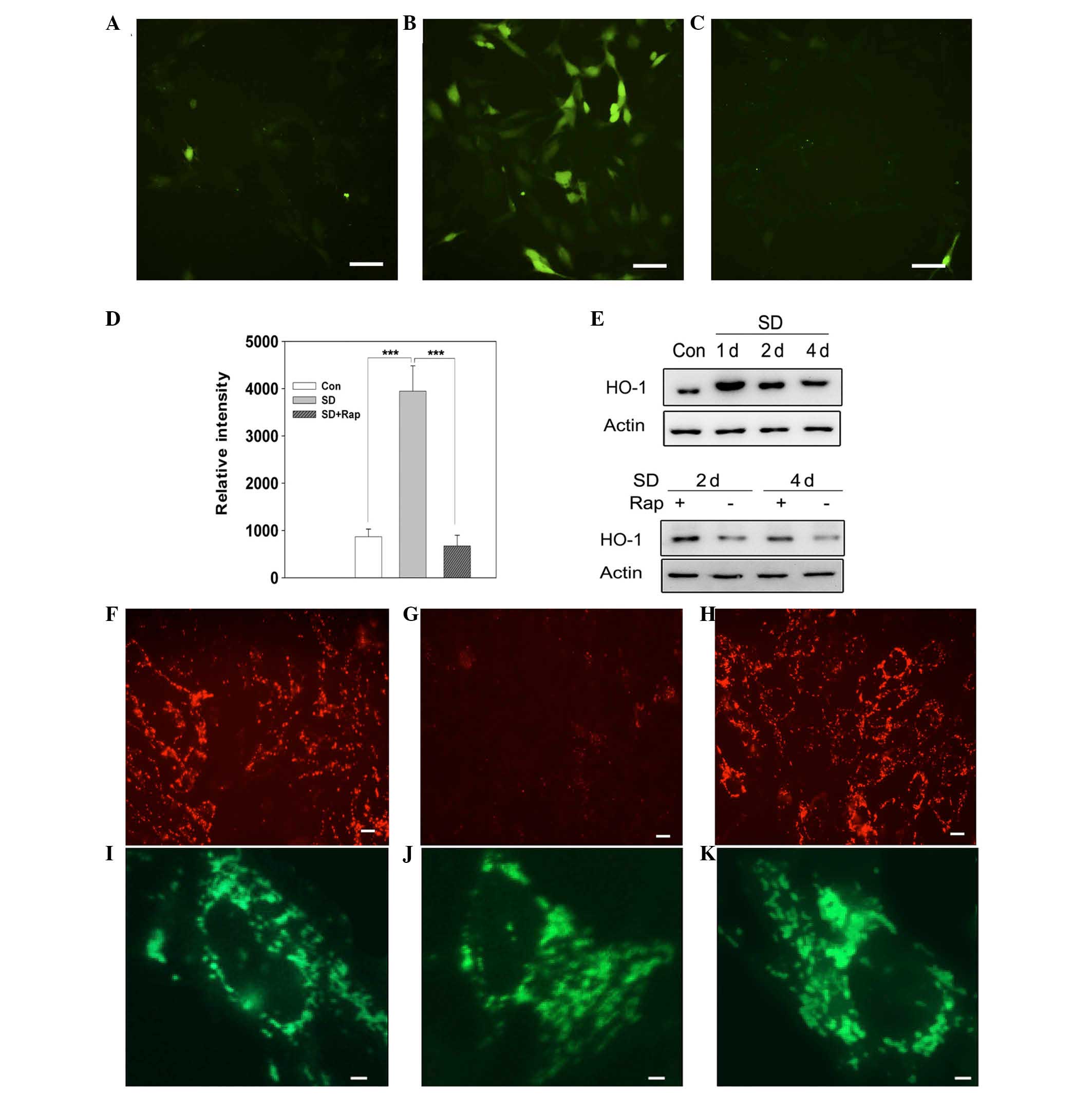
As shown in Fig.
2A–C, intracellular ROS increased after 2 days of serum
deprivation, as compared with in the normally cultured cells, as
measured using the oxidation-sensitive fluorescent probe DCFH-DA,
which fluoresces green under fluorescent microscopy. Quantitative
analysis indicated that cellular ROS were significantly upregulated
in serum-starved cells compared with normally cultured cells
(P<0.001). Conversely, treatment with rapamycin suppressed
cellular ROS generation in serum-deprived cells, and reduced
DCFH-DA fluorescence intensity was detected (Fig. 2D; P<0.001 vs. the serum-deprived
group). To evaluate oxidative stress in 661W cells, the expression
of HO-1, an enzyme modulating antioxidant defense, was detected. As
shown in Fig. 2E serum deprivation
upregulated HO-1 expression from day 1, whereas the addition of
rapamycin markedly attenuated serum starvation-induced increases in
HO-1. These results indicate that inhibiting the mTOR pathway may
suppress serum starvation-induced oxidative stress in 661W
cells.
Inhibition of mTOR helps attenuate
mitochondrial dysfunction
Mitochondrial membrane potential was analyzed using
JC-1 staining. The majority of 661W cells were stained fluorescent
red, indicating the presence of healthy mitochondria (Fig. 2F), whereas serum-deprived 661W
cells exhibited less fluorescence (Fig. 2G). Treatment with rapamycin
restored mitochondrial potential in serum-deprived cells, as
evidenced by increased red fluorescence (Fig. 2H). Mitochondrial swelling denotes
mitochondrial dysfunction, due to opening of the mitochondrial
permeability transition pore. Mitochondrial morphology was assessed
using the mitochondrial-specific MitoTracker Green. As shown in
Fig. 2I–K, mitochondria in
normal-cultured cells exhibited clear dot-shaped structures
(Fig. 2I), whereas the
serum-starved cells exhibited increased swelling with ambiguous
boundaries (Fig. 2J). As
predicted, treatment with rapamycin improved mitochondrial
morphology in serum-deprived cells, and they were able to maintain
a clear dot shape (Fig. 2K). These
results indicate that inhibition of mTOR may contribute to
restoration of mitochondrial function, improving mitochondrial
membrane potential and mitochondrial structure.
Inhibition of mTOR leads to
G2/M cell cycle arrest
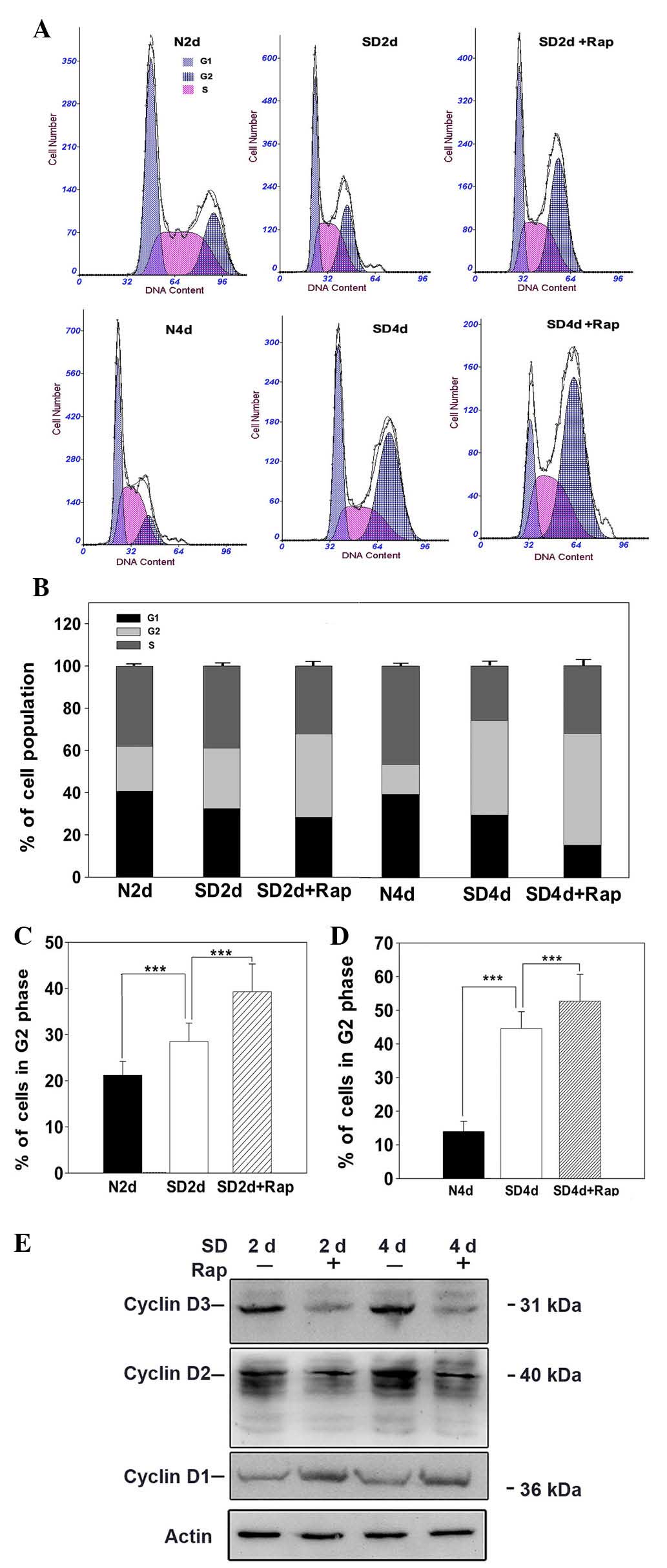
Cell cycle checkpoints and DNA repair systems
closely cooperate to maintain genomic integrity of cells damaged by
external or internal insults, including intracellular ROS (15). To investigate the neuroprotective
mechanisms underlying the effects of mTOR suppression, cell cycle
progression was investigated in photoreceptor cells. The 661W cells
were treated with rapamycin (100 nM) under serum-deprived
condition, and on days 2 and 4 cell cycle distribution was
assessed. As shown in Fig. 3A–D,
rapamycin significantly increased the number of serum-deprived
cells in G2/M phase; cells accumulated from 28.5 to
38.2% on day 2 (Fig. 3C), and from
45.1 to 54.5% by day 4 (Fig. 3D;
P<0.001 vs. the serum-deprived group). Notably, serum
deprivation as an external insult resulted in significant cell
cycle arrest at G2/M phase, compared with in the
normally cultured cells (P<0.001). Subsequently, the effects of
mTOR inhibition on the protein expression levels of cyclins (key
cell cycle regulatory molecules) were analyzed by western blotting.
Rapamycin markedly reduced cyclin D2 and D3 protein expression
levels on days 2 and 4 under serum-deprived conditions, whereas
cyclin D1 expression was markedly increased following treatment
with rapamycin for 2 and 4 days (Fig.
3E). These results suggest that mTOR inhibition may lead to
G2/M cell cycle arrest in serum-deprived 661W cells,
which may be due to the modulation of cyclin protein
expression.
Inhibition of mTOR reduces expression of
apoptotic markers
As shown in Fig.
4A, serum deprivation increased the expression of cleaved
caspase-3 from day 2 to 6, as evidenced by detection of a 19 kDa
protein band. Quantitative analysis revealed that the relative
amount of cleaved caspase-3 in the serum-deprived cell lysate was
significantly higher than in the normally cultured cell lysate
(P<0.001). As a substrate of caspase-3, PARP-1 is normally
cleaved into two protein forms while the caspase-dependent pathway
is activated. Western blot analysis confirmed that concentration of
cleaved PARP-1 increased in the serum-deprived cells compared with
the control cells (P<0.001). To illustrate serum
deprivation-induced cell death, a caspase-independent pathway
marker, AIF, was measured. The 57 kDa active form of AIF was
upregulated under serum deprivation conditions from days 2 to 6,
and was significantly higher than in normally cultured cells
(P<0.001). Western blotting confirmed that rapamycin
significantly attenuated upregulation of cleaved caspase-3 and
PARP-1 at days 2 and 4 and the active form of AIF was reduced
(Fig. 4B; P<0.001 vs. the
serum-deprived group). These results suggest that the
caspase-dependent and -independent pathways were activated by serum
starvation and mTOR inhibition may block apoptotic pathways by
suppressing the expression of apoptotic factors.
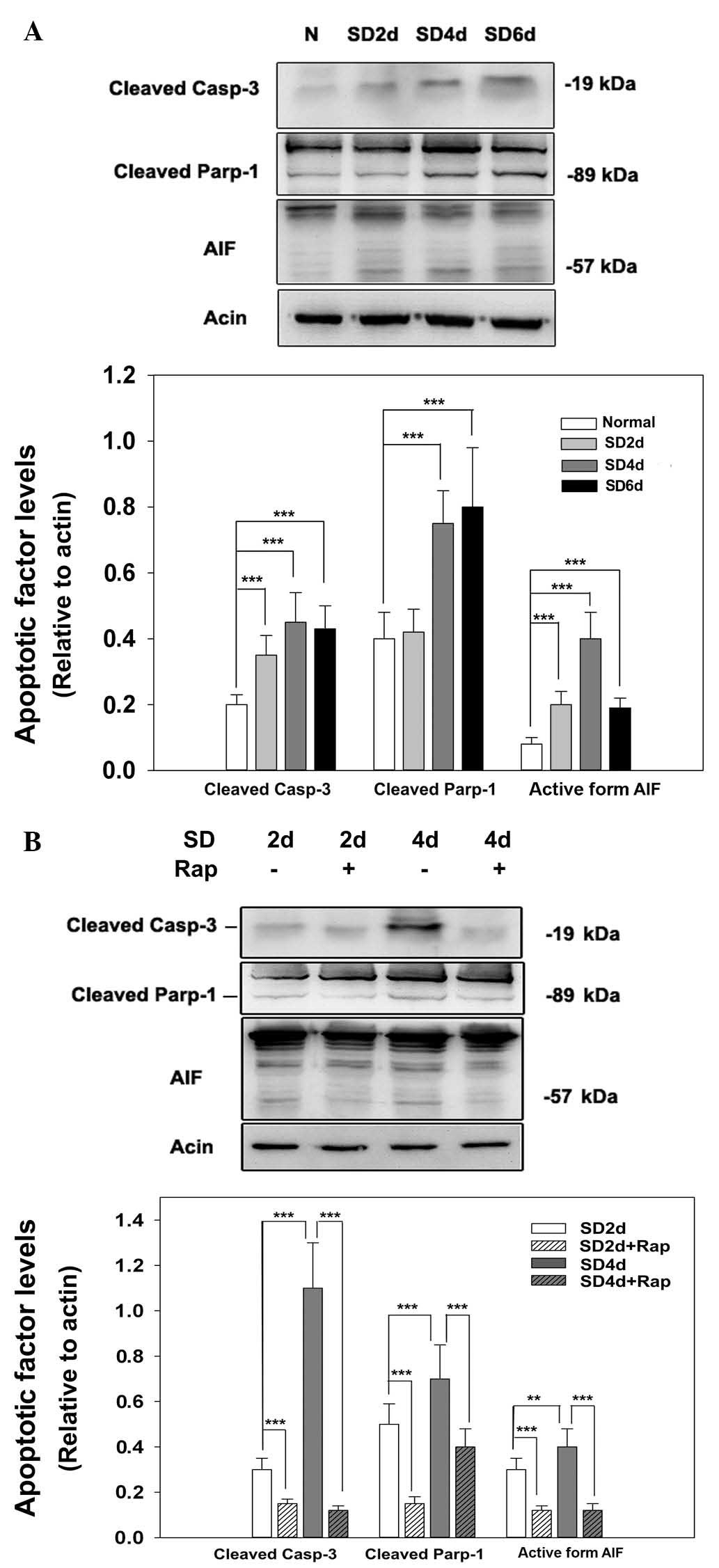 | Figure 4Inhibition of mammalian target of
rapamycin reduces the expression of apoptotic markers. (A) 661W
cells were cultured with serum-free medium for the designated
times. Cells were then harvested and lysed for immunoblot analysis.
N, normal medium; SD2d, serum deprivation for 2 days; SD4d, serum
deprivation for 4 days; SD6d, serum deprivation for 6 days. (B)
661W cells were cultured with/without rapamycin while
serum-deprived for the designated times. Cells were then harvested
and lysed for immunoblot analysis. SD, serum deprivation; Rap, 100
nM rapamycin. Each experiment was repeated at least 3 times, and a
representative blot is presented. Data were obtained from at least
three independent experiments and are presented as the mean ±
standard error of the mean. **P<0.05,
***P<0.001. Casp-3, caspase-3; Parp-1, poly
(ADP-ribose) polymerase 1; AIF, apoptosis inducing factor. |
Discussion
mTOR is an evolutionarily conserved serine/threonine
kinase, which responds to numerous stimuli, including growth
factors, nutrients, energy status and oxygen, to regulate cellular
proliferation and growth (16).
The present study demonstrated that serum deprivation, as an
external insult, mimics neurotrophin withdrawal and causes
photoreceptor cell death. Conversely, mTOR inhibition was
neuroprotective, and was able to rescue 661W cells from serum
starvation.
Mitochondria are chief producers of ROS via the
electron transport chain. A decline in mitochondrial function,
including damaged membrane potential, leads to enhanced ROS
production (17). Once ROS
production is increased, due to changes in oxidative mitochondrial
metabolism, damage to cellular components and cell death may occur
(18). The results of the present
study are consistent with those of a previous study, and indicated
that serum deprivation led to damaged mitochondrial membrane
potential and increased ROS production (19). After 2 days of serum deprivation,
the majority of 661W cells exhibited reduced JC-1 red fluorescence,
indicating the presence of impaired mitochondria. However, abnormal
mitochondrial activities were inhibited by treatment with
rapamycin. Consistently, increased ROS production was detected in
serum-starved cells, whereas rapamycin decreased cellular ROS
levels. Notably, mitochondrial structure appeared swollen in
response to serum deprivation, whereas following rapamycin
treatment mitochondria exhibited a clear dot pattern, according to
the results of MitoTracker Green staining. Mitochondria are
essential to multicellular life, therefore apoptotic pathways
target this organelle in various ways; the apoptotic stimuli may
cause mitochondrial swelling via membrane pore formation, or they
may increase mitochondrial membrane permeability, allowing leakage
of apoptotic effectors (13).
Cell growth and proliferation are major cellular
functions controlled by mTORC1 (20), which is reported to preferentially
drive cell growth through S6K1 and cell proliferation through 4EBP1
(21). The cell cycle consists of
four phases: G1 phase, S (or DNA synthesis) phase,
G2 phase, and M (or mitosis) phase. The results of the
present study demonstrated that inhibition of the mTOR pathway
induced 661W cell cycle arrest at G2/M stage, as
evidenced by flow cytometry. This cell cycle arrest occurred in
cells cultured under serum-deprived conditions. The G2
checkpoint prevents cells from entering mitosis when DNA is
damaged, providing an opportunity for repair and cessation of
damaged cell proliferation. DNA repair systems and cell cycle
checkpoints closely cooperate to maintain genomic integrity of
cells damaged by external or internal insults, including
intracellular ROS and ionizing radiation. Cells at G1/S
and S phases prevent replication of damaged DNA, and those at
G2/M phase prevent segregation of modified chromosomes.
Cells that lack, or have altered, genes involved in DNA
double-strand break repair and cell cycle checkpoints die early and
are genomically unstable (15).
Eukaryotes possess various types of cyclins, all of which exhibit
specialized functions. In animal cells, D-type cyclins are
fundamental for re-entry into the cell cycle in response to
extracellular signals (22). The
results of the present study indicated that inhibition of mTOR
signaling caused 661W cell cycle arrest at G2/M phase
and upregulated cyclin D1 expression; however, cyclin D2 and cyclin
D3 expression was downregulated. Cyclin D1 is critical for
regulating G1/S-phase transition. When cells are
arrested in G2/M phase, cyclin D1 is likely increased as
a compensatory response. In addition, cyclin D1 governs DNA damage
repair by recruiting DNA repair complexes. When nuclear DNA is
damaged by excessive intracellular ROS during oxidative stress,
cyclin D1 may be increased, in order to repair damaged DNA. The
observation that mTOR inhibition caused a decrease in cyclin D2 and
D3 expression was consistent with the results of a previous study,
thus indicating their importance in regulating G2/M
arrest (23).
Apoptosis is a well-orchestrated type of cell death,
which is responsible for maintenance of normal tissue homeostasis
and removal of damaged, old or infected cells. Caspase-dependent
apoptosis is finely regulated by a series of pro-apoptotic
proteins, including caspase-3 and PARP-1, which are markers of
apoptotic pathways (24).
Increased active caspase-3 and cleaved PARP-1 are products of the
activated caspase-dependent pathway. Decreased cleaved caspase-3
and PARP-1 expression was detected in the rapamycin-treated 661W
cells in the present study, thus suggesting that rapamycin may
prevent retinal neurons from subsequent pro-apoptotic insults. In
addition, a typical caspase-independent pathway was investigated.
AIF is a flavoprotein, which is confined to the mitochondrial
inter-membrane space in healthy cells. Upon lethal signaling, AIF
translocates, via the cytosol, to the nucleus where it binds to DNA
and provokes caspase-independent chromatin condensation (25). The present study revealed that
serum deprivation resulted in upregulation of the active form of
AIF (57 kDa), thus suggesting that the caspase-independent pathway
may be involved in 661W cell death. Conversely, treatment with
rapamycin reduced the expression of 57 kDa AIF. Therefore, a
proposed neuroprotective mechanism underlying the effects of mTOR
inhibition may be suppression of caspase-dependent and -independent
apoptotic pathways.
In conclusion, mTOR inhibition exerts protective
effects against serum deprivation in 661W cells, and induces a
stable G2/M cell cycle arrest, in which energy
consumption and cellular metabolism are largely decreased. In this
state, damaged nuclear DNA is primarily repaired, mitochondrial
dysfunction is restored, and the cell death cascade is suppressed,
which may contribute to its anti-apoptotic effects. These findings
suggested that rapamycin may be a potential therapeutic agent for
the prevention of retinal degenerative diseases and neurotrophin
defects. Inducing neurons into a lower and more stable bioenergetic
state by blocking the mTOR pathway may be considered a therapeutic
strategy to slow the progression of neurodegenerative diseases.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81100660,
30801271 and 81570864) and the Norman Bethune Program of Jilin
University (grant no. 2012208).
References
|
1
|
Wullschleger S, Loewith R and Hall MN: TOR
signaling in growth and metabolism. Cell. 124:471–484. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Loewith R, Jacinto E, Wullschleger S,
Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P and Hall MN:
Two TOR complexes, only one of which is rapamycin sensitive, have
distinct roles in cell growth control. Mol Cell. 10:457–468. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Manning BD and Cantley LC: Rheb fills a
GAP between TSC and TOR. Trends Biochem Sci. 28:573–576. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hay N and Sonenberg N: Upstream and
downstream of mTOR. Genes Dev. 18:1926–1945. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harada T, Harada C and Parada LF:
Molecular regulation of visual system development: More than meets
the eye. Genes Dev. 21:367–378. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
von Bartheld CS: Neurotrophins in the
developing and regenerating visual system. Histol Histopathol.
13:437–459. 1998.PubMed/NCBI
|
|
7
|
Wen R, Tao W, Li Y and Sieving PA: CNTF
and retina. Prog Retin Eye Res. 31:136–151. 2012. View Article : Google Scholar :
|
|
8
|
Tan E, Ding XQ, Saadi A, Agarwal N, Naash
MI and Al-Ubaidi MR: Expression of cone-photoreceptor-specific
antigens in a cell line derived from retinal tumors in transgenic
mice. Invest Ophthalmol Vis Sci. 45:764–768. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brem R, Li F, Montaner B, Reelfs O and
Karran P: DNA breakage and cell cycle checkpoint abrogation induced
by a therapeutic thiopurine and UVA radiation. Oncogene.
29:3953–3963. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Riethdorf S, Müller V, Zhang L, Rau T,
Loibl S, Komor M, Roller M, Huober J, Fehm T, Schrader I, et al:
Detection and HER2 expression of circulating tumor cells:
Prospective monitoring in breast cancer patients treated in the
neoadjuvant GeparQuattro trial. Clin Cancer Res. 16:2634–2645.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma GF, Chen SY, Sun ZR, Miao Q, Liu YM,
Zeng XQ, Luo TC, Ma LL, Lian JJ and Song DL: FoxP3 inhibits
proliferation and induces apoptosis of gastric cancer cells by
activating the apoptotic signaling pathway. Biochem Biophys Res
Commun. 430:804–809. 2013. View Article : Google Scholar
|
|
12
|
Qi Y, Li Y, Zhang Y, Zhang L, Wang Z,
Zhang X, Gui L and Huang J: IFI6 inhibits apoptosis via
mitochondrial-dependent pathway in Dengue virus 2 infected vascular
endothelial cells. PLoS One. 10:e01327432015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stojkovic M, Machado SA, Stojkovic P,
Zakhartchenko V, Hutzler P, Gonçalves PB and Wolf E: Mitochondrial
distribution and adenosine triphosphate content of bovine oocytes
before and after in vitro maturation: Correlation with
morphological criteria and developmental capacity after in vitro
fertilization and culture. Biol Reprod. 64:904–909. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li GY, Fan B and Jiao YY: Rapamycin
attenuates visible light-induced injury in retinal photoreceptor
cells via inhibiting endoplasmic reticulum stress. Brain Res.
1563:1–12. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Belli M, Sapora O and Tabocchini MA:
Molecular targets in cellular response to ionizing radiation and
implications in space radiation protection. J Radiat Res.
43(Suppl): S13–S19. 2002. View Article : Google Scholar
|
|
16
|
Morita M, Gravel SP, Hulea L, Larsson O,
Pollak M, St-Pierre J and Topisirovic I: mTOR coordinates protein
synthesis, mitochondrial activity and proliferation. Cell Cycle.
14:473–480. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Du H, Duanmu M, Witte D and Grabowski GA:
Targeted disruption of the mouse lysosomal acid lipase gene:
Long-term survival with massive cholesteryl ester and triglyceride
storage. Hum Mol Genet. 7:1347–1354. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hamanaka RB and Chandel NS: Mitochondrial
reactive oxygen species regulate cellular signaling and dictate
biological outcomes. Trends Biochem Sci. 35:505–513. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yan C, Ding X, Dasgupta N, Wu L and Du H:
Gene profile of myeloid-derived suppressive cells from the bone
marrow of lysosomal acid lipase knock-out mice. PLoS One.
7:e307012012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang H, Stallock JP, Ng JC, Reinhard C
and Neufeld TP: Regulation of cellular growth by the Drosophila
target of rapamycin dTOR. Genes Dev. 14:2712–2724. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dowling RJ, Topisirovic I, Alain T,
Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj
A, Liu Y, et al: mTORC1-mediated cell proliferation, but not cell
growth, controlled by the 4E-BPs. Science. 328:1172–1176. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sherr CJ: G1 phase progression: Cycling on
cue. Cell. 79:551–555. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weidner C, Rousseau M, Plauth A, Wowro SJ,
Fischer C, Abdel-Aziz H and Sauer S: Melissa officinalis extract
induces apoptosis and inhibits proliferation in colon cancer cells
through formation of reactive oxygen species. Phytomedicine.
22:262–270. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Siddiqui A, Hanson I and Andersen JK:
Mao-B elevation decreases parkin's ability to efficiently clear
damaged mitochondria: Protective effects of rapamycin. Free Radic
Res. 46:1011–1018. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Polster BM: AIF, reactive oxygen species,
and neurodegeneration: A 'complex' problem. Neurochem Int.
62:695–702. 2013. View Article : Google Scholar :
|