Introduction
The inflammatory responses of glomerular endothelial
cells (GECs) lead to proteinuria; however, the underlying mechanism
has yet to be fully elucidated (1). Thrombomodulin (TM), a type I
membrane-bound glycoprotein, is ubiquitously expressed in vascular
endothelial cells (VECs) and is a potential suppressor of
inflammatory processes (2). Thus,
upregulation of TM expression and/or function may be a potential
therapy for inflammation-induced microvascular diseases, including
proteinuria (2). The potential
therapeutic value of TM modulation motivated the search for
naturally abundant compounds that may regulate this vasoprotective
molecule. Notably, previous studies determined that various nuclear
receptors (NRs) were involved in the regulation of TM expression
(3–5). However, the regulatory mechanism
underpinning TM expression has yet to be fully elucidated.
Liver X receptors (LXRs) are important regulators of
cholesterol, free fatty acids and glucose metabolism (6,7). In
addition to their importance in lipid and glucose metabolism, the
activation of LXR was also reported to be of high importance in
regulating immune processes and in inhibiting inflammatory gene
expression (8–12). LXRs are ligand-activated
transcription factors of the nuclear receptor superfamily. There
are two LXR isoforms, termed LXR-α and LXR-β. Upon activation, they
form heterodimers with retinoid X receptor and bind to the LXR
response element located in the promoter region of the target genes
(13). Previous studies have
determined that TM may improve diabetic nephropathy (DN) via
anti-nuclear factor-κB (NF-κB)/NLR family pyrin domain containing 3
inflammasome-mediated inflammation or via complement inhibition
(14,15). Full transcriptional activity of
NF-κB requires physical interaction with the closely associated
transcriptional coactivators, p300 and cAMP response element
binding protein (CREB1) (16,17).
In addition, inflammation is also reported to regulate the
development of DN (16). Thus,
seeking effective targets to ameliorate inflammation of human GECs
(HUGECs) may provide a promising strategy to reduce the secretion
of urinary proteins.
In addition to the importance of LXRs in lipid and
glucose metabolism, synthetic LXR agonists have been determined to
inhibit inflammation in various animal models, including pulmonary
inflammation (18). A previous
study has reported that LXRs may antagonize the cytokine-mediated
expression of proinflammatory genes in macrophages via the
silencing of proinflammatory transcription factors, particularly
NF-κB (19). Cheng et al
(20) determined that LXR
activation may inhibit the transcription of various inflammatory
genes, including tumor necrosis factor (TNF), cytochrome c
oxidase subunit II, interleukin-1β (IL-1β), matrix metallopeptidase
9 and inducible nitric oxide synthase. Although they share similar
anti-inflammatory functions and location of expression in
endothelial cells, the association between LXR and TM remains to be
fully elucidated (21,22).
In the present study, the expression and activity of
TM were demonstrated to be upregulated by the LXR agonist,
T0901317, in HUGECs. Luciferase reporter assays indicated no effect
of LXR on the activity of the TM promoter (−2,494 to +160 bp).
However, activating LXR effectively inhibited IκB phosphorylation
and p65 translocation in HUGECs. LXR activation enhanced the
interaction between LXR and p300, which is a physical partner with
NF-κB in HUGEC extracts, as indicated by western blotting and
co-immunoprecipitation analyses. The current findings suggest that
NF-κB may be a negative regulator of TM expression, and that LXR
activation may indirectly modulate TM expression via inhibition of
NF-κB activation and/or restricting its availability for the
formation of the LXR/p300 complex.
Materials and methods
Reagents
T0901317 (a specific synthetic ligand agonist for
LXR) was purchased from Sigma-Aldrich (T2320; St. Louis, MO, USA).
Pierce BCA Protein Assay Reagent B kit was obtained from Thermo
Fisher Scientific, Inc. (cat. no. 23224; Waltham, MA, USA).
Lipofectamine 2000, TRIzol® and all reagents for cell
culture procedures were purchased from Invitrogen; Thermo Fisher
Scientific, Inc. (cat. no. 11668-019). Argatroban was obtained from
Tianjin Pharmaceuticals Research Institute Co., Ltd. (Tianjin,
China). D-Mannitol and D-glucose (cat. nos. M8140 and G8270) were
sourced from Beijing Solarbio Science & Technology Co., Ltd.
(Beijing, China).
Cell culture
The primary human glomerular endothelial cell line
was purchased from ScienCell Research Laboratories (cat. no. 4000;
Carlsbad, CA, USA) and cultured in Dulbecco's modified Eagle's
medium with 10% fetal bovine serum (FBS), 100 U/ml penicillin and
100 μg/ml streptomycin. Prior to being treated with
T0901317, the cells were cultured in Opti-MEM medium (Thermo Fisher
Scientific, Inc.) supplemented with 0.5% FBS (conditioned medium).
All experiments performed with HUGECs were performed at passages
4–6. The cells were grown on 0.2% gelatin-coated plates (Corning,
Inc., Wiesbaden, Germany) and maintained at 37°C in a humidified 5%
CO2 incubator using endothelial cell medium (ScienCell
Research Laboratories, Carlsbad, CA, USA; cat. no. 1001). Cells
were subcultured at confluence by trypsinization with 0.05% trypsin
and 0.02% EDTA. The medium was changed every other day.
Construction of recombinant
adenoviruses
The 72 nt oligonucleotide encoding mouse TM small
interfering (si) RNA was inserted into the BamHI and EcoRI sites of
pHBAd-U6-GFP to build the pHBAd-U6-GFP-siRNA/TM shuttle plasmid.
The resultant shuttle plasmid was identified by double
restriction-enzyme digestion and DNA sequencing was conducted by
Shanghai Sunny Biotech Co., Ltd. (Shanghai, China). The shuttle
plasmid was then co-transfected with a backbone plasmid [pBHGlox
(∆)E1, 3Cre] into HEK293 cells (CRL12108; American Type Culture
Collection, Manassas, VA, USA) as described below. The expression
of the TM protein in the infected endothelial cells was detected by
western blot analysis. A control adenovirus (Vector Biolabs,
Malvern, PA, USA; cat. no. 1060) containing the green fluorescence
protein (GFP) gene (Ad.GFP) was generated, purified, titrated, and
stored as described previously (23).
Transfection
Cells were seeded into 25 cm2 flasks and
were cultured until >60% confluent. HUGECs were successfully
transfected with LXR-α shRNA and treated with 2 μM T0901317
for 24 h. Cells were then exposed to 25 mM glucose or 25 mM
mannitol for 6 h. The recombinant adenovirus transfection was
performed following the protocol described in a previous study
(24). LXR-α and LXR-β siRNAs were
obtained from Invitrogen; Thermo Fisher Scientific, Inc. Cells were
washed once with OptiMEM medium, 2.5 ml OptiMEM medium was added to
each flask, and subsequently these flasks were placed in an
incubator at 37°C for 30 min prior to transfection. A transfection
mix was prepared by adding 4 μg recombinant adenovirus
plasmid DNA and 20 μl Lipofectamine 2000 to 500 μl
OptiMEM medium, according to manufacturer's protocol. For gene
silencing, cells were seeded into 6-well plates and were cultured
until >75% confluent. The cells were transiently transfected
with 25 nmol/l LXR-α siRNA, LXR-β siRNA or negative control siRNA
(scrambled siRNA) using Lipofectamine 2000 transfection reagents,
according to the manufacturer's protocol. After 24 h, fluorescence
images of transfected cells were observed under a Nikon T300
fluorescence microscope (Nikon Corporation, Tokyo, Japan).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA (2 μg) was extracted from HUGECs
using the TRIzol reagent (Takara Bio, Inc., Otsu, Japan), according
to the manufacturer's protocol. The first-strand cDNA was
synthesized using Moloney murine leukemia virus reverse
transcriptase (Toyobo Co., Ltd., Osaka, Japan) and an oligo (dT)
primer. The expression levels of human TM and human small
ATP-binding cassette transporter A1 (ABCA1), a target gene of LXR,
were examined using RT-qPCR, which was performed using the Bio-Rad
iQ5 Gradient Real Time PCR system with SYBR Premix Ex Taq II kit
(Takara Bio, Inc.; cat. no. RR820A) according to the manufacturer's
protocol in a 20 μl volume in triplicate. Briefly, 10
μl SYBR Green were mixed with 0.3 μl reverse primer
and 2 μl cDNA. The cycle parameters performed were as
follows: 94°C for 5 min, 40 cycles at 94°C for 30 sec, 57°C for 30
sec and 72°C 30 sec, and 72°C for 10 min. The primers used for TM
and ABCA1 were as follows: i) TM (173 bp), forward (F),
5′-CGGGCTCCTACTCGTGCATG-3′; reverse (R),
3′-GCCGTCCACCAGGTCGTAGTTA-5′; and ii) ABCA1 (100 bp), F,
5′-ATGTCCAGTCCAGTAATGGTTCTGT-3, R, 3′-CGAGATATGGTCCGGATTGC-5′. As
an internal control, β-actin (527 bp) was analyzed in parallel by
using the following primers: F, 5′-CTACAATGAGCTGCGTGTGG-3′, R,
3′-AAGGAAGGCTGGAAGAGTGC-5′. The Cq value, which is
inversely proportional to the initial template copy number, was the
calculated cycle number at which the emitted fluorescence signal
was first significantly higher than the background levels. The
separate well 2−ΔΔCq cycle quantification method was
used to determine relative quantitative levels of TM and ABCA1, and
these were expressed as the fold-change relative to β-actin
(25).
Western blot analysis
Briefly, HUGECs were successfully transfected with
LXR-α shRNA and treated with 2 μM T0901317 for 24 h. Cells
were then exposed to 25 mM glucose or 25 mM mannitol for 6 h to
induce high-glucose conditions. Cells were collected and lysed in
2X lysis buffer (200 μl/50 cm2; Beyotime
Institute of Biotechnology). Samples were resolved by sodium
dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE; 10%
gels; run at 100 Volts for 1 h) and electroblotted onto a
polyvinylidene difluoride membrane. The membranes were then blocked
with 5% bovine serum albumin (BSA; Hyclone; GE Healthcare Life
Sciences, Logan, UT, USA) in Tris-buffered saline and Tween 20
buffer, and incubated with mouse monoclonal TM (1:500),
phospho-NF-κB p65 (Ser536), NF-κB p65, p300 and ICAM-1 (1:1,000),
goat polyclonal LXR-α and mouse monoclonal LXR-β (1:200), and
rabbit monoclonal IκBα, phospho-IκBα (Ser32) and β-actin (1:1,000)
primary antibodies overnight at 4°C. Membranes were then incubated
with horseradish peroxidase-conjugated mouse anti-rabbit, goat
anti-mouse or rabbit anti-goat IgGs (1:5,000; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA; cat. nos. sc-2357, sc-2005
and sc-2299, respectively) for 1 h at room temperature. Primary
antibodies against TM, LXR-α, LXR-β, p300 and ICAM-1 were purchased
from Santa Cruz Biotechnology (cat. nos. sc-271804, sc-133221,
sc-1202, sc-8439 and sc-48343, respectively). Phospho-NF-κB p65,
NF-κB p65, IκBα, phospho-IκBα and β-actin were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA; cat. nos. 6956,
13346, 2859, 4812 and 8457, respectively). The proteins were
detected using the SignalFire ECL Reagent (Cell Signaling
Technology, Inc.; cat. no. 6883). Protein expression was quantified
by densitometry using the Quantity One 1-D analysis software,
version 4.6.2 (Bio-Rad Laboratories, Inc.).
Immunoprecipitation and
immunoblotting
HUGECs were treated with media containing 200
μM CaCl2 and dimethylsulfoxide or 2 μM
T0901317. The media covering the cells was removed after 24 h, and
the cells were washed once with phosphate-buffered saline
containing CaCl2. The cells were then scraped from the
plates in 200 μl ice-cold lysis buffer containing 25 mM Tris
(pH 8.0), 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, and protease and
phosphatase inhibitors (Nacalai USA, Inc., San Diego, CA, USA). The
lysed cells were pipetted into 1.5 ml tubes (Eppendorf, Hamburg,
Germany), incubated on ice for 30 min and microcentrifuged at
14,000 × g for 30 min at 4°C. Clarified lysates were incubated
overnight at 4°C with 0.2 μg p300 (sc-48343) or LXR-α
(sc-1202p) monoclonal antibody, and subsequently were incubated for
1 h with Protein A/G agarose (Life Technologies; Thermo Fisher
Scientific). The beads were extensively washed in lysis buffer 3
times for 5 min, and bound proteins were eluted in SDS sample
buffer prior to western blot analysis.
Enzyme-linked immunosorbent assay (ELISA)
for proinflammatory cytokines
Diabetes mellitus is characterized by a systemic
proinflammatory environment, exhibiting enhanced basal and
postprandial circulating levels of proinflammatory cytokines,
including IL-1β, IL-6 and TNF-α (11). To examine the quantity of IL-1β,
and TNF-α in the supernatant of HUGECs, commercially available
ELISA kits were used. IL-1β and TNF-α ELISA kits (EH2IL1B, EH3TNFA)
were obtained from Thermo Fisher Scientific, Inc. In accordance
with the manufacturer's protocol, all supernatants were stored at
−80°C prior to assessment. The supernatants and standards were
centrifuged at 1200 × g for 3 min at 4°C and run in triplicates.
Optical density (OD) at 450 nm was calculated by subtracting the
background, and standard curves were plotted.
Protein C (PC) activation assay
The first group of HUGECs were treated with 2
μM T0901317 for 0, 6, 12 or 24 h; the other two groups of
cells were transfected with pHBAd-U6-GFP-shRNA/TM (AdTMshRNA) or
AdControl first, and subsequently 2 μM T0901317 was
administered to the first group. TM activity was assessed by
determining PC activation in early confluent cells cultured in
96-well plates. The cells were rinsed with buffer containing 20 mM
Tris (pH 7.4), 0.15 M NaCl, 2.5 mM CaCl2 and 5 mg/ml BSA
and then incubated with 40 μl reaction mixture (37.5 nM
thrombin and 5 μg/ml PC in the washing buffer) at 37°C for
30 min. PC activity was terminated by adding 40 μl
argatroban (0.5 mg/ml) and heparin (24 International Units/ml). The
quantity of activated PC (APC) was determined by monitoring the
hydrolysis of chromogenic substrate at 405 nm in a Tecan Sunrise
microtiter plate reader (Tecan Australia Pty Ltd., Melbourne,
Australia). The results are expressed as the mean OD slope values
(ΔOD/Δt). The human PC and human thrombin, and chromogenic
substrate for APC were obtained from Merck Millipore (cat. no.
605190-100U, cat. no. 539215-50UG; Darmstadt, Germany).
Statistical analysis
Results from at least three independent experiments
were analyzed with one-way analysis of variance using SPSS
software, version 18 (SPSS, Inc., Chicago, IL, USA). All data are
expressed as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
LXRs are constitutively expressed in
HUGECs, and LXR agonist T0901317 further enhances their
function
To investigate the expression and function of LXRs
in HUGECs, HUGECs were treated with LXR agonist T0901317 (2 or 4
μM) for 24 h, and the expression of LXR-α and LXR-β was
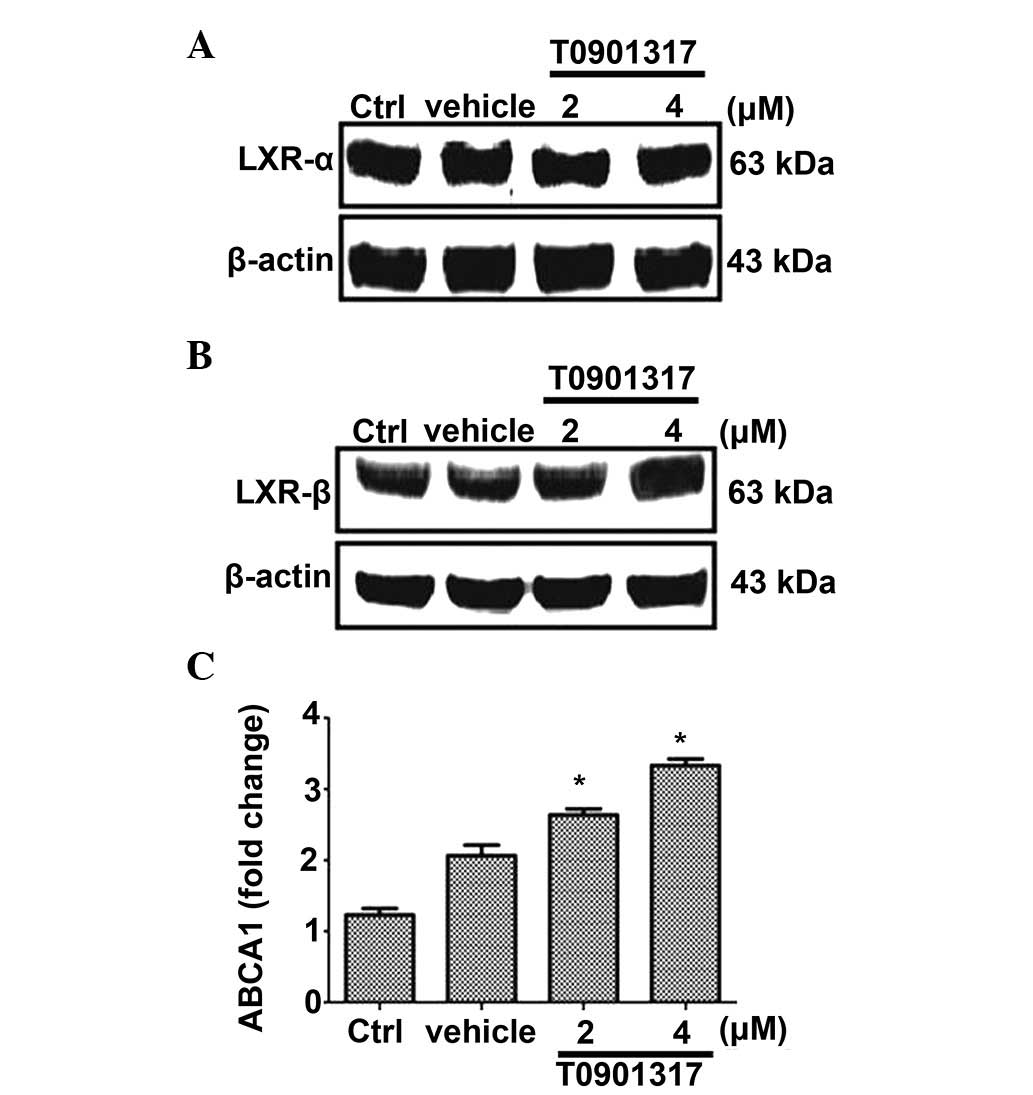
subsequently examined. Western blot analysis indicated that LXR-α
and LXR-β were constitutively expressed in HUGECs (Fig. 1A and B). Addition of the LXR
agonist, T0901317, did not result in an upregulation of the
expression of LXR-α or LXR-β following 48 h of treatment (data not
shown). Following confirmation of LXR expression, the functionality
of LXR in HUGECs was investigated by detecting the transcription
level of ABCA1, one of the LXR target genes (26,27).
RT-qPCR indicated that treatment with T0901317 significantly
increased the mRNA expression level of ABCA1 in HUGECs in a
dose-dependent manner (P<0.05; Fig.
1C).
LXR agonist T0901317 upregulates the
expression of TM in HUGECs
As it was determined that LXR is functional in
HUGECs, it was then investigated whether the activation of LXR may
be involved in the regulation of TM. RT-qPCR analysis indicated
that treatment of HUGECs with T0901317 led to significantly
increased mRNA expression levels of TM in a dose-dependent manner
(P<0.05; Fig. 2A). A similar
dose-dependent increase in protein expression levels was confirmed
by western blot analysis (P<0.05; Fig. 2B). Notably, RT-qPCR indicated that
only LXR-α-siRNA, and not LXR-β-siRNA or scrambled siRNA,
significantly inhibited T0901317-induced expression of TM in HUGECs
(P<0.05; Fig. 2C). These
results for the protein expression levels were confirmed by western
blot analysis (P<0.05; Fig.
2D). The present data suggest that activation of LXR by the
agonist, T0901317, induces upregulation of TM in HUGECs, more
specifically through LXR-α.
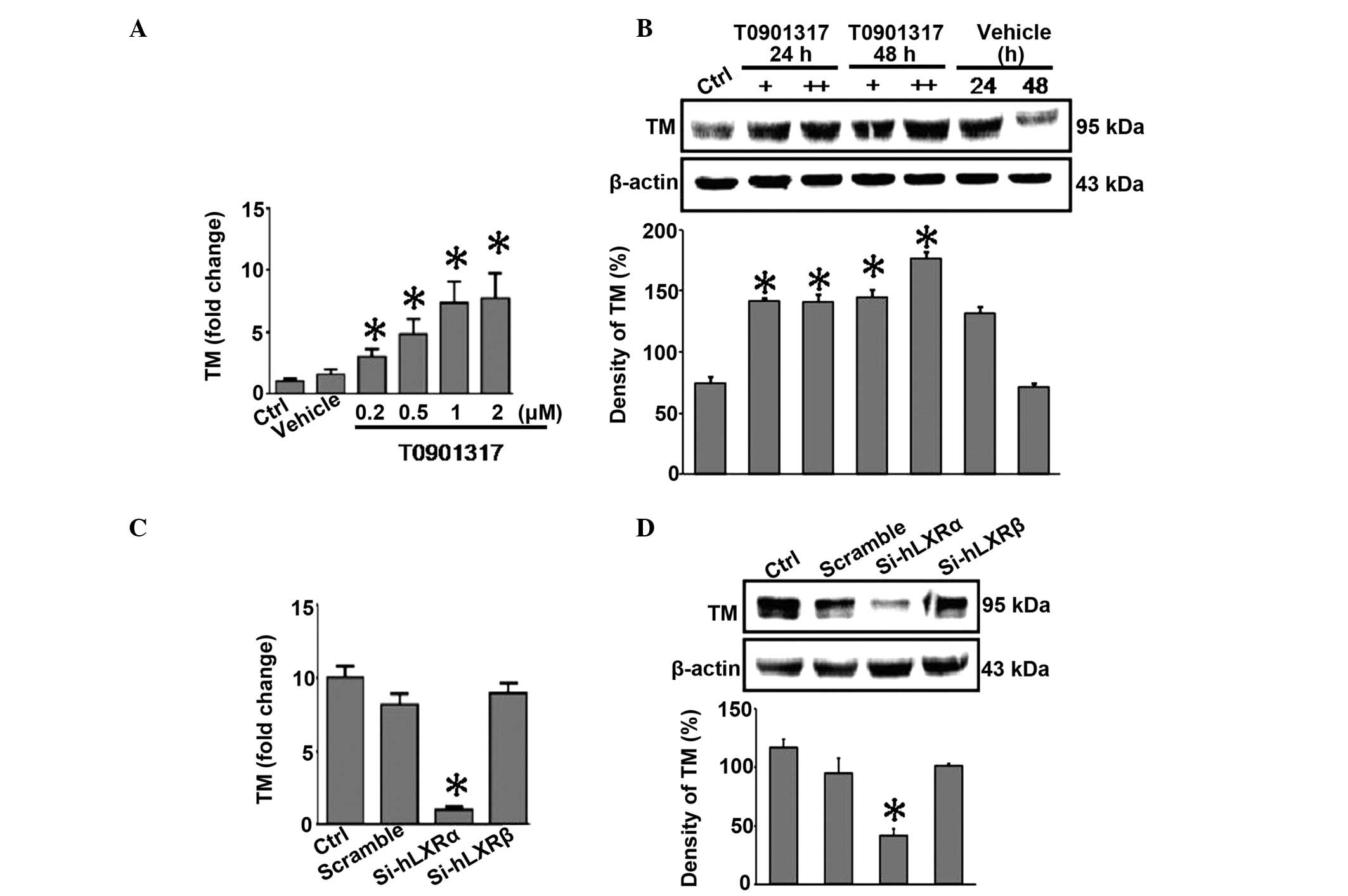 | Figure 2LXR agonist T0901317 upregulates TM
expression in HUGECs. (A) HUGECs were treated with T0901317 (0.2,
0.5, 1 or 2 μM) for 24 h. LXR agonist upregulated the level
of TM in a dose-dependent manner. (B) HUGECs were treated with 2 or
4 μM T0901317 for either a 24 or a 48 h time period. LXR
agonist upregulated the level of TM in a dose-dependent manner, as
determined by western blotting. (C) HUGECs were transfected with
Si-hLXRα or Si-hLXRβ, and the fold change in TM was monitored. (D)
HUGECs were transfected with Si-hLXRα or Si-hLXRβ. The protein
expression of TM in HUGECs was measured by western blot analysis.
Data are presented as the mean ± standard deviation
(*P<0.05). +, 1 μM T0901317; ++, 2 μM
T0901317; Ctrl, control; HUGECs, human glomerular endothelial
cells; TM, thrombomodulin; LXR-α, -β, liver X receptor-α, -β;
siRNA, small interfering RNA; Si-hLXRα, LXRα-siRNA; Si-hLXRβ,
LXRβ-siRNA. |
Upregulation of TM expression by LXR is
dependent on NF-κB inhibition
As LXR is a classical transcriptional factor, it was
hypothesized that LXR upregulates TM expression via directly
enhancing the activity of the TM promoter. To assess this,
luciferase reporter plasmids were constructed, driven by the human
TM promoter. A reporter assay indicated that T0901317 did not
increase the activity of the TM promoter (−2494 to +160 bp) when
transfected with various luciferase reporter plasmids (data not
shown). It is possible that LXR upregulates TM expression via
indirectly modulating the activity of other transcription factors.
To determine the role of NF-κB in mediating the LXR-α-induced
upregulation of TM, the effect of LXR activation on the NF-κB
pathway in HUGECs was investigated by western blot analysis.
High-glucose conditions induced the phosphorylation of IκBα and the
translocation of NF-κB p65 to the nucleus in HUGECs, compared with
the osmotic control (Fig. 3A).
However, the LXR agonist T0901317 suppressed the phosphorylation of
IκBα (Fig. 3A) and inhibited the
nuclear translocation of NF-κB p65 under high-glucose conditions
(Fig. 3B). The activity of NF-κB
was further increased by LXR-α silencing, despite the presence of
T0901317 (Fig. 3C and D). These
data highlight the possible involvement of LXR regulation of NF-κB
activation in TM induction.
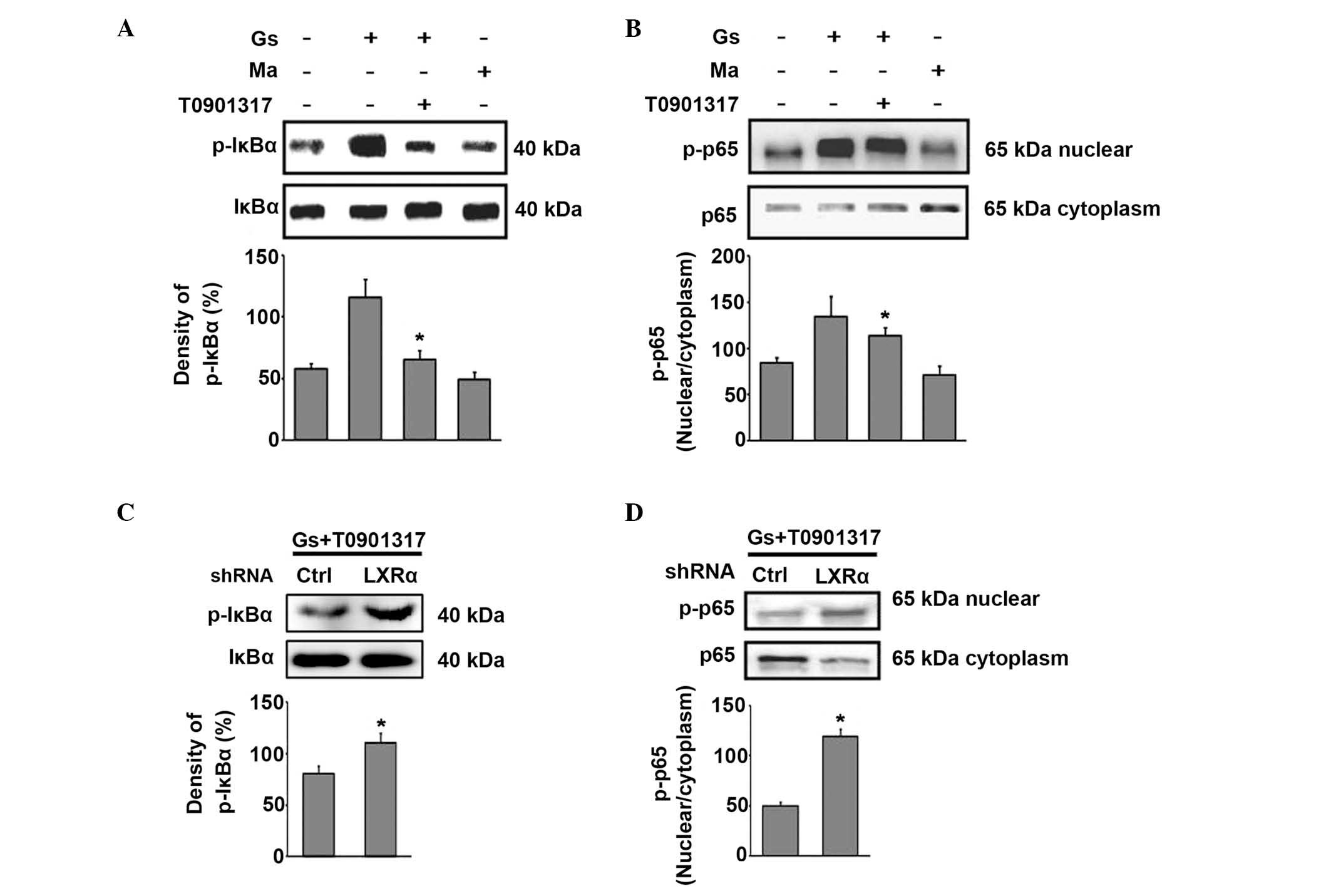 | Figure 3Upregulation of TM expression by LXR
is dependent on NF-κB inhibition. The HUGECs were treated with 2
μM T0901317 for 24 h. These cells were then exposed to 25 mM
glucose or 25 mM mannitol for 6 h. Prior to T0901317 treatment,
HUGECs were successfully transfected with LXR-α shRNA. (A) LXR
agonist T0901317 suppressed the phosphorylation of IκBα. (B) LXR
agonist T0901317 inhibited the nuclear translocation of NF-κB p65
in high-glucose conditions. (C) The expression of p-IκBα upon LXR-α
silencing, despite the presence of T0901317, as assessed by western
blotting. (D) The expression of p-p65 upon LXR-α silencing, despite
the presence of T0901317, as determined by western blotting. Data
are expressed as the mean ± standard deviation
(*P<0.05). Gs, glucose treatment; HUGECs, human
glomerular endothelial cells; LXR, liver X receptor; Ma, mannitol
treatment; NF-κB, nuclear factor-κB; p-IκBα, phospho-nuclear factor
of κ light polypeptide gene enhancer in B-cells inhibitor; shRNA,
small hairpin RNA; TM, thrombomodulin. |
Modulation of TM expression by
interaction of p300 with LXR-α
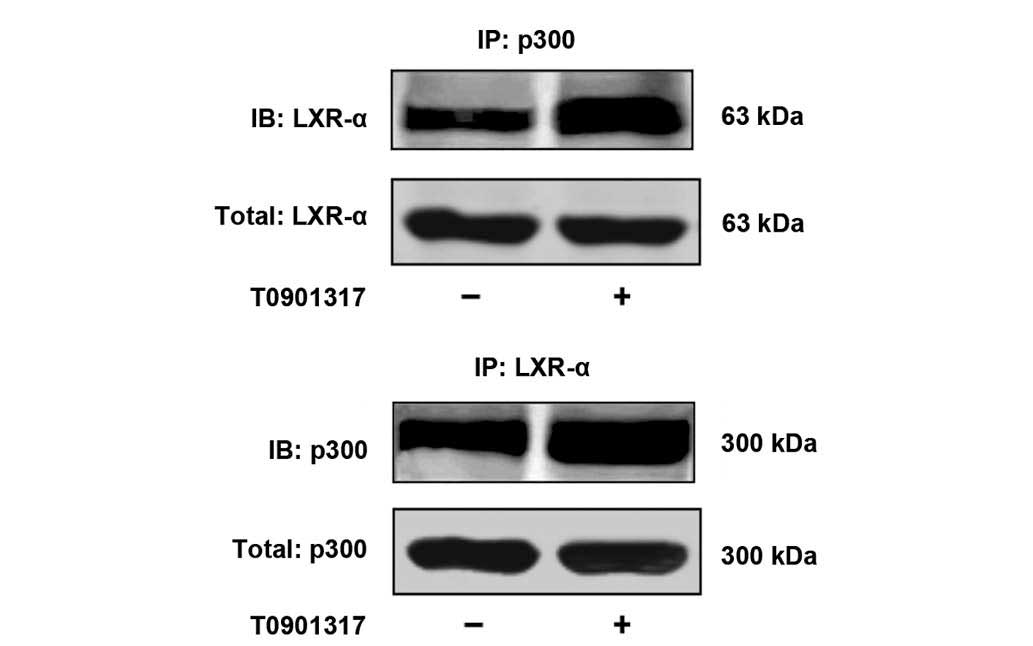
Subsequently, in vitro experiments were
performed to gain further insights into the mechanism through which
the LXR agonist upregulates the expression of TM via the NF-κB/p300
pathway. The effect of T0901317 stimulation on the interaction of
p300 with the LXR-α was also investigated. Therefore,
co-immunoprecipitation experiments were performed on extracts from
HUGECs with or without T0901317 stimulation for 24 h. An
interaction between p300 and LXR-α was demonstrated on the basis of
the co-immunoprecipitation experiment, and the treatment of cells
with 2 μM T0901317 enhanced this interaction (Fig. 4).
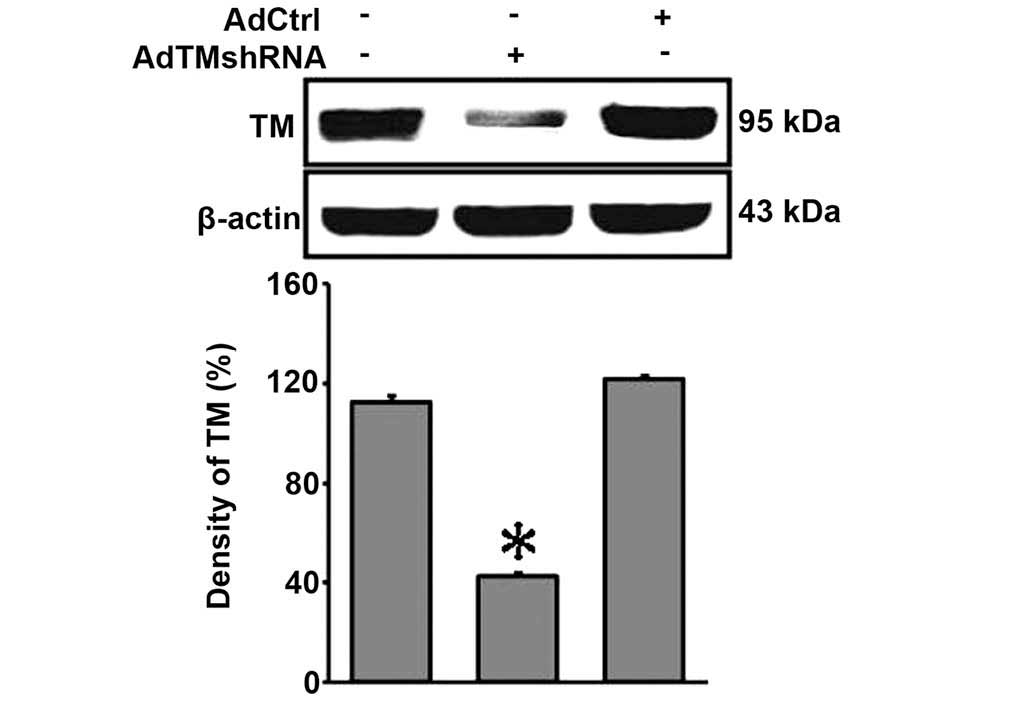
pHBAd-U6-GFP-siRNA/TM inhibits the
expression of TM in HUGECs
The evidence of endonuclease digestion and
sequencing indicated that the recombinant adenovirus vector,
pHBAd-U6-GFP-siRNA/TM, had been successfully constructed. The
expression of GFP was observed in HEK293 cells infected with
pHBAd-U6-GFP-siRNA/TM (data not shown). In the present study, it
was observed that transfection with the recombinant adenovirus
vector, pHBAd-U6-GFP-siRNA/TM, significantly reduced the protein
expression levels of TM (P<0.05; Fig. 5).
LXR agonist T0901317 inhibits the
secretion of inflammatory cytokines and increases APC activity in
HUGECs
As TM has anti-inflammatory effects on PC, the
secretion of inflammatory cytokines in HUGECs was examined upon
hyperglycemia. The cells were transfected with
pHBAd-U6-GFP-shRNA/TM prior to hyperglycemia stimulation. As
presented in Fig. 6A and B,
high-glucose conditions (25 mM glucose) significantly increased the
level of IL-1β and TNF-α in HUGECs, compared with the 25 mM glucose
treatment (P<0.05). The LXR agonist T0901317 (2 μM)
prevented glucose-induced inflammatory cytokine secretion. The
levels of TNF-α (Fig. 6A) and
IL-1β (Fig. 6B) were increased by
TM silencing, despite the presence of T0901317. The production of
APC was markedly increased in HUGECs following treatment with 2
μM T0901317 for 12 h; however, the production of APC was
reduced by TM silencing, despite the presence of T0901317 (Fig. 6C), indicating that LXR activation
may induce TM activity in HUGECs. An increased expression of ICAM-1
following high-glucose stimulation was not detected (data not
shown). The present findings suggest that LXR agonists may inhibit
the inflammatory response of HUGECs via TM activation.
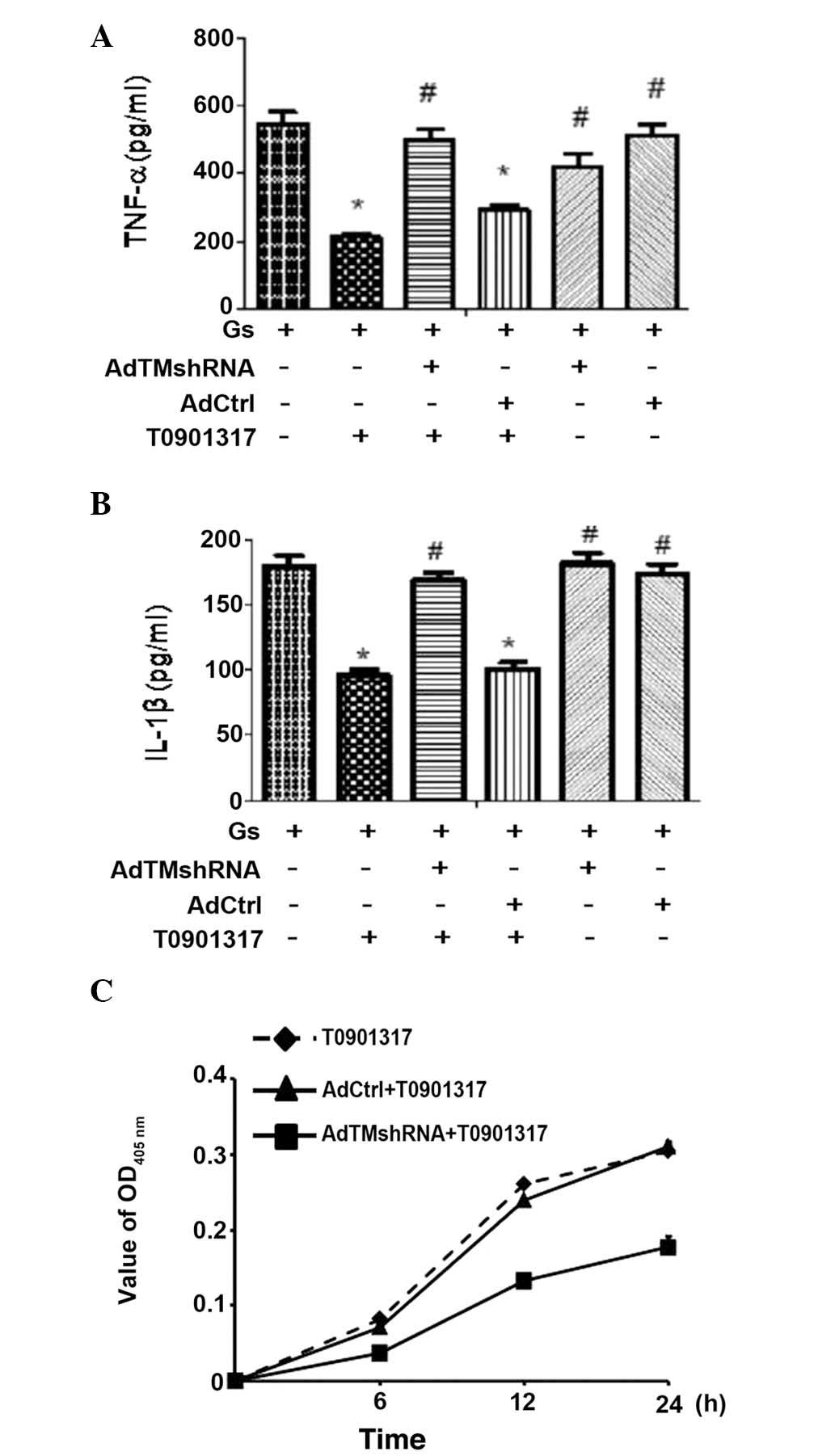 | Figure 6LXR agonist T0901317 inhibits the
secretion of inflammatory cytokines, and improves APC activity in
HUGECs. HUGECs were treated with 2 μM T0901317 for 24 h.
Subsequently, these cells were exposed to 25 mM glucose for 6 h.
Prior to T0901317 treatment, the cells were successfully
transfected with the recombinant adenovirus vector,
pHBAd-U6-GFP-siRNA/TM. Levels of (A) TNF-α and (B) IL-1β were
increased by TM silencing, despite the presence of T0901317. (C)
Production of APC was reduced by TM silencing, despite the presence
of T0901317. Data are presented as mean ± standard deviation
(*P<0.05; #P<0.01). AdCtrl, adenovirus
control; APC, activated protein C; TNF-α, tumor necrosis factor-α;
HUGECs, human glomerular endothelial cells; Gs, glucose treatment;
IL1-1β, interleukin-1β; OD, optical density; TM,
thrombomodulin. |
Discussion
As a key component of the PC anticoagulant pathway,
TM expressed by endothelial cells is an important anti-inflammatory
factor. The present study indicated that LXR-α and LXR-β were
expressed in HUGECs. Additionally, the LXR agonist, T0901317,
promoted transcription of ABCA1 in HUGECs, which was identified as
one of the LXR targets and upregulated by LXR activation (28–30).
In the present study, the LXR agonist T0901317 significantly
increased the expression of TM in HUGECs. Previous studies suggest
that LXR activation may enhance TM activity via the TM-thrombin-PC
system. Activated TM simultaneously binds to thrombin and PC to
form a complex on the surface of endothelial cells, which expresses
the endothelial cell PC receptor (EPCR) (31–33).
APC binds with EPCR to downregulate the production of inflammatory
cytokines (34). The current study
identified that the LXR agonist T0901317 inhibited the secretion of
inflammatory cytokines, and increased the TM activity in HUGECs.
Due to the close association between APC and TM activity, the
present results indicate that the LXR/TM pathway may be a novel
mechanism for LXR-mediated protection of the cells against
inflammation.
Previous studies have determined that NF-κB may
regulate the activities of certain transcription factors by
competitively binding with the p300/CBP complex within the nucleus
(35,36). As previous studies have reported,
NF-κB may be the target gene for the promoter region of LXR
(14,15). The present study determined that
inhibition of NF-κB may be a critical requirement for the LXR
agonist upregulating the expression of TM in vitro. T0901317
suppressed the phosphorylation of IκBα and inhibited the nuclear
translocation of NF-κB p65 protein following high-glucose
treatment. This result is consistent with a previous study, in
which an LXR agonist inhibited IκB phosphorylation and NF-κB p65
translocation into the nucleus, which resulted in an increase in
the protein expression of heme oxygenase-1 (37). The current findings suggest that
the inhibition of TM by NF-κB was possibly achieved indirectly by
competition for the coactivator, p300/CBP, whereas NF-κB
predominantly acts as a transcriptional activator (38). The mechanism by which NF-κB may
regulate gene expression is through competition for other
transcription factors. The transcriptional coactivator, p300,
functions almost identically with histone acetyltransferase, which
modulates the activities of NF-κB transcription factors (36). The current study determined that
treatment of the cells with T0901317 enhanced the interaction
between LXR-α and p300, compared with the vehicle control group. A
previous study reported that TM and p300 are capable of forming a
complex, and LXR may compete with p300 to form a complex with TM
(39). The co-immunoprecipitation
results of the current study suggested that the inhibition of TM
expression by NF-κB may be indirectly achieved through competition
with the coactivator, p300/CBP. Competition with p300/CBP may also
provide an explanation for the observation that
all-trans-retinoic acid (ATRA) is hypothesized to stimulate
the basal gene expression of TM, thus preventing the downregulation
of TM in response to TNF-α (40,41).
It is possible that ATRA or cAMP-induced phosphorylation of the
transcription factor Sp1 increases the affinity of p300/CBP for the
Sp1-Ets nucleosome, thereby preventing it from associating with
NF-κB. The mechanism by which LXRs regulate TM expression has yet
to be fully elucidated. Therefore, further investigation should be
conducted to elucidate the mechanism of LXR in modulating TM
expression.
It is generally accepted that endothelial
dysfunction is important for the development of diabetic
microvascular disease (42).
Microvascular disease may lead to organ damage through impaired
vascular function, increased inflammation or apoptosis (43,44).
Microvascular complications in DN have been associated with the
hyperglycemia-induced inflammatory response in mice and humans
(45–47). In unperturbed endothelial cells,
activation of PC that is dependent on TM inhibits coagulation,
inflammation and apoptosis (48).
The function of the endothelial TM-PC system is impaired in
diabetic individuals. This is indicated by the increased levels of
soluble TM, which reflects a loss of TM from the endothelium and
reduced levels of APC (49,50).
The ability to keep the endothelial balance by exogenous
administration of soluble TM via downregulation of specific
adhesion molecules and chemokines suggests a potential therapeutic
target for intervention in kidney disease associated with chronic
inflammation (51). Thus,
upregulation of TM in GECs may provide a potential intervention to
ameliorate inflammatory endothelial cell dysfunction in diabetes.
The synthesis of active TM in endothelial cells and mesangial cells
within the glomerulus stresses its importance in maintaining renal
hemostatic equilibrium (52). In
the current study, activation of LXR induced the activation of TM,
and reduced the secretion of proinflammatory mediators in
hyperglycemia-stimulated HUGECs. Following transfection with the
recombinant adenovirus vector and TM silencing in the HUGECs, the
secretion of proinflammatory cytokines was upregulated, and PC
activation was reduced compared with the normal control, even
subsequently to the administration of LXR agonists. Thus,
TM-dependent APC formation and the reduction of proinflammatory
mediator secretion may prevent endothelial dysfunction, glomerular
capillary injury and DN. The induction of adhesion molecules on
activated endothelial cells is crucial in monocyte recruitment
during the atherogenic process in DN (53). The current findings did not
determine whether high-glucose treatment increases the expression
of ICAM-1. This indicated that the elevation of extracellular
D-glucose levels may be insufficient to induce vascular
inflammation. A previous study has indicated that sera from
patients with diabetes contains components, which are capable of
inducing vascular cell adhesion molecule-1 expression in
endothelial cells independently of hyperglycemia (53). Further investigation is required to
elucidate the effect of LXR ligands on TM expression in diabetic
nephropathy.
LXR-β is expressed ubiquitously, whereas LXR-α is
expressed at high levels in cholesterol-metabolizing tissues
(54). In the current study, the
two isoforms of LXR, i.e., LXR-α and LXR-β, were expressed
constitutively in HUGECs. Transfection of LXR-α-siRNA was
sufficient to suppress TM expression; however, LXR-β did not
perform an identical function in HUGECs following LXR-β-siRNA
transfection. Additionally, the activity of the NF-κB pathway was
upregulated by LXR-α silencing, despite the presence of T0901317,
indicating LXR-α may be important in modulating TM expression in
HUGECs. T0901317 is a dual agonist that activates LXR-α and LXR-β
(55); however, the function of
endogenous LXR-β, which is involved in the regulation of TM
expression, should not be excluded. On the basis of the similarity
of locations in endothelial cells and anti-inflammatory functions
between LXR and TM, LXR may be a potential 'positive mediator' in
regulating the expression of TM.
In conclusion, the present study, to the best of our
knowledge is the first to report that LXR activation may upregulate
TM expression in HUGECs. It also identified that NF-κB inhibition
may be a critical mediator of TM activation in vitro,
providing a novel therapeutic target for the treatment of
inflammatory diseases characterized by a low expression of TM.
Acknowledgements
The current study was supported by a grant from the
National Natural Science Foundation of China (grant no. 81170680,
awarded to Hanlu Ding) and the Science & Technology Department
of Sichuan Province (grant no. 2013JY0141).
References
|
1
|
Goldberg RB: Cytokine and cytokine-like
inflammation markers, endothelial dysfunction, and imbalanced
coagulation in development of diabetes and its complications. J
Clin Endocrinol Metab. 94:3171–3182. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Conway EM: Thrombomodulin and its role in
inflammation. Semin Immunopathol. 34:107–125. 2012. View Article : Google Scholar
|
|
3
|
Mangan S, Clancy P and Golledge J:
Modulation of endothelial cell thrombomodulin by PPAR ligands -
variation according to environment. Thromb Res. 121:827–834. 2008.
View Article : Google Scholar
|
|
4
|
Kondo K, Ishida T, Yasuda T, Nakajima H,
Mori K, Tanaka N, Mori T, Monguchi T, Shinohara M, Irino Y, et al:
Trans-fatty acid promotes thrombus formation in mice by aggravating
antithrom-bogenic endothelial functions via Toll-like receptors.
Mol Nutr Food Res. 59:729–740. 2015. View Article : Google Scholar
|
|
5
|
He X, Xu Z, Wang B, Zheng Y, Gong W, Huang
G, Zhang L, Li Y and He F: Upregulation of thrombomodulin
expression by activation of farnesoid X receptor in vascular
endothelial cells. Eur J Pharmacol. 718:283–289. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Szanto A and Roszer T: Nuclear receptors
in macrophages: A link between metabolism and inflammation. FEBS
Lett. 582:106–116. 2008. View Article : Google Scholar
|
|
7
|
Calkin AC and Tontonoz P: Liver x receptor
signaling pathways and atherosclerosis. Arterioscler Thromb Vasc
Biol. 30:1513–1518. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Joseph SB, Castrillo A, Laffitte BA,
Mangelsdorf DJ and Tontonoz P: Reciprocal regulation of
inflammation and lipid metabolism by liver X receptors. Nat Med.
9:213–219. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peet DJ, Turley SD, Ma W, Janowski BA,
Lobaccaro JM, Hammer RE and Mangelsdorf DJ: Cholesterol and bile
acid metabolism are impaired in mice lacking the nuclear oxysterol
receptor LXR alpha. Cell. 93:693–704. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fowler AJ, Sheu MY, Schmuth M, Kao J,
Fluhr JW, Rhein L, Collins JL, Willson TM, Mangelsdorf DJ, Elias PM
and Feingold KR: Liver X receptor activators display
anti-inflammatory activity in irritant and allergic contact
dermatitis models: liver-X-receptor-specific inhibition of
inflammation and primary cytokine production. J Invest Dermatol.
120:246–255. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Joseph SB, Bradley MN, Castrillo A, Bruhn
KW, Mak PA, Pei L, Hogenesch J, O'connell RM, Cheng G, Saez E, et
al: LXR-dependent gene expression is important for macrophage
survival and the innate immune response. Cell. 119:299–309. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Valledor AF, Hsu LC, Ogawa S,
Sawka-Verhelle D, Karin M and Glass CK: Activation of liver X
receptors and retinoid X receptors prevents bacterial-induced
macrophage apoptosis. Proc Natl Acad Sci USA. 101:17813–17818.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baranowski M: Biological role of liver X
receptors. J Physiol Pharmacol. 59(Suppl 7): 31–55. 2008.
|
|
14
|
Yang SM, Ka SM, Wu HL, Yeh YC, Kuo CH, Hua
KF, Shi GY, Hung YJ, Hsiao FC, Yang SS, et al: Thrombomodulin
domain 1 ameliorates diabetic nephropathy in mice via
anti-NF-κB/NLRP3 inflammasome-mediated inflammation, enhancement of
NRF2 antioxidant activity and inhibition of apoptosis.
Diabetologia. 57:424–434. 2014. View Article : Google Scholar
|
|
15
|
Wang H, Vinnikov I, Shahzad K, Bock F,
Ranjan S, Wolter J, Kashif M, Oh J, Bierhaus A, Nawroth P, et al:
The lectin-like domain of thrombomodulin ameliorates diabetic
glomerulopathy via complement inhibition. Thromb Haemost.
108:1141–1153. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jayaraman G, Srinivas R, Duggan C,
Ferreira E, Swaminathan S, Somasundaram K, Williams J, Hauser C,
Kurkinen M, Dhar R, et al: p300/cAMP-responsive element-binding
protein interactions with ets-1 and ets-2 in the transcriptional
activation of the human stromelysin promoter. J Biol Chem.
274:17342–17352. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sheppard KA, Rose DW, Haque ZK, Kurokawa
R, McInerney E, Westin S, Thanos D, Rosenfeld MG, Glass CK and
Collins T: Transcriptional activation by NF-kappaB requires
multiple coactivators. Mol Cell Biol. 19:6367–6378. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Birrell MA, Catley MC, Hardaker E, Wong S,
Willson TM, McCluskie K, Leonard T, Farrow SN, Collins JL,
Haj-Yahia S and Belvisi MG: Novel role for the liver X nuclear
receptor in the suppression of lung inflammatory responses. J Biol
Chem. 282:31882–31890. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mogilenko DA, Kudriavtsev IV, Trulioff AS,
Shavva VS, Dizhe EB, Missyul BV, Zhakhov AV, Ischenko AM,
Perevozchikov AP and Orlov SV: Modified low density lipo-protein
stimulates complement C3 expression and secretion via liver X
receptor and Toll-like receptor 4 activation in human macrophages.
J Biol Chem. 287:5954–5968. 2012. View Article : Google Scholar :
|
|
20
|
Cheng O, Ostrowski RP, Liu W and Zhang JH:
Activation of liver X receptor reduces global ischemic brain injury
by reduction of nuclear factor-kappaB. Neuroscience. 166:1101–1109.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen S, Sorrentino R, Shimada K, Bulut Y,
Doherty TM, Crother TR and Arditi M: Chlamydia pneumoniae-induced
foam cell formation requires MyD88-dependent and-independent
signaling and is reciprocally modulated by liver X receptor
activation. J Immunol. 181:7186–7193. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Puddu GM, Cravero E, Arnone G, Muscari A
and Puddu P: Molecular aspects of atherogenesis: New insights and
unsolved questions. J Biomed Sci. 12:839–853. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang GX, Hu L, Zhang Z and Liu DP:
Construction of an adenoviral expression vector carrying FLAG and
hrGFP-1 genes and its expression in bone marrow mesenchymal stem
cells. Genet Mol Res. 13:1070–1078. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Luo J, Deng ZL, Luo X, Tang N, Song WX,
Chen J, Sharff KA, Luu HH, Haydon RC, Kinzler KW, et al: A protocol
for rapid generation of recombinant adenoviruses using the AdEasy
system. Nature Protoc. 2:1236–1247. 2007. View Article : Google Scholar
|
|
25
|
Edmunds RC, McIntyre JK, Luckenbach JA,
Baldwin DH and Incardona P: Toward enhanced MIQE compliance:
Reference residual normalization of qPCR gene expression data. J
Biomol Tech. 25:54–60. 2014.PubMed/NCBI
|
|
26
|
Khovidhunkit W, Moser AH, Shigenaga JK,
Grunfeld C and Feingold KR: Endotoxin down-regulates ABCG5 and
ABCG8 in mouse liver and ABCA1 and ABCG1 in J774 murine
macrophages: Differential role of LXR. J Lipid Res. 44:1728–1736.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stefulj J, Panzenboeck U, Becker T,
Hirschmugl B, Schweinzer C, Lang I, Marsche G, Sadjak A, Lang U,
Desoye G and Wadsack C: Human endothelial cells of the placental
barrier efficiently deliver cholesterol to the fetal circulation
via ABCA1 and ABCG1. Circ Res. 104:600–608. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ishibashi M, Filomenko R, Rébé C,
Chevriaux A, Varin A, Derangère V, Bessède G, Gambert P, Lagrost L
and Masson D: Knock-down of the oxysterol receptor LXRalpha impairs
cholesterol efflux in human primary macrophages: Lack of
compensation by LXRβ activation. Biochem Pharmacol. 86:122–129.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Repa JJ, Turley SD, Lobaccaro JA, Medina
J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM and Mangelsdorf
DJ: Regulation of absorption and ABC1-mediated efflux of
cholesterol by RXR heterodimers. Science. 289:1524–1529. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang N, Lan D, Chen W, Matsuura F and Tall
AR: ATP-binding cassette transporters G1 and G4 mediate cellular
cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci
USA. 101:9774–9779. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Feistritzer C and Riewald M: Endothelial
barrier protection by activated protein C through PAR1-dependent
sphin-gosine 1-phosphate receptor-1 crossactivation. Blood.
105:3178–3184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Matsumoto H, Yamakawa K, Ogura H, Koh T,
Matsumoto N and Shimazu T: Enhanced expression of cell-specific
surface antigens on endothelial microparticles in sepsis-induced
disseminated intravascular coagulation. Shock. 43:443–449. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Braach N, Frommhold D, Buschmann K, Pflaum
J, Koch L, Hudalla H, Staudacher K, Wang H, Isermann B, Nawroth P
and Poeschl J: RAGE controls activation and anti-inflammatory
signalling of protein C. PloS One. 9:e894222014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Esmon CT: Protein C anticoagulant
system–anti-inflammatory effects. Semin Immunopathol. 34:127–132.
2012. View Article : Google Scholar
|
|
35
|
Gires O, Kieu C, Fix P, Schmitt B, Münz M,
Wollenberg B and Zeidler R: Tumor necrosis factor alpha negatively
regulates the expression of the carcinoma-associated antigen
epithelial cell adhesion molecule. Cancer. 92:620–628. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ravi R, Mookerjee B, van Hensbergen Y,
Bedi GC, Giordano A, El-Deiry WS, Fuchs EJ and Bedi A: p53-mediated
repression of nuclear factor-kappaB RelA via the transcriptional
integrator p300. Cancer Res. 58:4531–4536. 1998.PubMed/NCBI
|
|
37
|
Wang Y, Li C, Cheng K, Cheng K, Zhang R,
Narsinh K, Li S, Li X, Qin X, Zhang R, Li C, et al: Activation of
liver X receptor improves viability of adipose-derived mesenchymal
stem cells to attenuate myocardial ischemia injury through
TLR4/NF-κB and Keap-1/Nrf-2 signaling pathways. Antioxid Redox
Signal. 21:2543–2557. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kaur U, Banerjee P, Bir A, Sinha M, Biswas
A and Chakrabarti S: Reactive oxygen species, redox signaling and
neuroinflammation in Alzheimer's disease: The NF-κB connection.
Curr Top Med Chem. 15:446–457. 2015. View Article : Google Scholar
|
|
39
|
Drdová B and Vachtenheim J: A role for p21
(WAF1) in the cAMP-dependent differentiation of F9 teratocarcinoma
cells into parietal endoderm. Exp Cell Res. 304:293–304. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ishii H, Horie S, Kizaki K and Kazama M:
Retinoic acid counteracts both the downregulation of thrombomodulin
and the induction of tissue factor in cultured human endothelial
cells exposed to tumor necrosis factor. Blood. 80:2556–2562.
1992.PubMed/NCBI
|
|
41
|
Koga S, Morris S, Ogawa S, Liao H,
Bilezikian JP, Chen G, Thompson WJ, Ashikaga T, Brett J, Stern DM,
et al: TNF modulates endothelial properties by decreasing cAMP. Am
J Physiol. 268:C1104–C1113. 1995.PubMed/NCBI
|
|
42
|
Goligorsky MS, Chen J and Brodsky S:
Workshop: Endothelial cell dysfunction leading to diabetic
nephropathy: Focus on nitric oxide. Hypertension. 37:744–748.
2003.§1. View Article : Google Scholar
|
|
43
|
Brownlee M: Biochemistry and molecular
cell biology of diabetic complications. Nature. 414:813–820. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Endemann DH and Schiffrin EL: Endothelial
dysfunction. J Am Soc Nephrol. 15:1983–1992. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Murata I, Takemura G, Asano K, Sano H,
Fujisawa K, Kagawa T, Baba K, Maruyama R, Minatoguchi S, Fujiwara T
and Fujiwara H: Apoptotic cell loss following cell proliferation in
renal glomeruli of Otsuka Long-Evans Tokushima Fatty rats, a model
of human type 2 diabetes. Am J Nephrol. 22:587–595. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kumar D, Robertson S and Burns KD:
Evidence of apoptosis in human diabetic kidney. Mol Cell Biochem.
259:67–70. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Baba K, Minatoguchi S, Sano H, Kagawa T,
Murata I, Takemura G, Hirano T, Ohashi H, Takemura M and Fujiwara
T: Involvement of apoptosis in patients with diabetic nephropathy:
A study on plasma soluble Fas levels and pathological findings.
Nephrology (Carlton). 9:94–99. 2004. View Article : Google Scholar
|
|
48
|
Esmon CT: Inflammation and the activated
protein C anticoagulant pathway. Semin Thromb Hemost. 32(Suppl 1):
49–60. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Borcea V, Morcos M, Isermann B, Henkels M,
Ziegler S, Zumbach M, Amiral J, Längst KD, Seiz W, Ziegler R, et
al: Influence of ramipril on the course of plasma thrombomodulin in
patients with diabetes mellitus. Vasa. 28:172–180. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fujiwara Y, Tagami S and Kawakami Y:
Circulating thrombomodulin and hematological alterations in type 2
diabetic patients with retinopathy. J Atheroscler Thromb. 5:21–28.
1998. View Article : Google Scholar
|
|
51
|
Rajashekhar G, Gupta A, Marin A, Friedrich
J, Willuweit A, Berg DT, Cramer MS, Sandusky GE, Sutton TA, Basile
DP, et al: Soluble thrombomodulin reduces inflammation and prevents
microalbuminuria induced by chronic endothelial activation in
transgenic mice. Am J Physiol Renal Physiol. 302:F703–F712. 2012.
View Article : Google Scholar :
|
|
52
|
Pruna A, Peyri N, Berard M and Boffa MC:
Thrombomodulin is synthesized by human mesangial cells. Kidney Int.
51:687–693. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Rasmussen LM, Schmitz O and Ledet T:
Increased expression of vascular cell adhesion molecule-1 (VCAM-1)
in cultured endothelial cells exposed to serum from type 1 diabetic
patients: No effects of high glucose concentrations. Scand J Clin
Lab Invest. 62:485–493. 2002. View Article : Google Scholar
|
|
54
|
Janowski BA, Willy PJ, Devi TR, Falck JR
and Mangelsdorf DJ: An oxysterol signalling pathway mediated by the
nuclear receptor LXR alpha. Nature. 383:728–731. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fiévet C and Staels B: Liver X receptor
modulators: Effects on lipid metabolism and potential use in the
treatment of atherosclerosis. Biochem Pharmacol. 77:1316–1327.
2009. View Article : Google Scholar
|