Introduction
Brugada syndrome (BrS) is a life-threatening,
inherited, primary arrhythmia disorder characterized by ST-segment
elevation in the right precordial leads (V1–V3) of the
electrocardiogram (ECG), which does not exhibit any clinical
cardiac abnormality (1,2). BrS patients may experience syncope
and sudden cardiac death (SCD) from episodes of polymorphic
ventricular tachycardia (PVT) and ventricular fibrillation (VF),
particularly in healthy young males (3). BrS accounts for 4% of all SCD and
≤20% of sudden deaths in patients without obvious structural heart
disease (3). BrS is an inherited
disease that shows an autosomal dominant pattern with incomplete
penetrance and an incidence ranging from ~5 per 10,000 in Western
countries to 12 per 10,000 in Southeast Asia (3–5).
To date, although 22 susceptibility genes have been
identified in BrS, SCN5A, which encodes the α subunit of the
major cardiac sodium channel (Nav1.5), is the most common
pathogenic gene and is responsible for 11–28% of BrS patients
(3,6–8).
However, mutations in other genes have rarely been observed in BrS
patients and account for the minority (<25%) of BrS
genotype-positive cases (8,9).
Furthermore, >370 mutations in the SCN5A gene have been
associated with BrS using a web database, The Gene Connection For
The Heart (http://triad.fsm.it/cardmoc/). Many of these
mutations, which have been characterized in cell lines expressing
BrS mutant channels revealed a loss-of-function effect on the
sodium current by certain mechanisms, such as reduced current
density or disrupted biophysical properties (10–12).
Thus, the genetic etiology of BrS remains unclear. Identifying
novel susceptibility genes will provide clinical benefit for early
diagnosis, risk stratification and personalized treatment of
BrS.
The present study investigated a Chinese Han patient
presenting with BrS carrying a novel heterozygous mutation,
p.D1690N [found in the S5–S6 linker of domain IV (DIV) of the
Nav1.5 channel] in the SCN5A gene. In addition, the
functional outcomes of the mutated Nav1.5 channel proteins were
examined in HEK293 cells. The results demonstrated that the
mutation reduced sodium current density and altered the biophysical
sodium channel characteristics.
Materials and methods
Clinical characteristics
A 37-year-old male was admitted to the emergency
department of The First Affiliated Hospital of Xiamen University
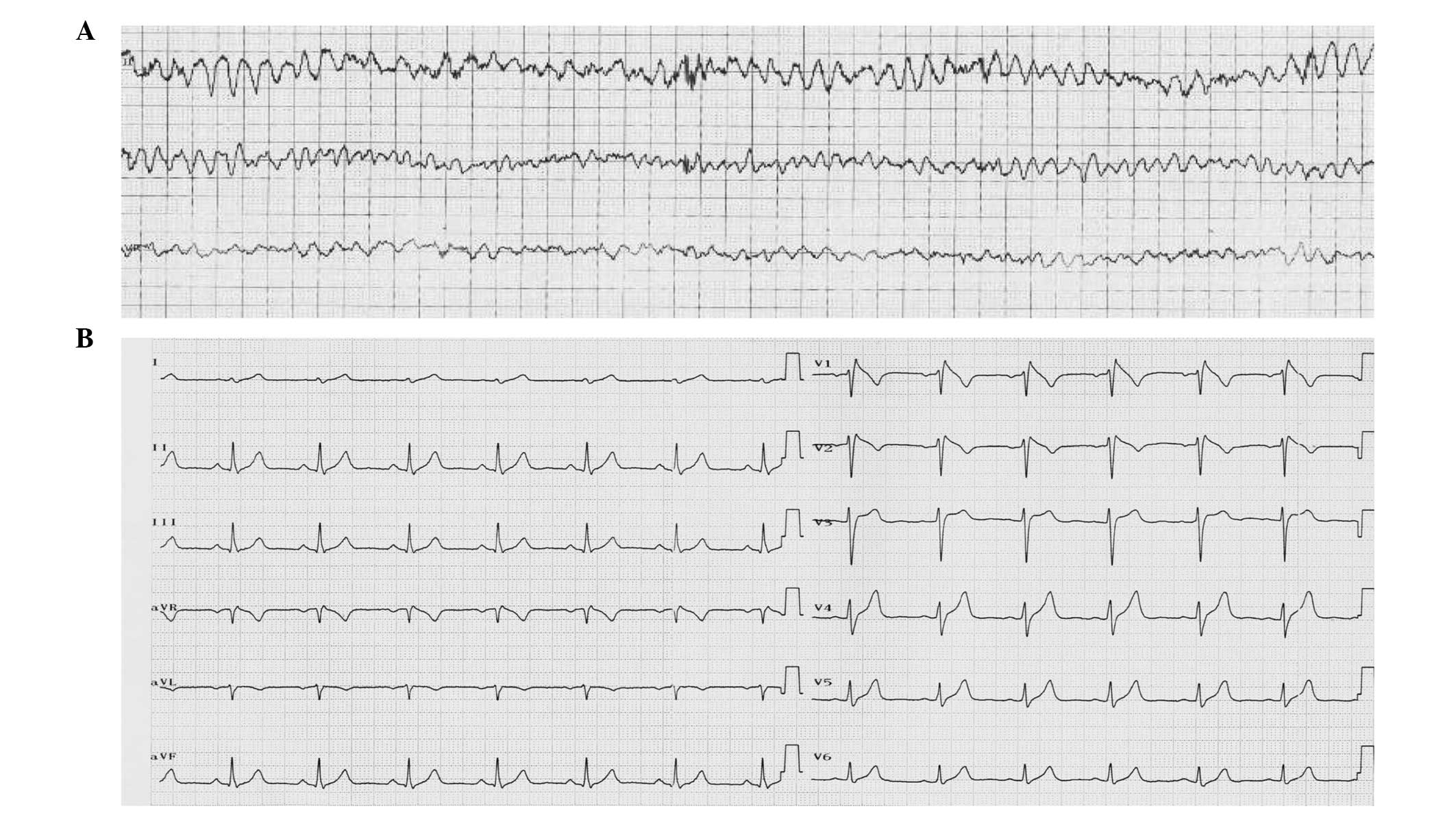
(Xiamen, China) in September 2009, due to sudden syncope from an
episode of VF (Fig. 1A). A
subsequent 12-lead ECG at rest was consistent with a type-1 Brugada
pattern, defined as a prominent coved-type ST-segment elevation,
displaying ST-segment elevation >2 mm at its peak followed by a
negative T wave (3) and incomplete
right bundle branch block (RBBB; Fig.
1B). The BrS patient demonstrated no evidence of structural
heart disease by exercise stress test, electrophysiological study
and echocardiography. Subsequently, the family of the patient (four
males and three females; mean age, 38.9±17.6 years) underwent
physical examination, 12-lead ECG, 24-h Holter ECG monitoring
(Nihon Kohden, Tokyo, Japan), echocardiography and genetic testing.
As ajmaline, flecainide and procainamide, recommended by the
consensus report (13), are
unobtainable in China, propafenone (Shanghai Xinyi Jinzhu
Pharmaceutical Co., Ltd., Shanghai, China) was administered, which
has been demonstrated to reveal BrS. Propafenone challenges were
performed and evaluated on two of the family members [the sister
(II.3) and nephew (III.2) of the proband] as previously described
(13). The subjects provided
written informed consent and, following a fasting period (~10 h
overnight), propafenone (1 mg/kg body weight; 10 mg/min) was
intravenously administered while the patient was continuously
monitored by 12-lead ECG and blood pressure. After 20 min, if the
reaction was negative (i.e. no change in ECG pattern compared with
the baseline, including absence of ST segment elevation and QRS
duration prolongation), an additional 0.5 mg/kg propafenone was
injected for 2.5 min. Propafenone infusion was terminated when the
diagnostic type I Brugada ECG pattern or ventricular arrhythmias
were apparent. After termination of propafenone administration,
monitoring was continued for a minimum of 24 h or until the ECG
normalized. The propafenone challenge was performed and evaluated
on two family members, II.3 and III.2, who were carriers of the
SCN5A mutation.
Genetic analysis
The current study conforms with the principles
outlined in the Helsinki Declaration and was approved by the
Medical Ethical Committee of The First Affiliated Hospital of
Xiamen University. All subjects provided written informed consent
following counseling. Genomic DNA was isolated from leukocyte
nuclei using a TIANamp Blood DNA kit (Tiangen Biotech Co., Ltd.,
Beijing, China), and the cardiac sodium channel gene, SCN5A
(transcript, NM_198056.2) was directly sequenced following
polymerase chain reaction (PCR) amplification, with the use of an
ABI PRISM 3730xl DNA sequencer (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) performed by Tsingke Biological Technology Co.,
Ltd. (Beijing, China), as previously described (12). All DNA-identified variants were
compared with a control group of 150 healthy and unrelated Chinese
Han individuals (300 alleles) after obtaining written informed
consent.
Site-directed mutagenesis and
heterologous expression
The p.D1690N mutation was introduced into
pcDNA3.1-hH1 using a PCR-based mutagenesis method, as previously
described (12). The mutated
plasmids were sequenced (Tsingke Biological Technology Co., Ltd.)
to ensure the presence of the p.D1690N mutation and the absence of
spurious mutations.
The human embryonic kidney 293 cell line, HEK293 was
cultured in an incubator in Gibco Dulbecco's modified Eagle's
medium (Thermo Fisher Scientific, Inc.) supplemented with Gibco 10%
fetal bovine serum (Thermo Fisher Scientific, Inc.), 4 mmol/l
glutamine (Invitrogen; Thermo Fisher Scientific, Inc.), 100 IU/ml
penicillin (Amresco LLC, Solon, OH, USA) and 100 μg/ml
streptomycin (Amresco LLC) at 37°C in a humidified atmosphere of 5%
CO2. Prior to over-expression, the cells were seeded in
6-well plates and, upon reaching 80% confluence, were
co-transfected with 0.8 μg pcDNA3.1-hH1 or p.D1690N mutant
and 0.8 μg pIRES2-DsReD-sodium voltage-gated channel β
subunit 1 (SCN1B; which served as a reporter gene) with
Invitrogen Lipofectamine 2000 (Thermo Fisher Scientific, Inc.),
according to the manufacturer's instructions and as previously
reported (12).
Electrophysiological analysis
Twenty-four to 48 h after transfection, the sodium
current (INa) was recorded in cells displaying red
fluorescence at room temperature (23–25°C) under whole cell
patch-clamp technique, as previously described (12). Briefly, the cells were continuously
superfused with a bath solution containing 70 mM NaCl, 80 mM CsCl,
5.4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM
HEPES and 10 mM glucose (pH adjusted to 7.3 using CsOH). The patch
pipette (tip resistance, 2–3 MΩ) was filled with intracellular
medium containing 20 mM NaCl, 130 mM CsCl, 10 mM HEPES and 10 mM
EGTA (pH adjusted to 7.2 with CsOH). All the above-mentioned items
were purchased from Sigma-Aldrich (St. Louis, MO, USA). The sodium
current was recorded using a MultiClamp™ 700B amplifier (Molecular
Devices, LLC, Sunnyvale, CA, USA) and was converted to digital data
using a Digidata® 1440A A/D converter (5 kHz filtering;
Molecular Devices, LLC).
Statistical analysis
Continuous results are expressed as means ± standard
error of the mean. Currents were analyzed using Clampfit 10.1
software (Molecular Devices, LLC, Sunnyvale, CA, USA) and Origin
Pro 8.5 (OriginLab Corporation,Northampton, MA, USA). Statistical
comparisons between the WT and mutation groups were evaluated using
the two-tailed Student's t-test and P<0.05 was considered
to indicate a statistically significant difference.
Results
Clinical studies
The present study included seven members of a
Chinese Han family with BrS and a history of SCD (the genetic
background is presented in Fig.
2A). The proband, a 37-year-old male, was admitted to the
emergency department of The First Affiliated Hospital of Xiamen
University due to sudden syncopes at night. An ECG demonstrated VF
(Fig. 1A), which was treated by
electric defibrillation. The clinical history revealed that the
patient's father (I.1) succumbed suddenly at the age 53 years. The
patient experienced PVT three times in hospital, each of which were
successfully treated by electric defibrillation.
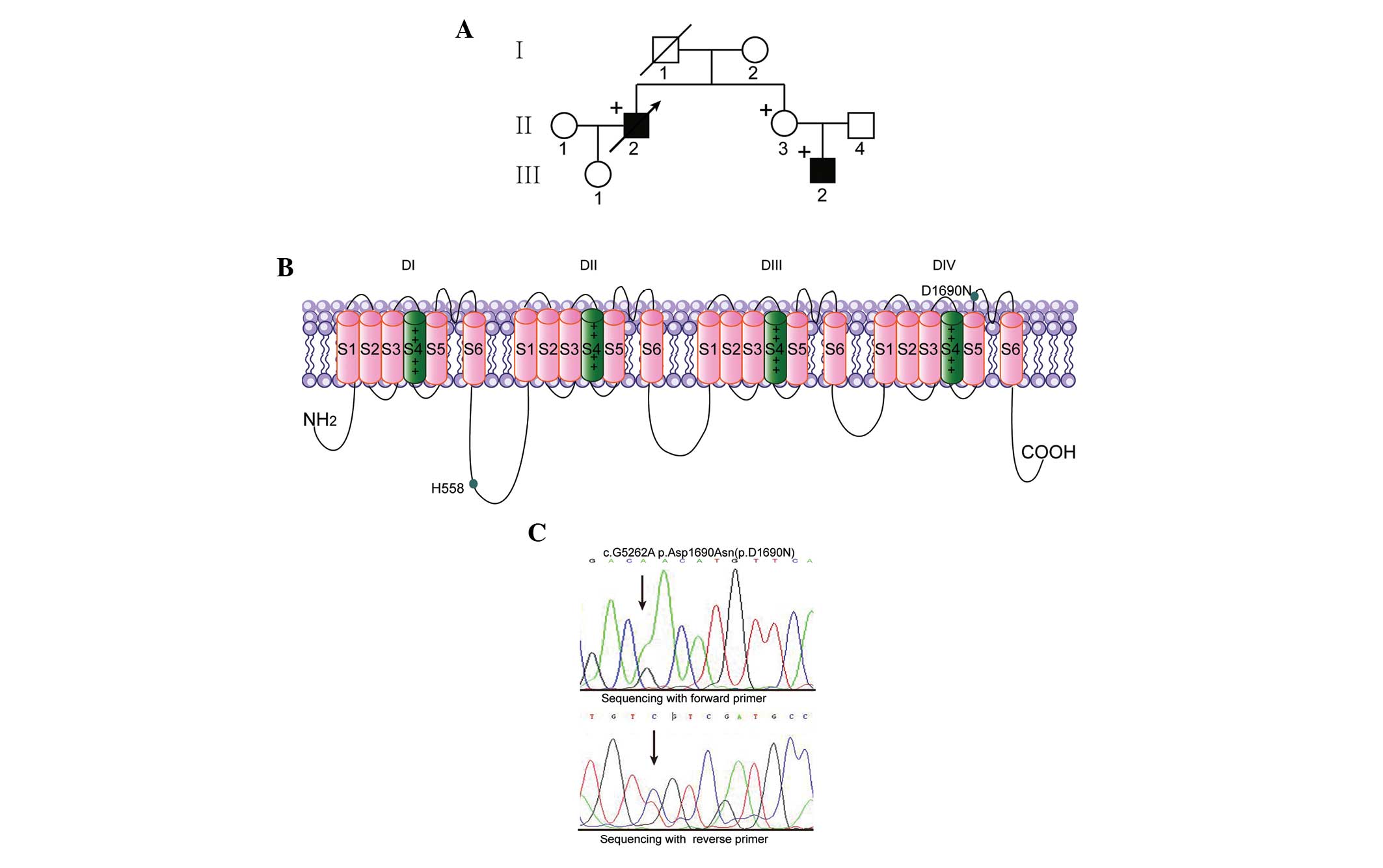 | Figure 2Genetic analysis and background of
the proband and family members. (A) The genetic background and
mutation status of the Chinese Han family: Males, squares; females,
circles; BrS phenotype, filled symbols; variation carrier for SCN5A
c.5262G > A, + symbol; proband, arrow. (B) Location of the
p.D1690N mutation and the polymorphism in the predicted topological
diagram of the Nav1.5 channel. (C) Polymerase chain reaction-based
sequence of SCN5A exon 28 demonstrating a heterozygous
nucleic acid substitution (c.5262G > A) in the proband, the
sister (II.3) and the nephew (III.2), resulting in a single amino
acid substitution, p.Asp1690Asn (p.D1690N). SCN5A, sodium
voltage-gated channel α subunit 5; D, domain. |
At rest, the ECG demonstrated sinus rhythm and was
compatible with the type-1 Brugada pattern (Fig. 1B), showing ST-elevation in V1–V3
followed by negative T wave and RBBB. No structural heart diseases
or valvular disease were observed by echocardiography. The proband
refused an implantable cardioverter defibrillator, as a result of
financial constraints, and succumbed two months later due to SCD.
Following complete cardiologic examination, the majority of the
patient's relatives exhibited normal ECG patterns and
echocardiograms, although the patient's nephew (III.2) exhibited a
BrS diagnostic ECG and the patient's sister (II.3) exhibited a
negative reaction following propafenone challenge.
Molecular genetic findings
By sequencing SCN5A, a novel missense
mutation, c.5262G > A, which was identified in three family
members (II.2, II.3 and III.2), is predicted to replace aspartic
acid (D) with asparagine (N) at position 1690 (D1690N) in the DIV
S5–S6 linker of the Nav1.5 channels (Fig. 2). The variant was not detected in
150 ethnically matched, healthy volunteers (300 alleles) and The
Human Gene Mutation Database (14)
ruled out the polymorphism.
Additionally, the common single nucleotide
polymorphism, c.1673A > G, which produces the p.H558R
polymorphism, was not detected in the family members.
Electrophysiology of p.D1690N Nav1.5
channels
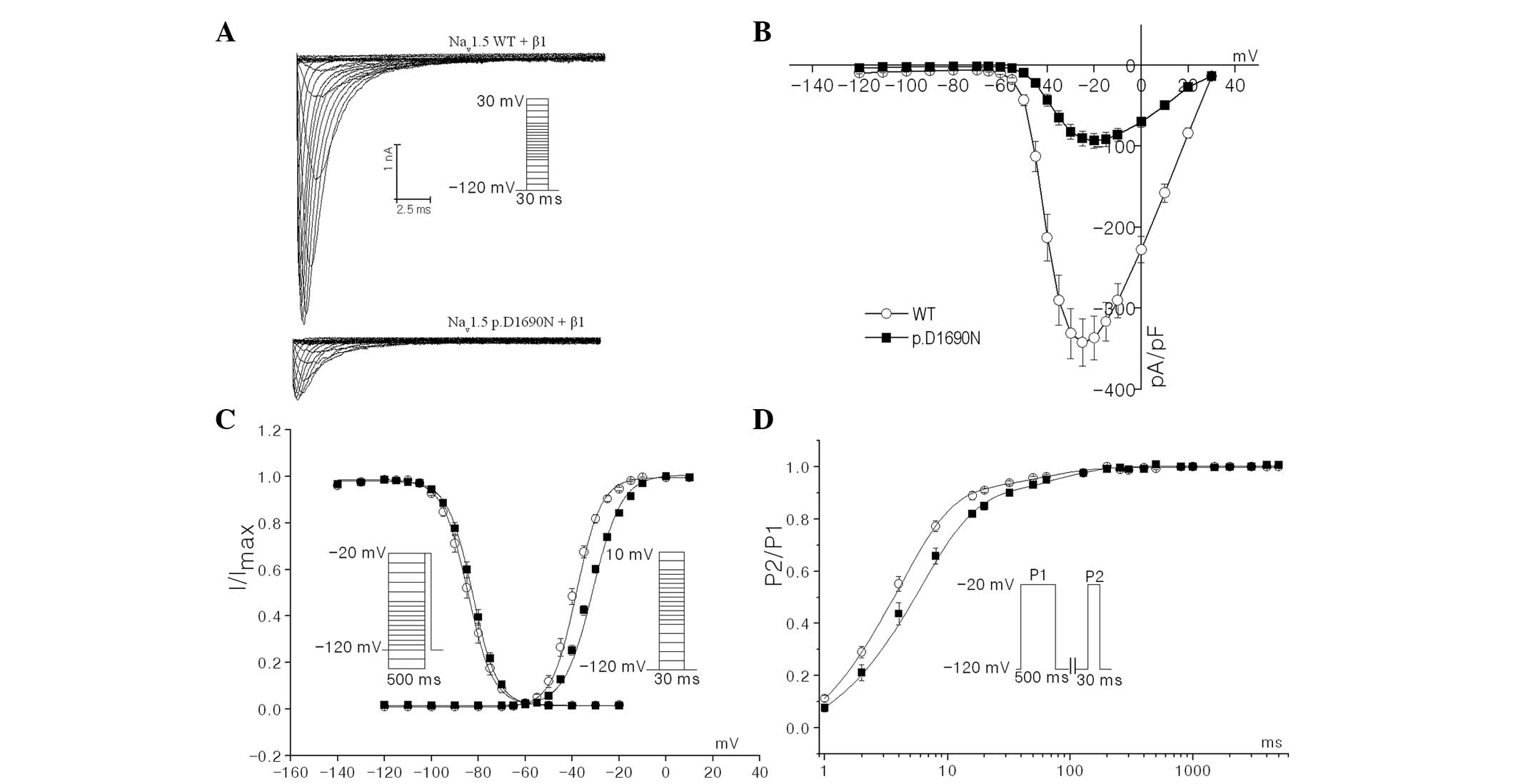
To characterize the electrophysiological
consequences of the p.D1690N mutation on Nav1.5 channel activity,
the biophysical properties of p.D1690N mutant with WT recombinant
human sodium channels, combined with the hβ1 subunit in HEK293 cell
lines, were compared using the whole-cell patch-clamp technique. As
shown in Fig. 3A and B, the
mutation significantly reduced the peak current densities of Nav1.5
[−20-mV current density: WT (n=31), −336.7±26.9 pA/pF vs. mutant
(n=34), −93.3±9.0 pA/pF; P<0.01]. Therefore, it was hypothesized
that the position, D1690 may be key in the trafficking and/or
stability of the channel.
As shown in Fig.
3C, the steady-state activation and inactivation curves of the
WT and mutant were obtained by plotting the normalized peak current
as the corresponding membrane voltage. The midpoint of activation
(V1/2 act) of the p.D1690N mutant was
markedly shifted by 7 mV to more positive potentials when compared
with the WT channels [V1/2 act: −38.2±1.7 mV,
(n=14) vs. −31.2±1.6 mV (n=12) for WT and mutant channels,
respectively; P<0.01, Fig. 3C].
By contrast, no significant differences were observed in the
voltage dependence of steady-state inactivation
[V1/2 inact: −86.0±1.5 mV (n=15) vs.
−82.2±1.6 mV (n=13) for WT and mutant channels, respectively;
Fig. 3C]. These results revealed
that the mutation accelerated activation kinetics. In addition, the
recovery constant from fast inactivation for mutant channels was
significantly delayed when compared with WT channels [τf=3.9±0.3
msec, τs=57.1±11.0 msec (n=14) vs. τf=5.9±0.4 msec (P<0.01 vs.
WT), τs=73.9±9.7 msec (n=15) for WT and mutant channels,
respectively; Fig. 3D].
Discussion
In the present study, the functional consequences of
a novel missense mutation, p.D1690N, which was identified in a
patient with repeated episodes of VF, and whose ECG presents the
typical features of BrS under basal conditions, was investigated.
The electrophysiological analysis revealed that the p.D1690N
mutation results in a significant loss of function in the sodium
channel, which was predominantly attributable to the reduction in
current density and abnormal kinetic properties. Numerous studies
indicate that SCN5A mutations associated with BrS result in
loss of function of sodium channels (10,15–17).
The present results indicate that the BrS in this particular family
was attributed to the novel missense mutation, p.D1690N in the
Nav1.5 channels.
BrS is an inherited channelopathy that is
characterized by ST-segment elevation in the right precordial leads
(V1–V3) of the electrocardiogram, but that does not demonstrate any
cardiac structural disease. To date, 22 BrS-susceptibility genes
have been identified (Table I),
primarily via candidate gene approaches (3,6–8).
These genes encode subunits of sodium channels and their
interacting proteins [SCN5A, sodium voltage-gated channel α
subunit 10 (SCN10A), SCN1B, sodium voltage-gated
channel β subunit 2, sodium voltage-gated channel β subunit 3,
glycerol-3-phosphate dehydrogenase 1-like, RAN guanine nucleotide
release factor, fibroblast growth factor 12, plakophilin 2,
sarcolemma associated protein], potassium channels and their
interacting proteins (potassium voltage-gated channel subfamily H
member 2, potassium voltage-gated channel subfamily E regulatory
subunit 3, potassium voltage-gated channel subfamily E regulatory
subunit 5, potassium voltage-gated channel subfamily D member 3,
potassium voltage-gated channel subfamily J member 8, semaphorin
3A, ATP binding cassette subfamily C member 9), L-type calcium
channels [calcium voltage-gated channel subunit α1 C
(CACNA1C), calcium voltage-gated channel auxiliary subunit β
and calcium voltage-gated channel auxiliary subunit α2δ 1] and
other channels (transient receptor potential cation channel
subfamily M member 4 and hyperpolarization activated cyclic
nucleotide gated potassium channel 4) (6,18,19).
The SCN5A mutations account for 11–28% of all BrS patients
(8) and the prevalence of the
CACNA1C mutation ranges from 2 to 12% in the literature
(20,21). Recently, Hu et al (9) identified that mutations in the
SCN10A gene, encoding the Nav1.8 channel located adjacent to
SCN5A on chromosome 3p21–22, contribute to 16.7% cases of
BrS. However, another study revealed that although SCN10A
variants demonstrated a loss-of-function during in vitro
electrophysiological experiments, those variants are not
significantly associated with BrS (22). In addition, novel burden testing
revealed that SCN5A alone is responsible for a significant
proportion of BrS cases, and other genes, including SCN10A
and CACNA1C, do not demonstrate enrichment in rare coding
variation (6). Thus, this should
be noted to avoid diagnosing false-positive cases of causality
during genetic counseling, as rare coding variations in
BrS-susceptibility genes are also common in healthy
individuals.
 | Table ISusceptibility genes for BrS. |
Table I
Susceptibility genes for BrS.
| Type of BrS | Gene | Ion-channel
component | Functional
effect | Carriers among BrS
patients (%) |
|---|
| BrS 1 | SCN5A | α subunit
INa | Loss of
function | 11–28 |
| BrS 2 | GPD1-L | INa
ChIP | Loss of
function | <1 |
| BrS 3 | CACNA1c | α subunit
ICa | Loss of
function | 3–4 |
| BrS 4 | CACNB2b | β subunit
INa | Loss of
function | 2–3 |
| BrS 5 | SCN1B | β subunit
INa | Loss of
function | <1 |
| BrS 6 | KCNE3 | β subunit
IKs/Ito | Gain of
function | <1 |
| BrS 7 | SCN3B | β subunit
INa | Loss of
function | <1 |
| BrS 8 | KCNH2 | α subunit
IKr | Gain of
function | <1 |
| BrS 9 | KCNJ8 | α subunit
IKATP | Gain of
function | <1 |
| BrS 10 |
CACNA2D1 | α2δ subunit
ICa | Loss of
function | <1 |
| BrS 11 | RANGRF | INa
ChIP | Loss of
function | <1 |
| BrS 12 | KCNE5 | β subunit
Ito | Gain of
function | <1 |
| BrS 13 | KCND3 | α subunit
Ito | Gain of
function | <1 |
| BrS 14 | HCN4 | If | Loss of
function | <1 |
| BrS 15 | SLMAP | INa
ChIP | Loss of
function | <1 |
| BrS 16 | TRMP4 | α subunit | Loss of
function | 6 |
| BrS 17 | SCN2B | β subunit
INa | Loss of
function | <1 |
| BrS 18 | FGF12 | INa
ChIP | Loss of
function | <1 |
| BrS 19 | ABCC9 | IKATP
CHIP | Gain of
function | 1 |
| BrS 20 | PKP2 | INa
ChIP | Loss of
function | 2.5 |
| BrS 21 | SEMA3A | Ito
CHIP | Gain of
function | 1 |
| BrS 22 | SCN10A | α subunit
INa | Loss of
function | 16.7 |
Genetic screening revealed that two family members
of the proband (II.3 and III.2) were carrying the mutation. BrS is
a genetically and clinically heterogeneous disease and presents in
individuals aged several-months to 80-years-old, although more
typically at the average age of 40 years (13). When the manifestations of BrS are
not shown during ECG diagnostics, drug challenge is recommended to
reveal asymptomatic first-degree relatives of BrS patients
(13). For the nephew of the
proband (III.2; age, 19 years), BrS was definitively diagnosed
following a positive propafenone challenge and in conjunction with
type 1 pattern ECGs of the family members. The ECG signature of BrS
is dynamic and can be undetected for a period of time. Numerous
factors, such as sex hormones, age and fever, have been proposed as
being potentially responsible for the phenomenon of BrS (23,24).
Thus, repeated drug challenges or 12-lead, 24-h Holter monitoring
are highly recommended for asymptomatic children with a family
history of BrS (25,26).
The p.D1690N mutation causes a negative charge to a
neutral amino acid substitution in the pore region between the
DIVS5 and DIVS6 transmembrane segments that significantly affects
channel expression and gating kinetics. A central ion-conducting
pore of Nav1.5 channels is formed by the S5 and S6 segments and
their linker extracellular loops, determining ion selectivity.
Núñez et al (15)
identified two p.D1690N and p.G1748D compound heterozygous
mutations in the Nav1.5 channel in a Caucasian family with a
history of BrS. Notably, p.D1690N completely restores the gating
defects presented by p.G1748D channels; however, has little impact
on its trafficking. However, the p.D1690N mutation alone has not
been identified in BrS patients. In the present study, a missense
variant, p.D1690N was identified in the Nav1.5 channel in three of
the proband's family members (II.2, II.3 and III.2). During
comparisons of the electrophysiological characteristics of the WT
and p.D1690N mutant in HEK293 cells, it was identified that the key
alteration of the p.D1690N mutation was the dramatic reduction in
INa density (~72%) predominantly attributable to a
significant impairment of channel trafficking toward the membrane.
In addition, the p.D1690N mutant delayed the time of channel
recovery from inactivation induced by depolarization. These changes
caused by the p.D1690N mutation may reduce the activity of the
Nav1.5 channel and contribute to BrS.
There have been various studies investigating the
underlying mechanisms of decreasing current density by missense
mutations. One mechanism is that the mutant channels fail to
properly traffic to the cytomembrane and are detained in the
endoplasmic reticulum (ER) by a quality control system, such as
R1432G (11). The quality control
system in the ER initially arrested and ultimately degraded the
mutation-induced incorrectly folded proteins (11). A second mechanism is that certain
mutations, localized in the ion-conducting pore, may block
Na+ permeation, inducing a significant reduction in the
sodium current through the channels (27). Consistent with previous results,
the present study identified that the mechanism underlying the
reduction in sodium current density is the trafficking disorders
due to the p.D1690N mutation.
A previous report demonstrated that pharmacological
interventions (such as administration of mexiletine) significantly
increased current density in a G1743R mutant by rescuing its
expression levels in the plasma membrane (28). Although pharmacological
interventions provide potential clinical benefit to BrS patients
carrying the mutation, whether the p.D1690N mutant channel may be
rescued by mexiletine administration requires further
investigation. In addition, various studies revealed that the
p.H558R polymorphism restores the biophysical defects caused by
numerous loss-of-function SCN5A mutations underlying sudden
infant death syndrome, cardiac conduction disease and BrS (29–31).
In the SCN5A mutations associated with BrS, p.H558R
increases the current density generated by p.R282H (32), p.D1690N (15), p.S216L (33) and p.K317N (34), predominantly by mitigating their
impaired trafficking. Thus, the absence of p.H558R in this family
fails to restore the trafficking defect in D1690N in the
SCN5A gene encoding cardiac Na+ channels (such as
Nav1.5).
In conclusion, a novel heterozygous human mutation
(D1690N), in the DIV S5–S6 linker of the Nav1.5 channel, was
identified in a Chinese Han patient with BrS and the patient's
family. The results indicate that the marked reduction of sodium
current density in the p.D1690N mutant is attributable to
trafficking disorders of the Nav1.5 channel proteins to the
sarcolemma. The decreased sodium current density and abnormal
activities caused by the p.D1690N mutation are consistent with the
hypothesis that a loss of function of Nav1.5 contributes to the
pathogenesis of BrS. Whether the characterizations of the p.D1690N
mutant exist in vivo and the precise mechanisms require
further research. However, the present study strengthens the
understanding of the association between the structure and function
of the Nav1.5 channel, and provides potential personalized
therapeutic approaches for inherited cardiac arrhythmia.
Acknowledgments
The authors would like to thank Dr Qing K. Wang
(Department of Molecular Cardiology, Lerner Research Institute,
Cleveland Clinic, Cleveland, OH, USA) for assistance with the
present study and for providing the pcDNA3.1-hH1 and
pIRES2-DsReD-SCN1B plasmids. The present study was supported
by the National Natural Science Foundation of China (grant nos.
81270277 and 81170090).
Abbreviations:
|
INa
|
depolarizing inward sodium current
|
|
ICa
|
depolarizing inward calcium
current
|
|
IKs
|
repolarizing outward slow rectifying
potassium current
|
|
Ito
|
transient outward potassium
current
|
|
IKr
|
repolarizing outward rapid rectifying
potassium current
|
|
IKATP
|
ATP-sensitive inward rectifying
potassium current
|
|
If
|
funny current
|
|
ChIP
|
channel-interacting protein
|
|
BrS
|
Brugada syndrome
|
|
ECG
|
electrocardiogram
|
|
SCD
|
sudden cardiac death
|
|
PVT
|
polymorphic ventricular
tachycardia
|
|
VF
|
ventricular fibrillation
|
|
RBBB
|
right bundle branch block
|
|
ER
|
endoplasmic reticulum
|
References
|
1
|
Chen PS and Priori SG: The Brugada
Syndrome. J Am Coll Cardiol. 51:1176–1180. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brugada P and Brugada J: Right bundle
branch block, persistent ST segment elevation and sudden cardiac
death: A distinct clinical and electrocardiographic Syndrome. A
multicenter report. J Am Coll Cardiol. 20:1391–1396. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Antzelevitch C, Brugada P, Borggrefe M,
Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K,
Perez Riera AR, et al: Brugada syndrome: Report of the second
consensus conference: Endorsed by the heart rhythm society and the
European heart rhythm association. Circulation. 111:659–670. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Probst V, Veltmann C, Eckardt L, Meregalli
PG, Gaita F, Tan HL, Babuty D, Sacher F, Giustetto C, Schulze-Bahr
E, et al: Long-term prognosis of patients diagnosed with brugada
syndrome: Results from the FINGER brugada syndrome registry.
Circulation. 121:635–643. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miyasaka Y, Tsuji H, Yamada K, Tokunaga S,
Saito D, Imuro Y, Matsumoto N and Iwasaka T: Prevalence and
mortality of the Brugada-type electrocardiogram in one city in
Japan. J Am Coll Cardiol. 38:771–774. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Le Scouarnec S, Karakachoff M, Gourraud
JB, Lindenbaum P, Bonnaud S, Portero V, Duboscq-Bidot L, Daumy X,
Simonet F, Teusan R, et al: Testing the burden of rare variation in
arrhythmia-susceptibility genes provides new insights into
molecular diagnosis for Brugada Syndrome. Hum Mol Genet.
24:2757–2763. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang Q, Ohno S, Ding WG, Fukuyama M,
Miyamoto A, Itoh H, Makiyama T, Wu J, Bai J, Hasegawa K, et al:
Gain-of-function KCNH2 mutations in patients with Brugada Syndrome.
J Cardiovasc Electrophysiol. 25:522–530. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kapplinger JD, Tester DJ, Alders M, Benito
B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A,
Harris-Kerr C, et al: An international compendium of mutations in
the SCN5A-encoded cardiac sodium channel in patients referred for
Brugada Syndrome genetic testing. Heart Rhythm. 7:33–46. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hu D, Barajas-Martínez H, Pfeiffer R, Dezi
F, Pfeiffer J, Buch T, Betzenhauser MJ, Belardinelli L, Kahlig KM,
Rajamani S, et al: Mutations in SCN10A are responsible for a large
fraction of cases of Brugada Syndrome. J Am Coll Cardiol. 64:66–79.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zimmer T and Surber R: SCN5A
channelopathies-an update on mutations and mechanisms. Prog Biophys
Mol Biol. 98:120–136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Baroudi G, Pouliot V, Denjoy I, Guicheney
P, Shrier A and Chahine M: Novel mechanism for Brugada Syndrome:
Defective surface localization of an SCN5A mutant (R1432G). Circ
Res. 88:E78–E83. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zeng Z, Zhou J, Hou Y, Liang X, Zhang Z,
Xu X, Xie Q, Li W and Huang Z: Electrophysiological characteristics
of a SCN5A voltage sensors mutation R1629Q associated with Brugada
syndrome. PLoS One. 8:e783822013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wilde AA, Antzelevitch C, Borggrefe M,
Brugada J, Brugada R, Brugada P, Corrado D, Hauer RN, Kass RS,
Nademanee K, et al: Proposed diagnostic criteria for the Brugada
Syndrome: Consensus report. Circulation. 106:2514–2519. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stenson PD, Mort M, Ball EV, Shaw K,
Phillips A and Cooper DN: The human gene mutation database:
Building a comprehensive mutation repository for clinical and
molecular genetics, diagnostic testing and personalized genomic
medicine. Hum Genet. 133:1–9. 2014. View Article : Google Scholar :
|
|
15
|
Núñez L, Barana A, Amorós I, de la Fuente
MG, Dolz-Gaitón P, Gómez R, Rodríguez-García I, Mosquera I,
Monserrat L, Delpón E, et al: p.D1690N Nav15 rescues pG1748D
mutation gating defects in a compound heterozygous Brugada Syndrome
patient. Heart Rhythm. 10:264–272. 2013. View Article : Google Scholar
|
|
16
|
Lin YJ, Higa S, Tai CT, Chang SL, Lee KT,
Lo LW, Ishigaki S, Tuan TC, Wongcharoen W, Hu YF, et al: Role of
the right atrial substrate in different types of atrial
arrhythmias. Heart Rhythm. 6:592–598. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chiang KC, Lai LP and Shieh RC:
Characterization of a novel Nav1.5 channel mutation, A551T,
associated with Brugada Syndrome. J Biomed Sci. 16:762009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Boczek NJ, Ye D, Johnson EK, Wang W,
Crotti L, Tester DJ, Dagradi F, Mizusawa Y, Torchio M, Alders M, et
al: Characterization of SEMA3A-encoded semaphorin as a naturally
occurring Kv4.3 protein inhibitor and its contribution to Brugada
Syndrome. Circ Res. 115:460–469. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saber S, Amarouch MY, Fazelifar AF,
Haghjoo M, Emkanjoo Z, Alizadeh A, Houshmand M, Gavrilenko AV,
Abriel H and Zaklyazminskaya EV: Complex genetic background in a
large family with Brugada Syndrome. Physiol Rep. 3:e122562015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Burashnikov E, Pfeiffer R,
Barajas-Martinez H, Delpón E, Hu D, Desai M, Borggrefe M,
Häissaguerre M, Kanter R, Pollevick GD, et al: Mutations in the
cardiac L-type calcium channel associated with inherited J-wave
Syndromes and sudden cardiac death. Heart Rhythm. 7:1872–1882.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wilde AA and Behr ER: Genetic testing for
inherited cardiac disease. Nat Rev Cardiol. 10:571–583. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Behr ER, Savio-Galimberti E, Barc J, Holst
AG, Petropoulou E, Prins BP, Jabbari J, Torchio M, Berthet M,
Mizusawa Y, et al UK10K Consortium; Jamshidi Y: Role of common and
rare variants in SCN10A: Results from the Brugada syndrome QRS
locus gene discovery collaborative study. Cardiovasc Res.
106:520–529. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shimizu W, Matsuo K, Kokubo Y, Satomi K,
Kurita T, Noda T, Nagaya N, Suyama K, Aihara N, Kamakura S, et al:
Sex hormone and gender difference-role of testosterone on male
predominance in Brugada Syndrome. J Cardiovasc Electrophysiol.
18:415–421. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Junttila MJ, Gonzalez M, Lizotte E, Benito
B, Vernooy K, Sarkozy A, Huikuri HV, Brugada P, Brugada J and
Brugada R: Induced Brugada-type electrocardiogram, a sign for
imminent malignant arrhythmias. Circulation. 117:1890–1893. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Conte G, de Asmundis C, Ciconte G, Julià
J, Sieira J, Chierchia GB and Brugada P: Follow-up from childhood
to adulthood of individuals with family history of Brugada syndrome
and normal electrocardiograms. JAMA. 312:2039–2041. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cerrato N, Giustetto C, Gribaudo E,
Richiardi E, Barbonaglia L, Scrocco C, Zema D and Gaita F:
Prevalence of type 1 brugada electrocardiographic pattern evaluated
by twelve-lead twenty-four-hour holter monitoring. Am J Cardiol.
115:52–56. 2015. View Article : Google Scholar
|
|
27
|
Amin AS, Verkerk AO, Bhuiyan ZA, Wilde AA
and Tan HL: Novel Brugada syndrome-causing mutation in
ion-conducting pore of cardiac Na+ channel does not affect ion
selectivity properties. Acta Physiol Scand. 185:291–301. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Valdivia CR, Tester DJ, Rok BA, Porter CB,
Munger TM, Jahangir A, Makielski JC and Ackerman MJ: A trafficking
defective, Brugada syndrome-causing SCN5A mutation rescued by
drugs. Cardiovasc Res. 62:53–62. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lizotte E, Junttila MJ, Dube MP, Hong K,
Benito B, DE Zutter M, Henkens S, Sarkozy A, Huikuri HV, Towbin J,
et al: Genetic modulation of brugada Syndrome by a common
polymorphism. J Cardiovasc Electrophysiol. 20:1137–1141. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Viswanathan PC, Benson DW and Balser JR: A
common SCN5A polymorphism modulates the biophysical effects of an
SCN5A mutation. J Clin Invest. 111:341–346. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shinlapawittayatorn K, Du XX, Liu H,
Ficker E, Kaufman ES and Deschênes I: A common SCN5A polymorphism
modulates the biophysical defects of SCN5A mutations. Heart Rhythm.
8:455–462. 2011. View Article : Google Scholar :
|
|
32
|
Poelzing S, Forleo C, Samodell M, Dudash
L, Sorrentino S, Anaclerio M, Troccoli R, Iacoviello M, Romito R,
Guida P, et al: SCN5A polymorphism restores trafficking of a
Brugada syndrome mutation on a separate gene. Circulation.
114:368–376. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Marangoni S, Di Resta C, Rocchetti M,
Barile L, Rizzetto R, Summa A, Severi S, Sommariva E, Pappone C,
Ferrari M, et al: A Brugada syndrome mutation (p.S216L) and its
modulation by pH558R polymorphism: Standard and dynamic
characterization. Cardiovasc Res. 91:606–616. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shinlapawittayatorn K, Dudash LA, Du XX,
Heller L, Poelzing S, Ficker E and Deschênes I: A novel strategy
using cardiac sodium channel polymorphic fragments to rescue
trafficking-deficient SCN5A mutations. Circ Cardiovasc Genet.
4:500–509. 2011. View Article : Google Scholar : PubMed/NCBI
|