Introduction
Cadmium (Cd) is a toxic heavy metal, which is
present in the air, soil, sediment and water. Cd causes injury to a
number of organs and tissues, and induces dysfunction, including in
the kidney, liver, lung, bone, cardiovascular system and immune
system. Cd pollution originates from mining, metallurgical
industries, and the manufacture of nickel-Cd batteries, pigments
and plastic stabilizers. The Agency for Toxic Substances and
Disease Registry has classified Cd as the sixth most toxic
substance to human health (1).
Numerous studies have revealed that Cd induces
apoptosis in several types of cells and tissues (2,3).
Mitochondria are the early and primary targets of Cd injury
(4), with exposure to Cd inducing
mitochondria-dependent apoptosis in oligodendrocytes, and the
mitochondria are crucial in coordinating caspase activation through
the release of cytochrome c (Cyt C). A previous study
(5) reported that Cd induces
apoptosis in tumor cells through the Fas/Fas ligand (FasL) pathway.
The Fas/FasL pathway is an important apoptosis signal transduction
pathway, in which ligand-receptor interaction activates the cell
death pathway. As a member of the tumor necrosis factor family, Fas
is a 45-kDa type I transmembrane protein, which induces apoptosis
in susceptible cells by crosslinking with its ligand. Following the
trimerization of Fas on the cell membrane by extracellular FasL,
the Fas-associated death domain and caspase-8 form a death-inducing
signal complex, which mediates Fas-induced cell death. Once
activated, caspase-8 activates effector caspases, including
caspase-3, caspase-6 and caspase-7, ultimately leading to the
hydrolysis of cytosolic and nuclear substrates.
N-acetylcysteine (NAC), formed of l-cysteine
and acetyl, is the precursor of reducing glutathione in cells. As a
thiol donor, NAC is an effective antioxidant has effects including
interference of free radical generation and scavenging of generated
free radicals, resistance to apoptosis, preventing DNA damage,
anti-angiogenesis, regulation of gene expression and signal
transduction, and inhibiting malignant tumor development (6). NAC has been widely used clinically
and experimentally, and exerts effects in the respiratory system
(7), cardiovascular system
(8) and central nervous system
(9). It is reported that NAC can
effectively inhibit the apoptosis induced by Cd in macrophage
(10) and human lens epithelial
cells (11). Repeated application
in severe early sepsis showed that NAC can effectively inhibit the
apoptosis of pulmonary dendritic cells, protecting the function of
the cells (7). In severe sepsis,
the early repeated application of NAC effectively inhibits lung
dendritic cell apoptosis. NAC can also effectively regulate
respiratory apoptosis in rats caused by smoking, and at as a cancer
chemopreventive agent (12).
Although there have been previous investigations on
Cd-induced injury, its mechanism remains to be fully elucidated.
The liver is the primary target organ of Cd injury (13,14).
Therefore, the present study used cytobiological and molecular
biological methods to examine the mechanism of Cd-induced apoptosis
involving the mitochondrial and FasL pathways, and investigated the
protective effect of NAC, in immortalized rat BRL 3A
hepatocytes.
Materials and methods
Materials
Cadmium acetate (CdAc2), penicillin,
streptomycin, gluteraldehyde, osmium textroxide, propylene oxide,
epoxy resin, uranyl acetate, lead citrate, sodium dodecyl sulfate
(SDS), tris-buffered saline with 0.1% Tween-20 (TBST) were
purchased from Sigma-Aldrich (St. Louis, MO, USA). Annexin
V-fluorescein isothiocyanate (FITC) Apoptosis Selection Kit I was
purchased from BD Pharmingen (San Diego, CA, USA). Dulbecco's
modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were
obtained from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Trypsin was purchased from Amresco LLC (Solon, OH, USA). The
Mitochondrial Membrane Potential Assay Kit with JC-1, and
bicinchoninic acid (BCA) protein assay kits were from Beyotime
Institute of Biotechnology (Jiangsu, China). Rabbit anti-rat
polyclonal caspase-3 (1:1,000; cat. no. 9662), rabbit anti-rat
polyclonal caspase-9 (1:1,000; cat. no. 9506), rabbit anti-rat
polyclonal poly(ADP-ribose)polymerase (PARP) (1:1,000; cat. no.
9542), rabbit anti-rat monoclonal caspase-8 (1:1,000; cat. no.
4790), rabbit anti-rat polyclonal FasL (1:1,000; cat. no. 4273),
and rabbit anti-rat polyclonal β-actin (1:5,000; cat. no. 4967)
antibodies, and horseradish peroxidase (HRP)-conjugated goat
anti-rabbit immunoglobulin G (IgG) (1:5,000; cat. no. 7074) were
obtained from Cell Signaling Technology, Inc. (Danvers, MA,
USA).
Radioimmunoprecipitation assay (RIPA) lysis buffer
was purchased from Beijing Solarbio Science & Technology
(Beijing, China). Kodak X-ray film was purchased from Eastman Kodak
(Rochester, NY, USA). Cell culture plates were obtained from
Corning Incorporated (New York, NY, USA). Nitrocellulose (NC)
filter membranes were purchased from Pall Gelman Sciences (Port
Washington, NY, USA). The enhanced chemiluminescence (ECL)
detection kit was from Thermo Fisher Scientific, Inc. (Pierce ECL
Plus Western Blotting Substrate). Other reagents used were
available locally and of analytical grade.
Cell culture and treatments
In the present study, BRL 3A immortalized rat liver
cells between passages 10 and 20 were used (Chinese Academy of
Sciences, Shanghai, China). The cells were cultured in DMEM
supplemented with 100 U/ml penicillin, 100 μg/ml
streptomycin and 10% FBS, and maintained at 37°C in a 5%
CO2 humid incubator (Thermo Fisher Scientific,
Inc.).
The BRL 3A cells (2×105 cells/ml) were
seeded in 6- or 96-well plates. CdAc2 was dissolved in
distilled deionized water as a stock solution (5 mM), and diluted
with serum-free culture medium to different concentrations prior to
being added to the cell cultures.
The cells were treated with 0, 10, 20 or 40
μmol/l Cd for 12 h (groups a-d, respectively). In another
two experiments, the cells were preincubated with 2 mmol/l NAC for
30 min, prior to 12 h incubation with 20 μmol/l Cd, or
incubation with 2 mmol/l NAC alone for 12 h (groups e and f,
respectively). At each stage, cells were incubated at 37°C in 5%
CO2.
Detection of apoptosis
Apoptosis was detected using the apoptosis detection
kits, according to the manufacturer's protocols. Following
treatment, the cells were harvested and resuspended in 100
μl binding buffer containing 5 μl annexin V-FITC and
5 μl propidium iodide (PI) solution. Following incubation in
the dark at 25°C for 15 min, 400 μl binding buffer was
added. The cells were analyzed using a FACSAria flow cytometer
(Becton Dickinson, San Jose, CA, USA) with excitation and emission
wavelengths of 488 and 605 nm, respectively. A minimum of 10,000
cells per sample were registered. Positioning of quadrants on the
annexin V-/PI dot plots was performed, and living (annexin
V−/PI−), early apoptotic (annexin
V+/PI−), late apoptotic (annexin
V+/PI+) and necrotic cells (annexin
V−/PI+) were distinguished. The total
proportion of apoptotic cells comprised the percentage of cells
with annexin V+/PI− and annexin
V+/PI+ fluorescence (15).
Each independent experiment included another three
samples: Unstained, annexin V-FITC staining only, and PI staining
only, as controls. Each experiment was repeated at least three
times.
Determination of mitochondrial membrane
potential (ΔΨm)
The fluorescent probe, JC-1, was used to measure the
ΔΨm of the BRL 3A cells. JC-1 is a cationic dye, which accumulates
in the mitochondria according to the ΔΨm. If the ΔΨm is high, JC-1
aggregates in the mitochondrial matrix, forming J-aggregates, which
generate red fluorescence (16).
If the ΔΨm is lost, JC-1 remains in a monomeric form, generating
green fluorescence. The relative ratio of red (590 nm) and green
(525 nm) fluorescence intensity provides the proportional
measurement of the ΔΨm.
The cells (5×105) were incubated with 0.5
ml 1X JC-1 at 37°C for 20 min in the dark, and were centrifuged at
2,000 × g for 4 min at 4°C. The cells were then washed twice with
1X JC-1 buffer, resuspended in 1 ml 1X JC-1 buffer, and analyzed
immediately using flow cytometry.
Observation of mitochondrial
ultrastructure
The changes in mitochondrial ultrastructure were
confirmed by transmission electron microscopy (TEM) examination
using the method previously described by Yuan et al
(17). Briefly, the cell
suspensions (1×107 cells/ml) were centrifuged at 2,000 ×
g for 5 min at 4°C following treatment and the medium was
discarded. The cells were then fixed with 200 μl 2.5%
glutaraldehyde in phosphate-buffered saline (PBS) at 25°C for 24 h.
Following fixing, the cells were washed three times with PBS and
post-fixed for 1.5 h in 1% osmium tetroxide. The specimens were
dehydrated using a graded series of ethanol (75, 85, 95 and 100%),
rinsed in propylene oxide, and impregnated with epoxy resin.
Ultrathin sections (70 nm) were obtained and contrasted with uranyl
acetate and lead citrate. Electron micrographs were captured using
a CM-100 TE microscope (Philips, Eindhoven, the Netherlands).
Western blotting
Following treatment, the cells were washed twice
with cold PBS, and lysed in RIPA lysis buffer on ice for 30 min,
followed by sonication at 3 W for 15 sec. The cell lysates were
then centrifuged at 12,000 × g for 10 min at 4°C. The protein
content was determined using the BCA protein assay kit, and the
absorbance was measured at 560 nm using a microplate reader (BioTek
Instruments, Inc., Winooski, VT, USA). Aliquots of the lysate were
diluted in 6X SDS sample buffer and boiled for 10 min. The protein
(30 μg) from each treatment group was separated on a 12%
SDS-polyacrylamide gel and electrophoretically transferred onto NC
membranes. Following blocking at room temperature for 2 h with 5%
non-fat milk in 0.1% TBST, the membranes were incubated overnight
at 4°C with the corresponding primary antibodies to caspase-3,
caspase-9, PARP, caspase-8, FasL (1:1,000) and β-actin (1:5,000).
Following washing with TBST (six times, 5 min each), the membranes
were incubated with HRP-conjugated goat anti-rabbit IgG (1:5,000)
at room temperature for 2 h. Following washing with TBST (six time,
5 min each), the blots were visualized using the ECL detection kit,
according to the manufacturer's protocol, and were then exposed to
X-ray film.
Statistical analysis
The results are presented as the mean ± standard
deviation. Significance was assessed using one-way analysis of
variance, following appropriate transformation to normalized data
and equalized variance, where necessary. Statistical analyses were
performed using SPSS version 17.0 (SPSS Inc., Chicago, IL, USA);
P<0.05 was considered to indicate a statistically significant
difference. All assays were performed in triplicate.
Results
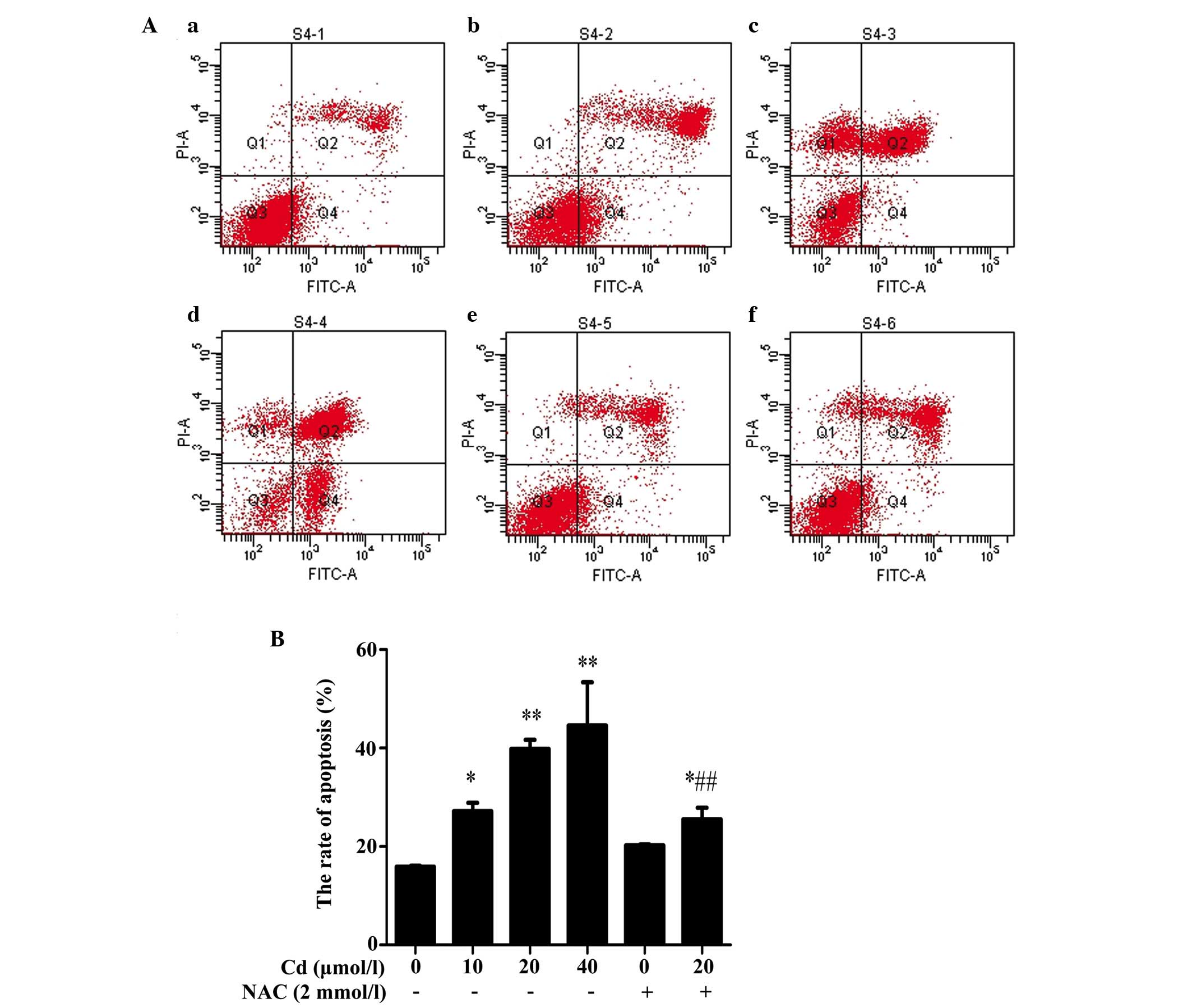
Cd induces apoptosis and is reversed
following preincubation with NAC
In the present study, flow cytometry was used to
distinguish the effects of Cd on apoptosis following annexin
V-FITC/PI double staining. Following 12 h incubation with Cd (10,
20 and 40 μmol/l), the percentages of total (early+late)
apoptotic cells increased significantly to 27.2±1.65, 39.83±1.82
and 44.57±8.81%, respectively, compared with the control
(15.87±0.21%), indicating that Cd induced apoptosis in the BRL 3A
cells. NAC alone did not alter the rate of apoptosis significantly,
compared with the control. However, preincubation with NAC reduced
the rate of Cd-induced apoptosis, compared with that induced by Cd
alone (Fig. 1).
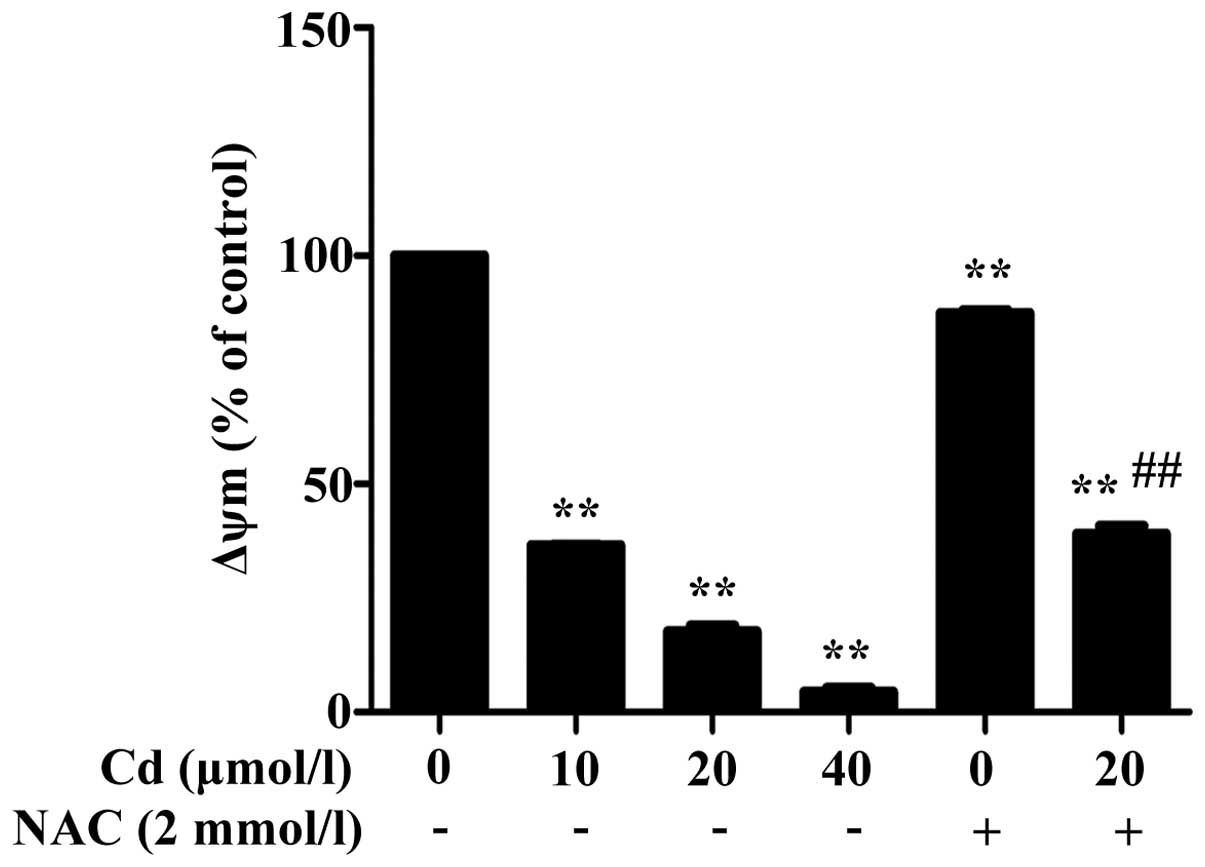
Cd reduces ΔΨm and is increased by
preincubation with NAC
The ΔΨm decreased following 12 h exposure to Cd,
which occurred in a dose-dependent manner (Fig. 2). Preincubation with NAC improved
the Cd-induced decrease in ΔΨm, compared with the decrease induced
by Cd alone.
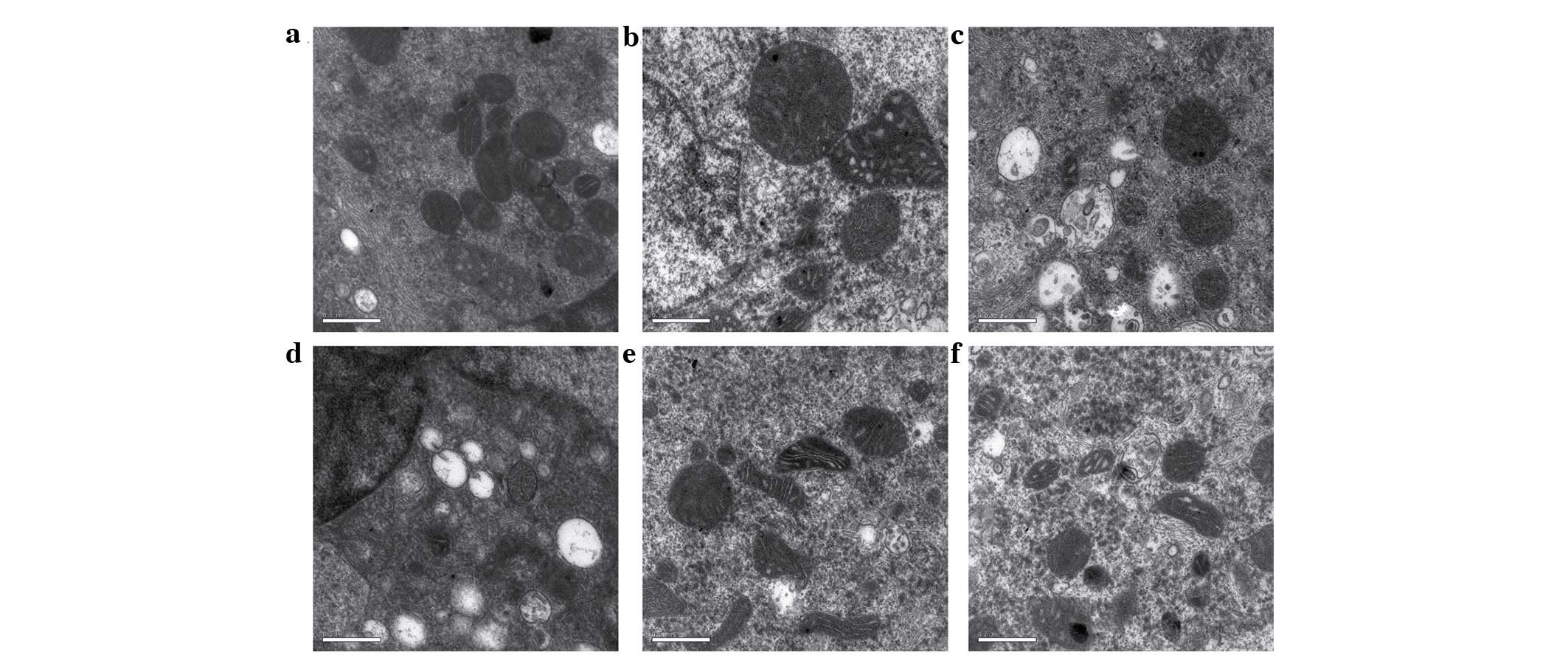
Effects of Cd and NAC on BRL 3A cell
ultrastructure
The results of the TEM examination showed that, 12 h
following exposure to Cd, the control cells had a well-defined
outline and contained spherical or oval mitochondria, with
well-defined transversal cristae. By contrast, the cells incubated
with Cd exhibited changes in mitochondrial ultrastructure,
including disruption and loss of cristae, swelling and
degeneration, vacuole formation in the cytoplasm, and cell plasma
membrane disruption. NAC alone did not alter the cell
ultrastructure significantly, compared with the control. However,
preincubation with NAC reduced the mitochondrial swelling induced
by Cd, compared with that induced by Cd alone (Fig. 3).
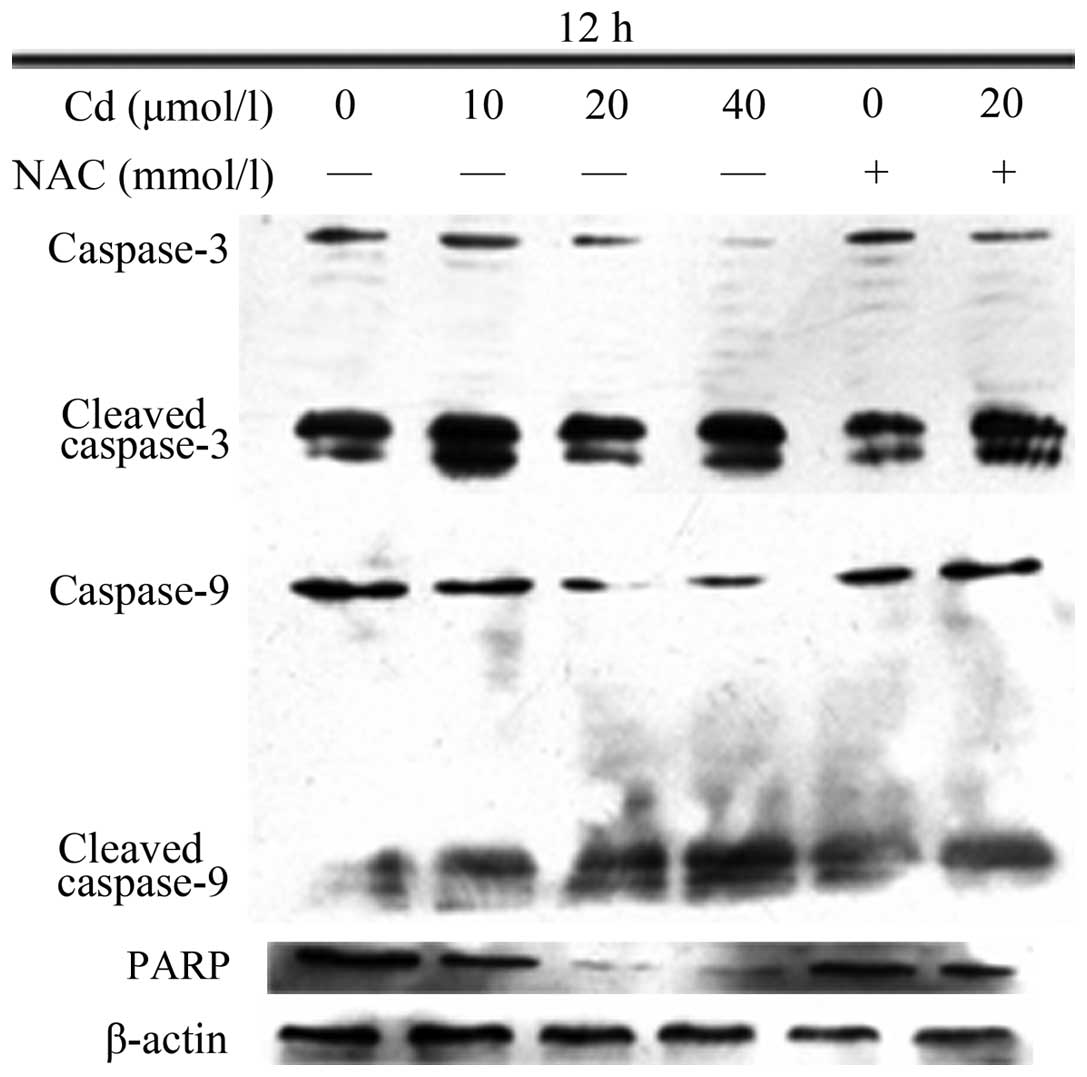
Cd decreases the protein expression
levels of caspase-3, caspase-9 and PARP, which is attenuated by
preincubation with NAC
The protein levels of caspase-3, caspase-9 and PARP
decreased as the Cd dose increased, whereas the levels of cleaved
caspase-3 and cleaved caspase-9 increased (Fig. 4). NAC alone did not affect the
protein levels; preincubation with NAC prior to Cd treatment
inhibited the tendency of the protein levels to change, compared
with the changes induced by Cd alone.
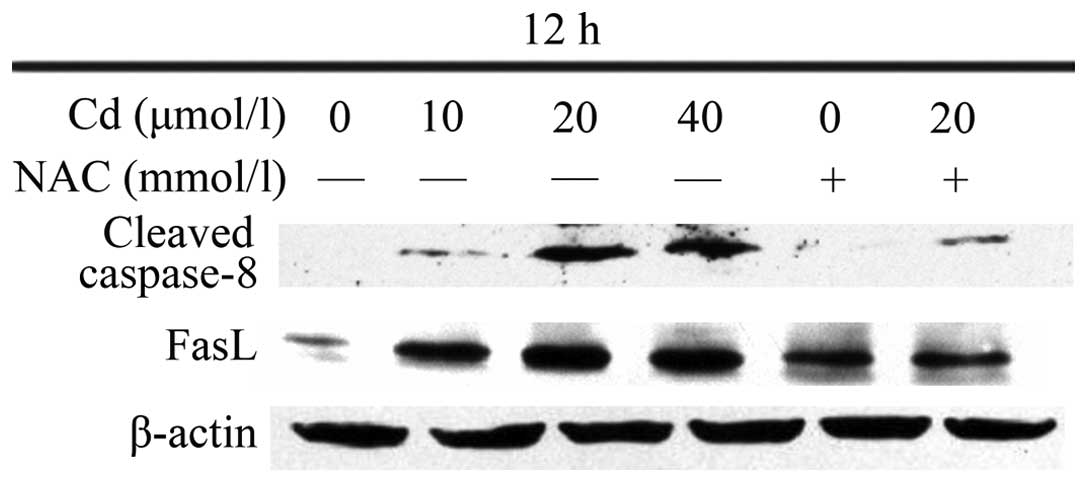
Cd increases the protein expression
levels of caspase-8 and FasL, which is attenuated by preincubation
with NAC
The results of the Western blotting revealed that 12
h treatment with Cd significantly elevated the protein levels of
cleaved caspase-8 and FasL in the BRL 3A cells (Fig. 5), whereas preincubation with NAC
prior to Cd treatment inhibited the tendency of the protein levels
to increase.
Discussion
It has been suggested that Cd can cause apoptosis in
a variety of cells, and can occur in a dose- and time-dependent
manner (18). Pathak and
Khandelwal (19) reported that 6 h
exposure to 25 μmol/l Cd induced apoptosis in rat thymus
cells. Chen et al (20)
reported that Cd caused apoptosis in PC12 and SH-SY5Y nerve cells,
in a dose-dependent manner. In the present study, flow cytometry
demonstrated that all concentrations of Cd induced a significantly
higher rate of apoptosis, compared with the control, and that NAC
decreased the rate of apoptosis in a dose-dependent manner,
suggesting that it had a protective effect against Cd-induced
apoptosis.
The mitochondria are an important target of Cd, as
mitochondrial dysfunction can cause cellular damage (21). Decreased ΔΨm is vital in the
process of apoptosis, and Cd can lead to decreased ΔΨm and Cyt C
release from the mitochondrial membrane, which in turn leads to
uncoupling of the mitochondrial respiratory chain. This produces a
large quantity of active oxygen species, which are released into
the cytoplasm, triggering the downstream apoptotic pathway
(22–24). Mao et al suggested that Cd
can increase HEK293 cell permeability, reducing ΔΨm and triggering
mitochondrial dysfunction (25).
The mitochondria are central in the process of apoptosis, and loss
of ΔΨm is an important mechanism of inducing apoptosis.
Mitochondrial permeability transition pore (MPTP) opening is an
important aspect of apoptosis (26). Dorta et al (27) treated extracted liver mitochondria
with 5 μmol/l Cd for 2.5 min, which triggered loss of ΔΨm,
reflecting the fact that the ΔΨm is sensitive to Cd. In the present
study, the ΔΨm of the Cd-exposed cells decreased significantly, in
a dose-dependent manner, compared with the control. Preincubation
with NAC effectively inhibited this tendency. Cd exposure causes
mitochondrial membrane damage, reducing ΔΨm and leading to MPTP
opening, enhanced membrane permeability, ΔΨm loss, and
mitochondrial release of apoptosis-inducing factor and Cyt C,
triggering the caspase cascade and leading to apoptosis (28–30).
As an energy source, mitochondria are important in
cells; mitochondrial damage can lead to disordered cell structure
and function. The mitochondria are involved in apoptosis triggered
by several stimuli. Yan et al found that Cd causes
mitochondrial swelling, deformation, crest fracture or
disappearance, membrane rupture, matrix outflow and vacuole
degeneration in nerve cells, and that NAC confers certain
protective effects (18). The
present study found that Cd caused similar mitochondrial changes,
as 12 h exposure to 10 μmol/l Cd caused marginal
mitochondrial cristae fracture, and 12 h exposure to 20–40
μmol/l Cd eventually caused mitochondrial collapse and
vacuolation. NAC preincubation reduced mitochondrial deformation
and damage, indicating that Cd may have caused mitochondrial injury
through oxidative damage in the BRL 3A cells.
PARP is a post-translational modification enzyme,
which is predominantly present in eukaryotic cell nuclei. Numerous
studies have demonstrated that multiple stimuli can lead to
activation of the caspase family, cleaving the substrate (PARP) and
leading to apoptosis (31,32). Guégan and Sola (33) demonstrated that long-term focal
cerebral ischemia in mice following middle cerebral artery
obstruction always involves caspase-3 activation; caspase-3 cleaves
PARP, leading to the loss of PARP activity, followed by DNA
fragmentation and apoptosis. Using a rat acute cerebral ischemia
model, Benchoua et al (34)
reported that caspase-8 cleaves PARP and induces apoptosis. Pacher
et al (35,36) demonstrated, that in oxidative
stress, PARP activation by oxidation damages DNA, and is an
important mechanism in promoting cell dysfunction an inhibiting
tissue function. The present study demonstrated that Cd activated
PARP precursor cleavage, and that the combined application of NAC
reversed this tendency, suggesting that Cd-induced apoptosis in the
BRL 3A cells involved PARP activation. As an antioxidant, NAC can
alleviate cell damage caused by oxidative stress. Studies have
shown that mitochondria coordinate caspase activation through the
release of Cyt C due to the outer mitochondrial membrane becoming
permeable (37,38). Following its release into the
cytoplasm, Cyt C combines with Apaf-1 to form a polyadenylic
complex (39). The domain
structure can recruit the cytoplasmic caspase-9 precursor and
self-activate, and caspase-9 can cleave and activate the downstream
caspases, cleaving their substrates and triggering apoptosis.
Caspase-9 is an important initiator enzyme, which can cleave
caspase-3 and caspase-7, leading to apoptosis. Caspase-3 is a key
factor in apoptosis, and is directly involved in the chromosome
condensation and DNA fragmentation processes. The present study
demonstrated that caspase-3 and caspase-9 activities were
significantly enhanced, and that there was increased caspase-3
cleavage and protein expression of caspase-9 as the Cd
concentrations increased. Preincubation with NAC effectively
reversed the caspase activation, suggesting that the mitochondrial
pathway was an important channel through which Cd induced apoptosis
in the BRL 3A cells.
Fas/FasL are membrane proteins, which are closely
associated with apoptosis (40).
The induction of caspase-mediated apoptosis through the Fas pathway
is an important mechanism by which apoptosis is induced.
Fas/FasL-mediated alveolar epithelial cell apoptosis is involved in
the process of pulmonary fibrosis (41) and the treatment of pulmonary
fibrosis and tumors in rats (42,43).
In osteosarcoma cells treated with matrine, cell proliferation is
inhibited, triggering apoptosis, increasing the protein expression
of Fas/FasL and the activation of caspase-3, caspase-8 and
caspase-9 in a dose-dependent manner, indicating that Fas/FasL
protein activation may be involved in inducing apoptosis in tumor
cells (5). The present study
demonstrated that Cd increased the protein expression levels of
cleaved caspase-8 and FasL, and that NAC inhibited this increase.
Caspase-8 may be involved in the death receptor pathway of
apoptosis induced by Cd. and may be involved in BRL 3A cell
apoptosis.
Cd was found to cause the uncoupling of the
mitochondrial respiratory chain, decreased ΔΨm, and led to
mitochondrial swelling, degeneration, cristae blurring, deformation
and eventual collapse. It also stimulated the activation of
caspase-3 and caspase-9, PARP cleavage, and increased the protein
expression levels of cleaved caspase-8 and FasL, leading to
apoptosis. NAC exerted an inhibitory effect on the mitochondrial
and death receptor pathways involved in Cd-induced apoptosis in the
BRL 3A cells, suggesting that it exerts a protective effect.
Acknowledgments
This study was supported by grants from the Nation
Natural Science Foundation of China (grant nos. 31101866 and
31302058) and a project funded by the Priority Academic Program
Development of Jiangsu Higher Education Institutions.
References
|
1
|
Bertin G and Averbeck D: Cadmium: Cellular
effects, modifications of biomolecules, modulation of DNA repair
and genotoxic consequences (a review). Biochimie. 88:1549–1559.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pham TN, Marion M, Denizeau F and Jumarie
C: Cadmium-induced apoptosis in rat hepatocytes does not
necessarily involve caspase-dependent pathways. Toxicol In Vitro.
20:1331–1342. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li Y and Lim SC: Cadmium-induced apoptosis
of hepatocytes is not associated with death receptor-related
caspase-dependent pathways in the rat. Environ Toxicol Pharmacol.
24:231–238. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hossain S, Liu HN, Nguyen M, Shore G and
Almazan G: Cadmium exposure induces mitochondria-dependent
apoptosis in oligodendrocytes. Neurotoxicology. 30:544–554. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liang CZ, Zhang JK, Shi Z, Liu B, Shen CQ
and Tao HM: Matrine induces caspase-dependent apoptosis in human
osteosarcoma cells in vitro and in vivo through the upregulation of
Bax and Fas/FasL and downregulation of Bcl-2. Cancer Chemother
Pharmacol. 69:317–331. 2012. View Article : Google Scholar
|
|
6
|
De Flora S, Izzotti A, D'Agostini F and
Balansky RM: Mechanisms of N-acetylcysteine in the prevention of
DNA damage and cancer, with special reference to smoking-related
end-points. Carcinogenesis. 22:999–1013. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang HW, Yang W, Lu JY, Li F, Sun JZ,
Zhang W, Guo NN, Gao L and Kang JR: N-acetylcysteine administration
is associated with reduced activation of NF-kB and preserves lung
dendritic cells function in a zymosan-induced generalized
inflammation model. J Clin Immunol. 33:649–660. 2013. View Article : Google Scholar
|
|
8
|
Rowbotham DS, Wendon JA and Harrison PM:
N-acetylcysteine infusion in viral myocarditis: A case report. Int
J Cardiol. 60:315–316. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen G, Shi J, Hu Z and Hang C: Inhibitory
effect on cerebral inflammatory response following traumatic brain
injury in rats: A potential neuroprotective mechanism of
N-acetylcysteine. Mediators Inflamm. 2008:7164582008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim J and Sharma RP: Cadmium-induced
apoptosis in murine macrophages is antagonized by antioxidants and
caspase inhibitors. J Toxicol Environ Health A. 69:1181–1201. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kalariya NM, Wills NK, Ramana KV,
Srivastava SK and van Kuijk FJ: Cadmium-induced apoptotic death of
human retinal pigment epithelial cells is mediated by MAPK pathway.
Exp Eye Res. 89:494–502. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
D'Agostini F, Balansky RM, Izzotti A,
Lubet RA, Kelloff GJ and De Flora S: Modulation of apoptosis by
cigarette smoke and cancer chemopreventive agents in the
respiratory tract of rats. Carcinogenesis. 22:375–380. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
El-Sokkary GH, Nafady AA and Shabash EH:
Melatonin administration ameliorates cadmium-induced oxidative
stress and morphological changes in the liver of rat. Ecotoxicol
Environ Saf. 73:456–463. 2010. View Article : Google Scholar
|
|
14
|
Li R, Yuan C, Dong C, Shuang S and Choi
MM: In vivo antioxidative effect of isoquercitrin on
cadmium-induced oxidative damage to mouse liver and kidney. Naunyn
Schmiedebergs Arch Pharmacol. 383:437–445. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jia Y, Lin J, Mi Y and Zhang C: Quercetin
attenuates cadmium-induced oxidative damage and apoptosis in
granulosa cells from chicken ovarian follicles. Reprod Toxicol.
31:477–485. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
White RJ and Reynolds IJ: Mitochondrial
depolarization in glutamate-stimulated neurons: An early signal
specific to excitotoxin exposure. J Neurosci. 16:5688–5697.
1996.PubMed/NCBI
|
|
17
|
Yan Y, Bian JC, Zhong LX, Zhang Y, Sun Y
and Liu ZP: Oxidative stress and apoptotic changes of rat cerebral
cortical neurons exposed to cadmium in vitro. Biomed Environ Sci.
25:172–181. 2012.PubMed/NCBI
|
|
18
|
Dong S, Shen HM and Ong CN:
Cadmium-induced apoptosis and phenotypic changes in mouse
thymocytes. Mol Cell Biochem. 222:11–20. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pathak N and Khandelwal S: Modulation of
cadmium induced alterations in murine thymocytes by piperine:
Oxidative stress, apoptosis, phenotyping and blastogenesis. Biochem
Pharmacol. 72:486–497. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen L, Liu L, Luo Y and Huang S: MAPK and
mTOR pathways are involved in cadmium-induced neuronal apoptosis. J
Neurochem. 105:251–261. 2008. View Article : Google Scholar
|
|
21
|
Li M, Kondo T, Zhao QL, Li FJ, Tanabe K,
Arai Y, Zhou ZC and Kasuya M: Apoptosis induced by cadmium in human
lymphoma U937 cells through Ca2+-calpain and
caspase-mitochondria-dependent pathways. J Biol Chem.
275:39702–39709. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ludovico P, Rodrigues F, Almeida A, Silva
MT, Barrientos A and Côrte-Real M: Cytochrome c release and
mitochondria involvement in programmed cell death induced by acetic
acid in Saccharomyces cerevisiae. Mol Biol Cell. 13:2598–2606.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Anuradha CD, Kanno S and Hirano S:
Oxidative damage to mitochondria is a preliminary step to caspase-3
activation in fluoride-induced apoptosis in HL-60 cells. Free Radic
Biol Med. 31:367–373. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mao WP, Zhang NN, Zhou FY, Li WX, Liu HY,
Feng J, Zhou L, Wei CJ, Pan YB and He ZJ: Cadmium directly induced
mitochondrial dysfunction of human embryonic kidney cells. Hum Exp
Toxicol. 30:920–929. 2011. View Article : Google Scholar
|
|
26
|
Li M, Xia T, Jiang CS, Li LJ, Fu JL and
Zhou ZC: Cadmium directly induced the opening of membrane
permeability pore of mitochondria which possibly involved in
cadmium-triggered apoptosis. Toxicology. 194:19–33. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dorta DJ, Leite S, DeMarco KC, Prado IM,
Rodrigues T, Mingatto FE, Uyemura SA, Santos AC and Curti C: A
proposed sequence of events for cadmium-induced mitochondrial
impairment. J Inorg Biochem. 97:251–257. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li M, Kondo T, Zhao QL, Li FJ, Tanabe K,
Arai Y, Zhou ZC and Saduya M: Apoptosis induced by cadmium in human
lymphoma U937 cells through Ca2+-calpain and
caspase-mitochondria-dependent pathways. J Biol Chem.
275:39702–39709. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ly JD, Grubb D and Lawen A: The
mitochondrial membrane potential (Δψm) in apoptosis; an update.
Apoptosis. 8:115–128. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yuan Y, Jiang CY, Xui H, Sun Y, Hu FF,
Bian JC, Liu XZ, Gu JH and Liu ZP: Cadmium-induced apoptosis in
primary rat cerebral cortical neurons culture is mediated by a
calcium signaling pathway. PLoS One. 8:e643302013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Coutant A, Lebeau J, Bidon-Wagner N,
Levalois C, Lectard B and Chevillard S: Cadmium-induced apoptosis
in lymphoblastoid cell line: Involvement of caspase-dependent and
-independent pathways. Biochimie. 88:1815–1822. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shen HM, Dong SY and Ong CN: Critical role
of calcium overloading in cadmium-induced apoptosis in mouse
thymocytes. Toxicol Appl Pharmacol. 171:12–19. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guégan C and Sola B: Early and sequential
recruitment of apoptotic effectors after focal permanent ischemia
in mice. Brain Res. 856:93–100. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Benchoua A, Couriaud C, Guégan C, Tartier
L, Couvert P, Friocourt G, Chelly J, Ménissier-de Murcia J and
Onténiente B: Active caspase-8 translocates into the nucleus of
apoptotic cells to inactivate poly (ADP-ribose) polymerase-2. J
Biol Chem. 277:34217–34222. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pacher P, Liaudet L, Soriano FG, Mabley
JG, Szabó E and Szabó C: The role of poly (ADP-ribose) polymerase
activation in the development of myocardial and endothelial
dysfunction in diabetes. Diabetes. 51:514–521. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pacher P, Mabley JG, Soriano FG, Liaudet L
and Szabó C: Activation of poly (ADP-ribose) polymerase contributes
to the endothelial dysfunction associated with hypertension and
aging. Int J Mol Med. 9:659–664. 2002.PubMed/NCBI
|
|
37
|
Mao WP, Ye JL, Guan ZB, Zhao JM, Zhang C,
Zhang NN, Jiang P and Tian T: Cadmium induces apoptosis in human
embryonic kidney (HEK) 293 cells by caspase-dependent and
-independent pathways acting on mitochondria. Toxicol In Vitro.
21:343–354. 2007. View Article : Google Scholar
|
|
38
|
Kondoh M, Araragi S, Sato K, Higashimoto
M, Takiguchi M and Sato M: Cadmium induces apoptosis partly via
caspase-9 activation in HL-60 cells. Toxicology. 170:111–117. 2002.
View Article : Google Scholar
|
|
39
|
Desagher S and Martinou JC: Mitochondria
as the central control point of apoptosis. Trends Cell Biol.
10:369–377. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gehring S, Rottmann S, Menkel AR,
Mertsching J, Krippner-Heidenreich A and Luscher B: Inhibition of
proliferation and apoptosis by the transcriptional repressor Mad1.
Repression of Fas-induced caspase-8 activation. J Biol Chem.
275:10413–10420. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Martin TR, Hagimoto N, Nakamura M and
Matute-Bello G: Apoptosis and epithelial injury in the lungs. Proc
Am Thorac Soc. 2:214–220. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Villa-Morales M and Fernández-Piqueras J:
Targeting the Fas/FasL signaling pathway in cancer therapy. Expert
Opin Ther Targets. 16:85–101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang W, Zheng Z, Yu W, Lin H, Cui B and
Cao F: Polymorphisms of the FAS and FASL genes and risk of breast
cancer. Oncol Lett. 3:625–628. 2012.PubMed/NCBI
|