Introduction
Cardiac hypertrophy is a general adaptive process in
response to almost all types of cardiac disease (including pressure
overload, cardiac arrhythmias, exercise training and endocrine
disorders) (1–3). Various extrinsic physiological or
pathological factors stimulate the development of cardiac
hypertrophy (1,4). Although various treatments are
available, advances in therapeutic strategies that are suitable for
preventing the progression of cardiac hypertrophy have been
limited, as the pathophysiology of cardiac hypertrophy remains to
be elucidated.
The calcium-sensing receptor (CaSR) stimulates the
phospholipase C system to release intracellular calcium
([Ca2+]i) (5). The expression of CaSR has previously
been observed in the kidney, bone, intestine and other tissues
(6,7). The functional expression of CaSR was
first described in rat cardiac tissue in 2003 (8). One of the crucial functions of CaSR
is regulating systemic Ca2+. A change in the
[Ca2+]i is an initiating factor in cardiac
hypertrophy (9,10). However, the regulation of CaSR
during the hypertrophic process remains poorly characterized. It
was previously reported that the expression of CaSR is increased in
hypertrophic cardiomyocytes (8).
Cardiac hypertrophy is a complex pathophysiological process
regulated by various signal pathways and gene networks. Thus,
identifying the underlying molecular mechanism of cardiac
hypertrophy is crucial for developing improved therapeutic
strategies.
Pathologically, autophagy, necrosis and apoptosis
are the predominant modes of cell death (11). Autophagy is a catabolic process
mediated by lysosomes leading to degradation of damaged organelles
and macromolecules to achieve effective cell recycling (12). This pathophysiological process is
involved in immune defense, cell differentiation and cell death
(12). Previous studies have
demonstrated that autophagy participates in pathological cardiac
hypertrophy (2,13,14).
It is well known that the level of physiological autophagy is
important for cellular homeostasis, whereas autophagic cell death
is a result of excessive autophagy (14). Numerous studies have indicated that
the autophagy may be upregulated in response to pathological
stress, including endoplasmic reticulum stress, cardiac hypertrophy
and heart failure (11,13). Upregulated autophagy may induce
ventricular hypertrophy by facilitating protein degradation during
the development from cardiac hypertrophy to heart failure (13).
However, the effect of CaSR modulation on autophagy
in cardiac hypertrophy remains to be elucidated. The present study
investigated whether the expression of CaSR was changed during
isoproterenol (ISO)-induced hypertrophy and whether CaSR was
involved in cardiac hypertrophy via the modulation of
autophagy.
Materials and methods
Materials
ISO, Calhex231, GdCl3,
compound C and 3-methyladenine (3-MA) were purchased from
Sigma-Aldrich (St. Louis, MO, USA). The following primary
antibodies were obtained from Cell Signaling Technology, Inc.
(Danvers, MA, USA): Rabbit polyclonal
anti-calcium/calmodulin-dependent protein kinase II (CaMKII; cat.
no. CST-3362), rabbit polyclonal anti-phosphorylated (p)-CaMKII
(cat. no. CST-3361); rabbit polyclonal anti-sequestosome 1 (p62;
cat. no. CST-5114); rabbit monoclonal anti-Beclin-1 (cat. no.
CST-3495); rabbit monoclonal anti-microtubule-associated protein
light chain 3 (LC3; cat. no. CST-3868); rabbit monoclonal
anti-caspase-3 (cat. no. CST-9664); rabbit monoclonal
anti-AMP-activated protein kinase (AMPK; cat. no. CST-2535); rabbit
monoclonal anti-p-AMPK (cat. no. CST-5832); rabbit monoclonal
anti-p-CaMKKβ (cat. no. CST-12818); rabbit monoclonal
anti-mammalian target of rapamycin (mTOR; cat. no. CST-2972);
rabbit monoclonal anti-p-mTOR (cat. no. CST-2971); and rabbit
monoclonal anti-GAPDH (cat. no. CST-5174). Mouse monoclonal
anti-CaMKKβ (cat. no. SC-100364) was obtained from Santa
Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Rabbit polyclonal
anti-CaSR (cat. no. ACR-004) was purchased from Alpha Diagnostic
International Inc. (San Antonio, TX, USA). Secondary antibody
(alkaline phosphatase-conjugated anti-rabbit IgG; cat. no. S3731)
was obtained from Promega Corporation (Madison, WI, USA).
Polyvinylidene difluoride membranes were purchased from Whatman, GE
Healthcare Life Sciences (Little Chalfont, UK) and alkaline
phosphatase-conjugated horse anti-mouse IgG (cat. no. ZB-2310;
Zhongshan Golden bridge Biotechnology, Co., Ltd., Beijing,
China).
ISO-induced cardiac hypertrophy in
vivo
Male Wistar rats (age, 10–12 weeks; n=70) were kept
at 21±2°C, 60±5% humidity and a 12-h light-dark cycle. The rats
were housed together and had free access to food and water. They
were injected subcutaneously with ISO once a day to activate
β-adrenergic receptors, according to the previously described
method (15). Wistar rats (weight,
200–250 g) were randomly assigned to seven treatment groups, as
follows: i) Control group (Control, n=10), rats were subcutaneously
injected with saline; ii) ISO-1d group (ISO-1d; n=10), rats were
subcutaneously injected with ISO (5 mg/kg in saline) for 1 day to
induce cardiac hypertrophy; iii) ISO-3d group (ISO-3d; n=10), rats
were subcutaneously injected with ISO for 3 days; iv) ISO-5d group
(ISO-5d; n=10), rats were subcutaneously injected with ISO for 5
days; v) ISO-7d group (ISO-7d; n=10), the rats were subcutaneously
injected with ISO for 7 days; vi) ISO group (ISO, n=10), the rats
were subcutaneously injected with normal saline for 2 weeks
following the administration of ISO for 7 days; and vii) ISO +
Calhex231 group (ISO + Calhex231; n=10), rats
were intravenously injected with the specific CaSR inhibitor
Calhex231 (10 µmol/kg/day in saline) for 2 weeks
following the administration of ISO for 7 days. Subsequently, three
rats were sacrificed in each group by overdose of 10% chloral
hydrate (0.75–1 ml/100 g). All animals were obtained from the
Experimental Animal Center of Harbin Medical University (Harbin,
China) and the present study was approved by the Institutional
Animal Research Committee of Harbin Medical University.
Echocardiographic assessment
Cardiac function was noninvasively monitored by
transthoracic echocardiography with a Vivid 7 Dimension
echocardiographic system (GE Healthcare Life Sciences). Briefly,
the rats were anesthetized with 10% chloral hydrate (0.25–0.35
ml/100 g; Shanghai Fanke Biotechnology Co. Ltd., Shanghai, China)
as described previously (1), and
echocardiograms were obtained and analyzed as reported
previously.
Histological analysis
Following anesthesia with 10% chloral hydrate
(0.25–0.35 ml/100 g), the hearts were excised and immediately
placed in 4% paraformaldehyde (Shanghai Fanke Biotechnology Co.
Ltd.) at room temperature for 24 h. The myocardial specimens were
embedded in paraffin (Shanghai Fanke Biotechnology Co. Ltd.), cut
into 4-µm sections and stained with hematoxylin and eosin
(H&E; Zhuhai Baso Biotechnology Co., Ltd., Zhuhai, China) and
Masson's trichrome reagent (Zhuhai Baso Biotechnology Co., Ltd.).
The fibrotic areas stained blue, and the normal tissues stained
red. Tissues were analyzed using an E2000 Nikon microscope (Tokyo,
Japan).
Electron microscopy
Hearts were removed from three mice in the control,
ISO and ISO+Calhex231 groups. Cardiac tissue was cut
into 1-mm cubes and fixed with 2.5% glutaraldehyde (Shanghai Fanke
Biotechnology Co. Ltd.) in 0.1 M phosphate buffer (Shanghai Fanke
Biotechnology Co. Ltd.) (pH 7.4) overnight at 4°C. Following
fixation, the sections were immersed in 1% osmium tetroxide
(Shanghai Fanke Biotechnology Co. Ltd.) for 2 h, dehydrated in
graded ethanol solutions graded ethanol (Shanghai North Connaught
Biotechnology Co, Ltd., Shanghai, China), embedded in epoxy resin
(Shanghai Fanke Biotechnology Co. Ltd.) and then cut into ultrathin
sections (60–70 nm) with an ultramicrotome (Leica Microsystems,
Shanghai, China). Sections were then post-stained with uranyl
acetate and lead citrate (Yuanye Technology Co., Ltd., Shanghai,
China) prior to examination under a JEM-1010 transmission electron
microscope (JEOL, Ltd., Tokyo, Japan).
Establishing an in vitro model of
ISO-induced cardiomyocyte hypertrophy
As previously described (8), neonatal rat cardiomyocytes were
prepared from 60 2-to-3 day-old neonatal Wistar rats (obtained from
the Experimental Animal Center of Harbin Medical University,
Harbin, China; wieght, 20–30 g). The rats were immersed in 70%
(v/v) alcohol (Shanghai North Connaught Biotechnology Co., Ltd.),
and placed on a flat board. Then the mice were sacrificed by
decapitation with scissors. The ventricles were removed and washed
three times in D-Hank's balanced salt solution (BosterBio, Beijing,
China) (0.4 g/l KCl; 0.06 g/l KH2PO4; 8.0 g/l
NaCl; 0.35 g/l NaHCO3; and 0.06 g/l
Na2HPO4·7H2O, pH 7.2) at 4°C. They
were then homogenized and incubated with 0.25% (w/v) trypsinase for
10 min at 37°C. Next, an equal volume of cold Dulbecco's modified
Eagle's medium (DMEM) (HyClone, Logan, UT, USA) containing 10%
(v/v) newborn calf serum was added to terminate the digestion. The
supernatant was discarded and cells were incubated with fresh 0.25%
trypsinase (Beyotime Institute of Biotechnology, Jiangsu, China)
for 15 min at 37°C, and the supernatant was then collected. The
latter digestion step was repeated four times. Cells in the
supernatant were isolated by centrifugation at 286 × g and room
temperature for 10 min, then resuspended in DMEM containing 20%
(v/v) newborn calf serum (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), 100 U/ml penicillin and 100 mg/ml streptomycin
(Yuanye Technology Co., Ltd.). The cells were cultured in a
monolayer at a density of 5×104 cells/cm2 at
37°C in a humidified atmosphere containing 5% (v/v) CO2.
The medium contained 2 µM fluorodeoxyuridine (Shanghai Fanke
Biotechnology Co. Ltd.) to prevent proliferation of
nonmyocytes.
Three days after seeding, the neonatal rat
cardiomyocytes were starved in serum-free DMEM for 24 h then
divided randomly into six groups as follows: i) Normal control
group; ii) ISO group, cardiomyocytes treated with 10 µM ISO
for 48 h; iii) GdCl3 + ISO group, cardiomyocytes
preincubated with 30 µM GdCl3 (specific CaSR
agonist) for 1 h and then treated with 10 µM ISO for 48 h;
iv) GdCl3 + Calhex231+ISO group, the
cardiomyocytes were preincubated with 3 µM
Calhex231 (specific CaSR inhibitor) for 30 min prior to
the addition of ISO; v) GdCl3 + 3-MA + ISO group, the
cardiomyocytes were preincubated with 5 mM 3-MA (specific autophagy
inhibitor) for 30 min prior to the addition of ISO; and vi)
GdCl3 + compound C + ISO group, the cardiomyocytes were
preincubated with 5 µM compound C (AMPK inhibitor) for 30
min prior to the addition of ISO.
Measurement of
[Ca2+]i in cardiomyocytes
Following the described treatments, cardiomyocytes
were loaded with 1 µM Fluo-4/AM (Sigma-Aldrich) at 37°C for
30 min. The cells were washed twice with Ca2+-free
phosphate-buffered saline to remove the remaining dye and then
further incubated in DMEM. Changes in [Ca2+]i
were measured by the fluorescence intensity induced by Fluo-4 in
the cardiomyocytes recorded for 5 min using an IX-70 confocal laser
scanning microscope (Olympus Corporation, Tokyo, Japan; ×600
magnification) with excitation and emission at 488 and 530 nm,
respectively. Image Pro Plus 6.0 (Media Cybernetics, Rockville, MD,
USA) was used for analysis.
Western blotting
Protein was isolated from rat cardiac tissues and
neonatal rat cardiomyocytes, which were homogenized in 0.5 ml of
RIPA buffer prior to being transferred into small tubes and rotated
1 h at 4°C. Protein concentrations were determined by the Coomassie
method (Beyotime Institute of Biotechnology) using bovine serum
albumin as the standard. All samples (containing 80 µg
protein) were mixed with loading buffer (Beyotime Institute of
Biotechnology) and subjected to 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis. Proteins in the samples
from different experimental groups were separated and transferred
onto polyvinylidene difluoride membranes (GE Healthcare Life
Sciences) by electroblotting (300 mA for 2 h). Membranes were
blocked in Tris-buffered saline with 0.1% (v/v) Tween 20 [TBS-T;
137 mM NaCl, 20 mM Tris (pH 7.6)] (Applygen Technologies Inc.,
Beijing, China) containing 5% (w/v) skimmed milk at 37°C for 1 h.
Membranes were then incubated overnight at 4°C with antibodies
against CaSR (1:800), and CaMKII, p-CaMKII, p62, Beclin-1, LC3,
caspase-3, AMPK, p-AMPK, CaMKKβ, p-CaMKKβ,
mTOR, p-mTOR and GAPDH (all 1:1,000). Membranes were then washed
with TBST three times for 5 min and incubated with an alkaline
phosphatase-conjugated goat anti-rabbit secondary antibody
(alkaline phosphatase-conjugated IgG; 1:5,000) and alkaline
phosphatase-conjugated anti-mouse IgG (Zhongshan Golden bridge
Biotechnology, Co., Ltd.) in TBS-T for 1 h at room temperature. The
densities of the protein bands were quantified using a Chemi Doc EQ
densitometer and Quantity One 4.6.2 software (Bio-Rad Laboratories,
Inc., Hercules, CA, USA), and GAPDH served as an internal control
for the semi-quantitative assay.
Statistical analysis
All data were obtained from at least three
independent experiments that were replicated two to four times
under each condition. All values are expressed as the mean ±
standard error of the mean. Comparisons between the groups were
performed using Kruskal-Wallis two-way analysis of variance.
P<0.05 was considered to indicate a statistically significant
difference.
Results
ISO induced cardiac hypertrophy in
rats
An increase in cardiomyocyte volume and
extracellular matrix deposition is characteristic of myocardial
hypertrophy. To investigate the in vivo effects of CaSR in
hypertrophic hearts, a model of cardiac hypertrophy model was
established by administering ISO to rats for 7 days. Myocardial
function was assessed using echocardiography. At 1, 3 and 5 days
after ISO injection, animals injected with ISO exhibited an
increase in interventricular septum (IVS) thickness, diastolic left
ventricle posterior wall (LVPWd) thickness and a decrease in left
ventricle ejection fraction (LVEF) compared with the control group
(Table I). However, there was no
statistical difference between the control and ISO-1d, -3d and -5d
animals with the exception of LVIDd. At 7 days after ISO injection,
the myocardial dysfunction was further exacerbated, with a
significant increase in diastolic and systolic IVS (IVSd and s),
LVPWd and diastolic left ventricular internal dimension (LVIDd),
and a significant decrease in LVEF, compared with the control group
(all P<0.05). The results indicated that cardiac hypertrophy was
occurring at 7 days post-ISO injection (Table I).
 | Table IEchocardiographic analysis of left
ventricular wall and chamber dimension in control, ISO-1d, ISO-3d,
ISO-5d, ISO-7d, and ISO + Calhex231-treated rats. |
Table I
Echocardiographic analysis of left
ventricular wall and chamber dimension in control, ISO-1d, ISO-3d,
ISO-5d, ISO-7d, and ISO + Calhex231-treated rats.
| Parameter | Control | ISO-1d | ISO-3d | ISO-5d | ISO-7d | ISO +
Calhex231 |
|---|
| IVSd (cm) | 0.17±0.02 | 0.17±0.01 | 0.19±0.01 | 0.19±0.02 | 0.22±0.02a | 0.19±0.01b |
| IVSs (cm) | 0.22±0.03 | 0.23±0.02 | 0.26±0.02 | 0.27±0.02 | 0.32±0.06a | 0.25±0.02b |
| LVPWd (cm) | 0.16±0.02 | 0.17±0.01 | 0.19±0.02 | 0.21±0.01 | 0.23±0.03a | 0.18±0.02b |
| LVIDd (cm) | 0.51±0.07 | 0.56±0.07 | 0.52±0.05 | 0.68±0.04a | 0.75±0.06a | 0.65±0.04b |
| LVEF (%) | 86.20±4.68 | 83.37±5.58 | 77.13±12.78 | 73.93±10.96 | 57.55±6.46a | 70.76±5.70b |
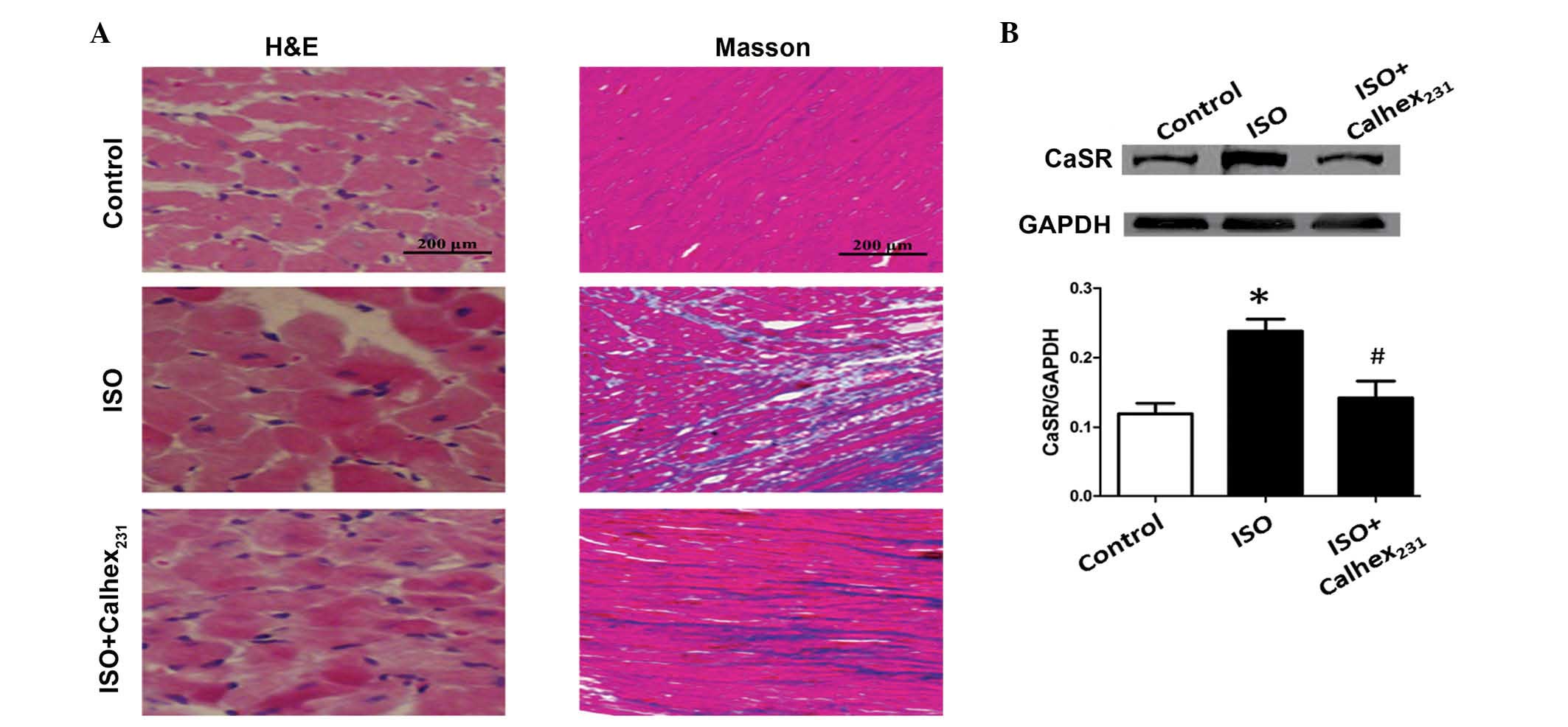
H&E staining demonstrated that ISO markedly
increased the cell cross-sectional area of the myocardial tissue
compared with the control group (Fig.
1A). Morphological analysis of the control group demonstrated
that the cardiomyocytes exhibited a clear arrangement into defined
rows, intercalated discs and transverse stripes with loose nuclear
chromatin. However, following ISO injection, the rat cardiac
tissues were expanded, and the muscle fibers became thickened and
disorganized. These changes were the most marked in the ISO group
(Fig. 1A).
Masson's staining demonstrated that the hearts of
ISO treated rats exhibited extensive interstitial fibrosis in the
ventricular wall compared with control hearts. Morphological
analysis revealed that the cytoplasm, collagen fibers and red blood
cells were stained blue, and the nuclei stained blue-brown. The
control group exhibited normal myocardial fibers, whereas, the ISO
group exhibited a large number of blue-stained collagen fibers
(Fig. 1A).
As demonstrated by western blot analysis, the
protein expression level of CaSR in the rat heart was significantly
increased in the ISO-7d group compared with the control group
(P<0.01; Fig. 1B).
Calhex231 ameliorates cardiac
hypertrophy induced by ISO in rats
The CaSR inhibitor, Calhex231, binds to
the transmembrane domains of CaSR to compete with Ca2+
molecules (16,17). Echocardiography demonstrated that
cardiac systolic and diastolic functions were improved following
Calhex231 administration compared with the ISO group.
Calhex231 treatment ameliorated cardiac functions, with
IVSd, IVSs, LVPWd and LVIDd significantly decreased, and LVEF
increased compared with the ISO group (all P<0.05; Table I). Calhex231 markedly
attenuated the levels of ventricular hypertrophy and fibrosis, as
well as cardiomyocyte apoptosis induced by ISO (Fig. 1A). In addition, the protein
expression level of CaSR was significantly decreased following
Calhex231 treatment compared with the ISO group
(P<0.05; Fig. 1B), suggesting
that Calhex231 treatment ameliorated cardiac hypertrophy
induced by ISO.
Levels of autophagy increased during
ISO-induced cardiac hypertrophy in rats
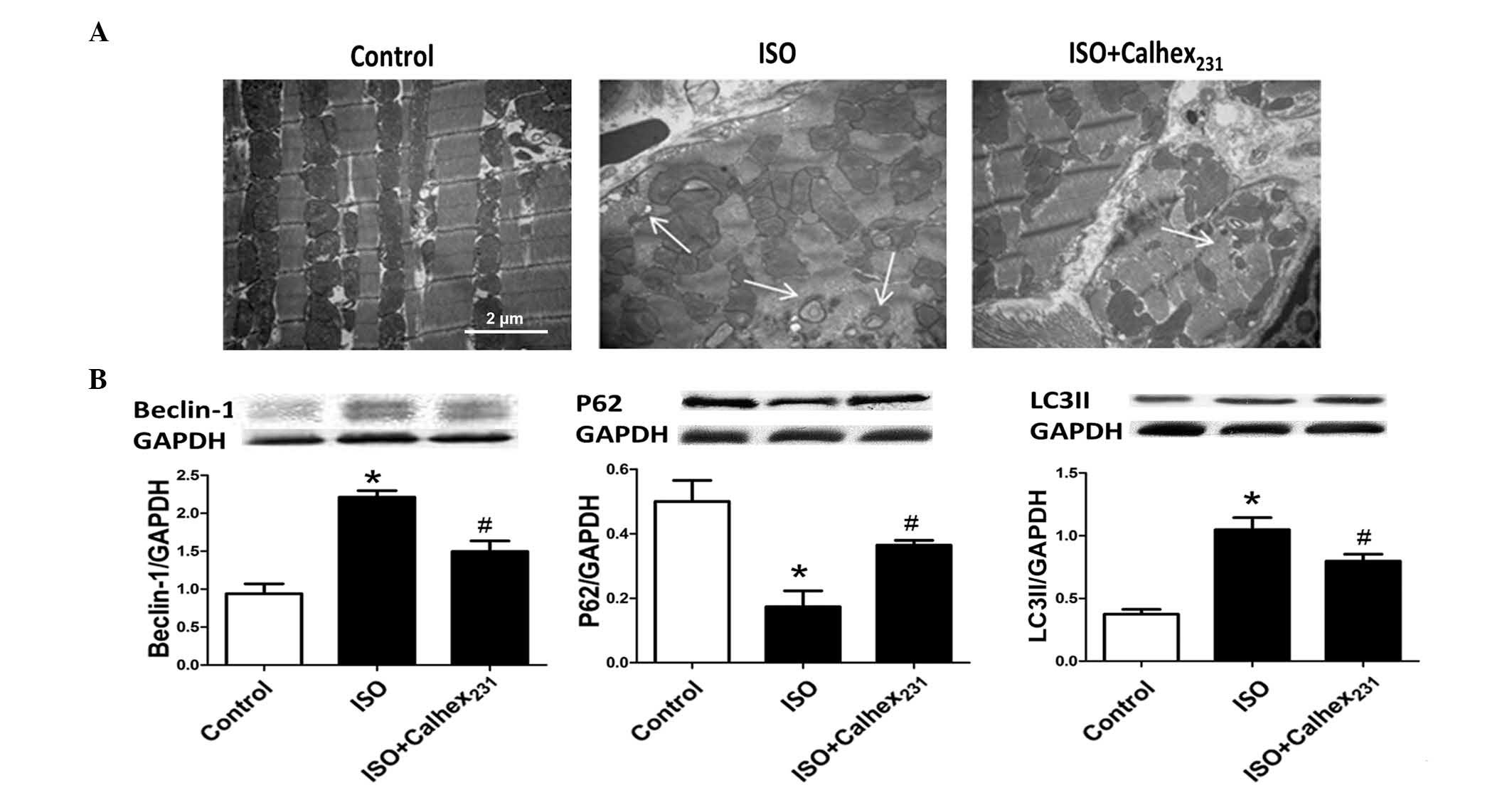
Transmission electron microscopy indicated that the
administration of Calhex231 attenuated the disorganized
sarcomere structure and mitochondrial disarray observed in
ISO-induced hypertrophic hearts, which was consistent with the
observation of increased autophagosomes. The electron microscopy
results prompted the present study to further investigate the
effect of Calhex231 on autophagy (Fig. 2A).
Autophagy is a bulk degradation mechanism for
damaged cytosolic organelles and proteins with long half lives. The
protein expression levels of Beclin-1 and LC3II were measured as
markers of autophagy (18) and
autophagosome formation (19),
respectively. Beclin-1 and LC3II levels were increased by ISO
treatment compared with control (P<0.05), whereas the expression
of P62, which transports a number of ubiquitinated substrates to
autophagosomes (20), was reduced
in the ISO group (P<0.05). Furthermore, the administration of
Calhex231 decreased the protein expression levels of
Beclin-1 and LC3II, and increased P62 expression levels compared
with that of ISO group (all P<0.05) (Fig. 2B).
Administration of CaSR inhibitor
ameliorated hypertrophy in neonatal cardiomyocytes
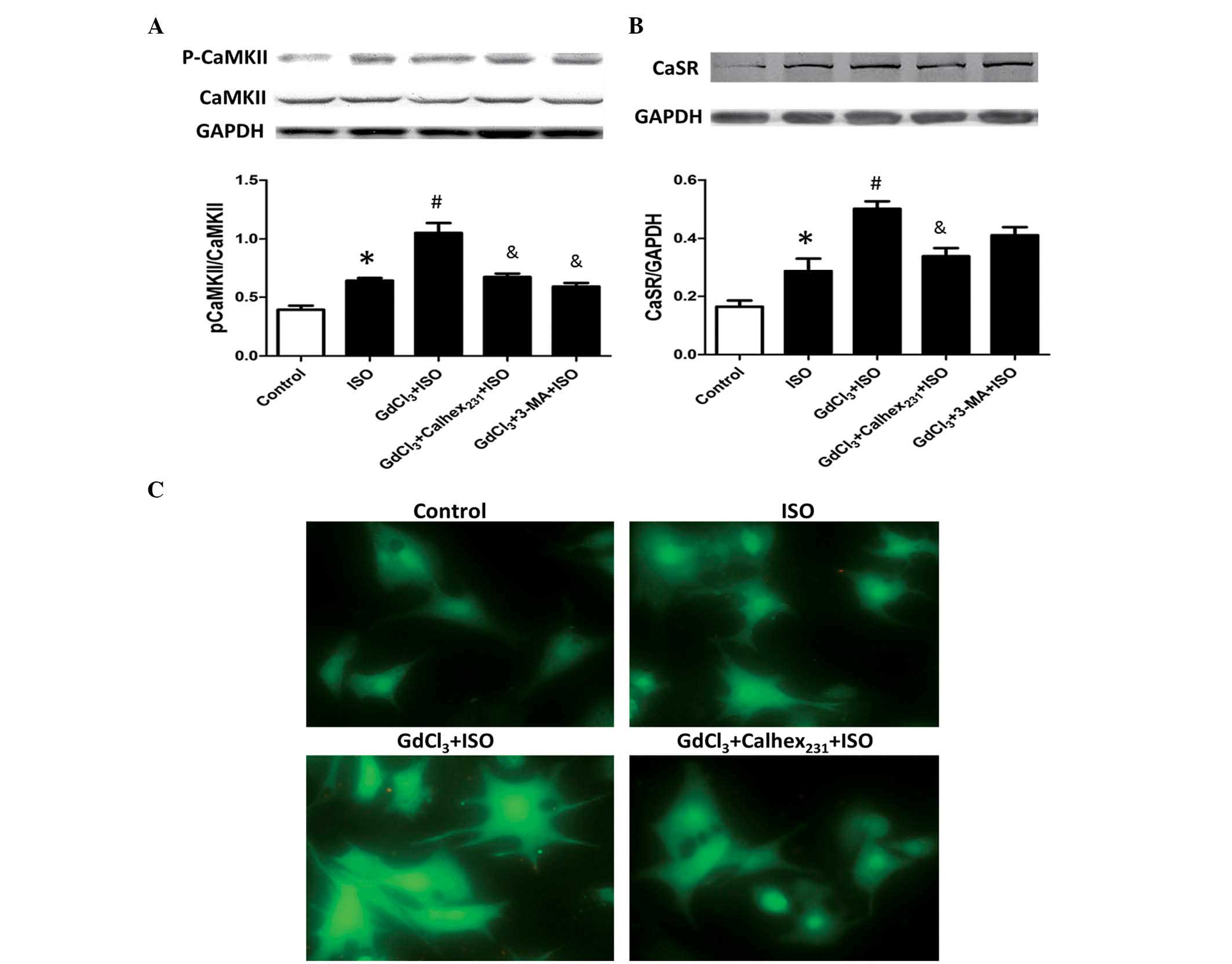
CaMKII is an essential signaling molecule involved
in cardiac hypertrophy. The present study demonstrated that the
protein content and expression of p-CaMKII were increased in
cardiomyocytes of the ISO group compared with the control group
(P<0.05; Table II; Fig. 3A). In addition, the protein content
and p-CAKKII levels were significantly increased in the
GdCl3 + ISO group compared with the ISO group
(P<0.05). Calhex231 treatment also attenuated these
increases, as the phosphorylation of p-CAMKII level were
significantly reduced compared with the GdCl3 + ISO
group (P<0.05). Notably, treatment with 3-MA, an autophagy
inhibitor, also significantly decreased the level of p-CaMKII
compared with that of the GdCl3 + ISO group
(P<0.05).
| Table IIChanges of protein content in the
control, ISO, GdCl3 + ISO, GdCl3 +
Calhex231 + ISO and GdCl3 + 3-MA + ISO group
rats. |
Table II
Changes of protein content in the
control, ISO, GdCl3 + ISO, GdCl3 +
Calhex231 + ISO and GdCl3 + 3-MA + ISO group
rats.
| Treatment
group | Rats | Protein
content |
|---|
| Control | 6 | 3.06±0.96 |
| ISO | 6 | 5.61±1.83a |
| GdCl3 +
ISO | 6 | 8.45±1.97b |
| GdCl3 +
Calhex231 + ISO | 6 | 5.07±2.11c |
| GdCl3 +
3-MA + ISO | 6 | 4.82±2.01c |
The protein expression level of CaSR in
cardiomyocytes was significantly increased in the ISO and
GdCl3 + ISO groups compared with the control group
(P<0.05). Calhex231 treatment attenuated the increase
of CaSR expression induced by ISO and GdCl3 ; the levels
were significantly decreased compared with the GdCl3 +
ISO group (P<0.05; Fig.
3B).
The current study demonstrated that ISO markedly
increased [Ca2+]i compared with the control
group, and that GdCl3 exerted a synergistic effect to
induce a further increase of [Ca2+]i in the
GdCl3 + ISO group compared with the ISO group
(P<0.05). Furthermore, this effect was blocked by
Calhex231 treatment compared with the GdCl3 +
ISO group (Fig. 3C).
CaSR inhibition ameliorated hypertrophic
cardiomyocytes via suppression of autophagy
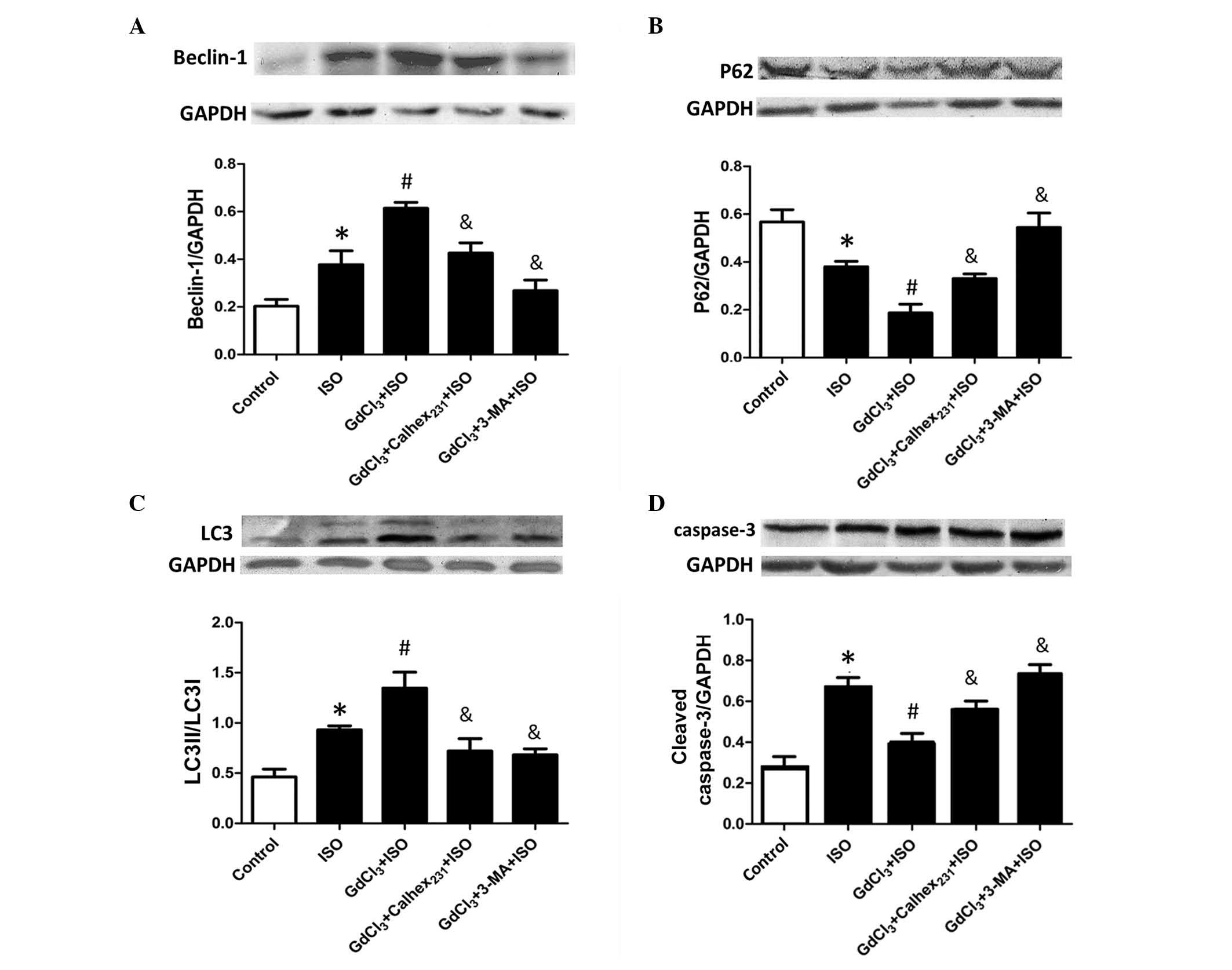
In agreement with the results from the in
vivo ISO-induced hypertrophy model, the protein expression
levels of Beclin-1 and p62 in the in vitro cardiomyocyte
model were significantly increased and decreased, respectively, in
the ISO group compared with the control (P<0.05). In addition,
the expression levels of Beclin-1 and p62 in the GdCl3 +
ISO group were significantly increased and decreased, respectively,
compared with the ISO group (P<0.05). Calhex231
inhibited these effects; the levels of Beclin-1 and p62 were
significantly decreased and increased, respectively, in the
GdCl3 + Calhex231 + ISO group compared with
the GdCl3 + ISO group (P<0.05). Furthermore,
autophagy was significantly inhibited by 3-MA treatment (P<0.05)
(Fig. 4A and B).
To investigate the functional association between
apoptosis and autophagy, the current study analyzed the protein
expression level of cleaved caspase-3 in the in vitro
cardiomyocyte model. The LC3 isoform, LC3I, is soluble and exists
in the cytosol, whereas LC3II is membrane-bound. The level of LC3II
increases when autophagy is induced, reflecting the enhanced
lipidation reaction. Thus, the level of LC3II provides a measure of
autophagy induction, however the rate of the LC3II increase depends
on the cell type (21). ISO
treatment increased the cleaved caspase-3 and LC3 protein
expression levels in the ISO group compared with the control group
(P<0.05; Fig. 4C and D). In
addition, the current study demonstrated that GdCl3
significantly increased the LC3II/LC3I ratio (P<0.05), whereas
it decreased the expression of caspase-3 (P<0.05) in the
GdCl3 + ISO group compared with the ISO group. Notably,
although 3-MA inhibited CaSR-augmented autophagy, the apoptotic
index was markedly increased. Compared with the GdCl3 +
ISO group, pretreatment with 3-MA significantly decreased the
LC3II/LC3I ratio (P<0.05) and increased the expression of
cleaved caspase-3 (P<0.05) in the GdCl3 + 3-MA + ISO
group. These results indicated that Calhex231 suppresses
autophagy against hypertrophic stimuli to ameliorate cardiomyocyte
survival.
CaSR inhibition reduced autophagy via
suppressing the CaMKKβ-AMPK-mTOR signaling pathway
To elucidate the underlying molecular mechanisms
involved in CaSR-induced autophagic responses, the expression
levels of CaMKKβ, p-CaMKKβ, AMPK, p-AMPK,
mTOR and p-mTOR were measured. In the in vitro cardiomyocyte
model, increased p-CaMKKβ and p-AMPK levels, and
decreased p-mTOR levels were demonstrated in the ISO group compared
with the control group (P<0.05). Furthermore, compared with the
ISO group, these effects were significantly enhanced in the
GdCl3 + ISO group (P<0.05). However,
Calhex231 blocked these effects; compared with the
GdCl3 + ISO group, p-CaMKKβ and p-AMPK levels
were significantly decreased, and p-mTOR levels increased by
Calhex231 (P<0.05; Fig.
5A). The results of the current study indicated that CaSR
stimulates autophagy and the effect is mediated by
CaMKKβ-AMPK-mTOR signaling.
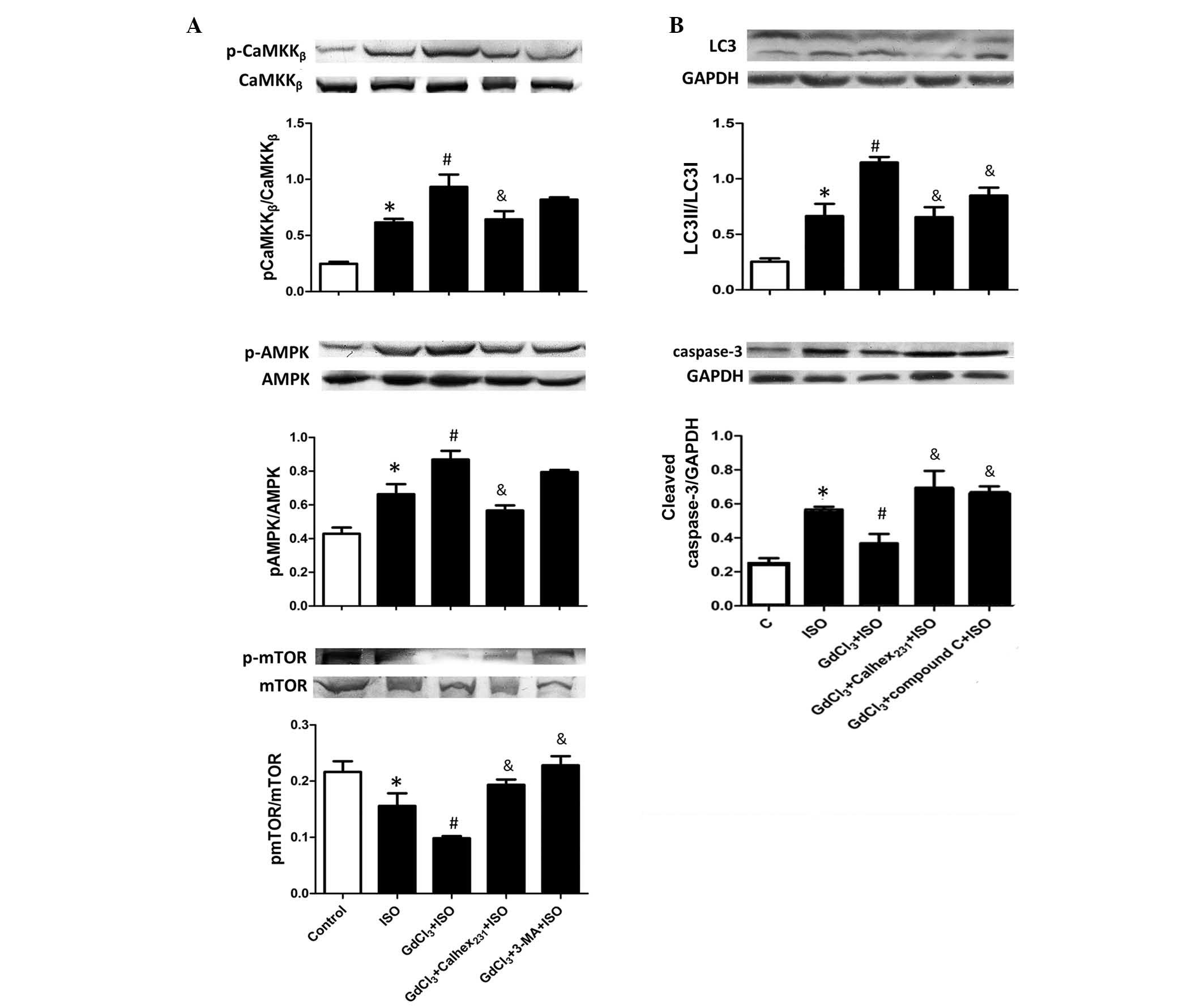 | Figure 5Effect of calcium-sensing receptor on
autophagy initiation signaling during ISO-induced cardiac
hypertrophy. Protein expression levels of (A) CaMKKβ,
AMPK and mTOR, and (B) LC3 and cleaved caspase-3 were determined by
western blot analysis of neonatal rat cardiomyocytes. The intensity
of each band was quantified by densitometry and the data were
normalized to the GAPDH protein band intensity. The fold change
values are represented as the mean ± standard error of the mean
from three independent determinations. *P<0.05 vs.
the control group, #P<0.05 vs. the ISO group and
&P<0. 05 vs. the GdCl3 + ISO group.
p-, phosphorylated-; CaMKKβ,
Ca2+/calmodulin-dependent-protein kinase kinase 2 β,
LC3, microtubule-associated protein light chain 3; AMPK,
AMP-activated protein kinase; ISO, isoproterenol; 3-MA,
3-methyladenine; mTOR, mechanistic target of rapamycin. |
The effect of compound C (a specific AMPK inhibitor)
on the Calhex231-induced suppression of autophagy was
investigated. Fig. 5B demonstrates
the cellular molecular signaling pathways induced by CaSR to
increase autophagy and decrease apoptosis. Compared with the
control group, ISO treatment significantly increased the LC3II/LC3I
ratio and the expression level of cleaved caspase-3 (P<0.05). In
addition, GdCl3 treatment increased the LC3II/LC3I ratio
compared with the ISO group, however, the cleaved caspase-3 level
was decreased. A negative association between autophagy and
apoptosis induced by CaSR was observed. However, compared with the
GdCl3 + ISO group, the LC3II/LC3I ratio was
significantly reduced and the caspase-3 level was increased in the
GdCl3 + compound C+ ISO group, suggesting that CaSR
regulates the autophagy level, which is mediated by the
CaMKKβ-AMPK-mTOR signaling pathway (P<0.05).
Calhex231 reduced the CaSR-augmented autophagy via
suppressing the CaMKKβ-AMPK-mTOR signaling pathway
(Fig. 5B).
Discussion
In the present study, a rat cardiac hypertrophy
model was established using ISO. The levels of CaSR expression and
autophagy were markedly increased in hypertrophic hearts.
Furthermore, CaSR inhibition significantly reduced autophagy
signaling and ameliorated cardiac hypertrophy (P<0.05). In
addition, the experimental results of the neonatal rat hypertrophic
cardiomyocytes induced by ISO in vitro were consistent with
the results obtained from the animal model, supporting the findings
of the present study.
Previous studies have demonstrated that CaSR is
expressed throughout the cardiovascular system and is important in
cardiac physiology and pathophysiology (22,23).
CaSR releases [Ca2+]i by accumulation of
inositol phosphate. Increased [Ca2+]i activates certain
Ca2+-dependent signaling pathways, which result in
myocardial hypertrophy (24). CaSR
is activated by Gd3+ and Mg2+ (type I
activators), which are present in extracellular fluids, whereas,
calcilytics, including Calhex231 inhibit the effect of
Ca2+ on CaSR (25).
Myocardial hypertrophy is characterized by an
increase in cardiomyocyte size and protein content (26,27).
The manifestation of cardiac hypertrophy involves increases in the
heart size and interstitial fibrosis (28). Numerous reports have indicated that
ISO is important for mediating load-induced myocardial hypertrophy
(4,9). In the present study, when rats were
administered ISO for 7 days, the heart size, quantity of
interstitial collagen and the cardiomyocyte size were markedly
increased, indicating dysfunction of the heart. The current study
demonstrated that ISO induced cardiac hypertrophy. Furthermore, the
protein expression level of CaSR was markedly increased in
hypertrophic myocardium. A similar effect was observed in
cardiomyocytes treated with ISO in vitro, with a notable
increase in the cardiomyocyte size, [Ca2+]i,
p-CaMKII expression level, protein content and expression of CaSR
observed. CaSR inhibitor markedly protected cardiomyocytes against
cardiac hypertrophy induced by ISO injection in vivo and
in vitro. The present study demonstrated that treatment with
CaSR inhibitor decreased cardiomyocyte size, CaMKII expression and
protein content in hypertrophic hearts and cardiomyocytes, and
markedly improved the cardiac functions. These results indicate
that CaSR inhibition effectively reduced myocardial hypertrophic
remodeling.
Autophagy maintains cellular homeostasis and
degrades proteins with long half-lives or damaged organelles to
avoid apoptosis initiation (29,30).
Consistent with the results of the present study, autophagy is
markedly increased during cardiac dysfunction resulting from
hypertensive heart disease, ischemic heart disease and dilated
cardiomyopathy (31,32). The current study demonstrated that
autophagy was markedly increased in ISO-induced hypertrophic rat
hearts and cardiomyocytes. CaSR inhibition significantly suppressed
autophagy to aid the survival of cardiomyocytes under hypertrophic
conditions (P<0.05). In addition, ISO treatment markedly
increased apoptosis in vivo and in vitro. However,
ISO treatment following pre-incubation with a CaSR activator
significantly increased the level of autophagy and decreased
apoptosis in vitro, compared with the ISO treatment alone
(P<0.05). By contrast, treatment with a CaSR inhibitor
significantly increased ISO-induced apoptosis and decreased
autophagy in cardiomyocytes. As the balance between autophagy and
apoptosis maintains homeostasis, inactivation of autophagy may
result in the accumulation abnormal proteins and organelles, thus,
promoting apoptosis (33).
CaSR-induced autophagy was investigated further by treatment of
cardiomyocytes with 3-MA. Compared with the GdCl3 + ISO
group, the autophagy level was decreased and cardiomyocyte
apoptosis was significantly increased following 3-MA treatment
(P<0.05), consistent with a previous report (34).
To investigate the potential underlying mechanisms
of CaSR-induced autophagy, the current study examined intracellular
signaling pathways regulated by CaSR that upregulate autophagy.
CaMKKβ has previously been demonstrated to be activated
by increased [Ca2+]i and stimulates AMPK
(35). AMPK is responsible for
sensing energy and nutrients, and is involved in promoting
autophagy by directly activating the mammalian autophagy-initiating
kinase unc-51 like autophagy activating kinase 1 via
phosphorylation of Ser317 and Ser777 (36). Thus, CaMKKβ may induce
autophagy by activating AMPK and inhibiting the mTOR signaling
pathway. ISO treatment increased the phosphorylation of
CaMKKβ and AMPK, and decreased mTOR. Notably, compared
with the ISO only, treatment with CaSR activator and ISO increased
CaMKKβ and AMPK phosphorylation, whereas mTOR
phosphorylation was decreased. However, the CaSR inhibitor blocked
these effects. The results of the current study indicate that
inhibition of CaSR in the CaMKKβ-AMPK-mTOR signaling
pathway may contribute to cardiomyocyte protection. Furthermore,
the present study demonstrated that compound C inhibits AMPK and
significantly decreases CaSR-induced autophagy in cardiomyocytes
treated with ISO (P<0.05). These data demonstrate that CaSR
stimulates autophagy in hypertrophic cardiomyocytes via activation
of the CaMKKβ-AMPK-mTOR signaling pathways.
In conclusion, the result of the present study
indicate that the expression of CaSR is upregulated in ISO-induced
cardiac hypertrophy. Furthermore, inhibition of CaSR may ameliorate
ISO-induced cardiac hypertrophy. This effect may be associated with
inhibition of autophagy and suppression of the
CaMKKβ-AMPK-mTOR signaling pathway. Cardiac hypertrophy
is induced by multiple factors, including pressure overload, ISO,
swimming and exercise. The present study investigated cardiac
hypertrophy in ISO-induced models, thus, the results require
further validation in other models of cardiac hypertrophy. The
results of the current study may support the potential use of a
CaSR inhibitor as a novel therapeutic agent for the treatment of
cardiac hypertrophy.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81100163,
81100170, 81170289 and 81170178) and the Natural Science Foundation
of Heilongjiang province (grant nos. H201415 and H2016015).
References
|
1
|
Sun B, Huo R, Sheng Y, Li Y, Xie X, Chen
C, Liu HB, Li N, Li CB, Guo WT, et al: Bone morphogenetic protein-4
mediates cardiac hypertrophy, apoptosis and fibrosis in
experimentally pathological cardiac hypertrophy. Hypertension.
61:352–360. 2013. View Article : Google Scholar
|
|
2
|
Yin X, Peng C, Ning W, Li C, Ren Z, Zhang
J, Gao H and Zhao K: miR-30a downregulation aggravates pressure
overload-induced cardiomyocyte hypertrophy. Mol Cell Biochem.
379:1–6. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
You J, Wu J, Jiang G, Guo J, Wang S, Li L,
Ge J and Zou Y: Olmesartan attenuates cardiac remodeling through
DLL4/Notch1 pathway activation in pressure overload mice. J
Cardiovasc Pharmacol. 61:142–151. 2013. View Article : Google Scholar
|
|
4
|
Lu F, Xing J, Zhang X, Dong S, Zhao Y,
Wang L, Li H, Yang F, Xu C and Zhang W: Exogenous hydrogen sulfide
prevents cardiomyocyte apoptosis from cardiac hypertrophy induced
by isoproterenol. Mol Cell Biochem. 381:41–50. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Loot AE, Pierson I, Syzonenko T,
Elgheznawy A, Randriamboavonjy V, Zivković A, Stark H and Fleming
I: Ca2+-sensing receptor cleavage by calpain partially accounts for
altered vascular reactivity in mice fed a high-fat diet. J
Cardiovasc Pharmacol. 61:528–535. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brown EM and MacLeod RJ: Extracellular
calcium sensing and extracellular calcium signaling. Physiol Rev.
81:239–297. 2001.PubMed/NCBI
|
|
7
|
Cifuentes M and Rojas CV: Antilipolytic
effect of calcium-sensing receptor in human adipocytes. Mol Cell
Biochem. 319:17–21. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang LN, Wang C, Lin Y, Xi YH, Zhang WH,
Zhao YJ, Li HZ, Tian Y, Lv YJ, Yang BF and Xu CQ: Involvement of
calcium-sensing receptor in cardiac hypertrophy-induced by
angiotensinII through calcineurin pathway in cultured neonatal rat
cardiomyocytes. Biochem Biophys Res Commun. 369:584–589. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu FH, Fu SB, Leng X, Zhang X, Dong S,
Zhao YJ, Ren H, Li H, Zhong X, Xu CQ and Zhang WH: Role of the
calcium-sensing receptor in cardiomyocyte apoptosis via the
sarcoplasmic reticulum and mitochondrial death pathway in cardiac
hypertrophy and heart failure. Cell Physiol Biochem. 31:728–743.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bisping E, Wakula P, Poteser M and Heinzel
FR: Targeting cardiac hypertrophy: Toward a causal heart failure
therapy. J Cardiovasc Pharmacol. 64:293–305. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin L, Xu J, Ye Y, Ge J, Zou Y and Liu X:
Isosorbide dinitrate inhibits mechanical stress-induced cardiac
hypertrophy and autophagy through downregulation of angiotensin II
type 1 receptor. J Cardiovasc Pharmacol. 65:1–7. 2015. View Article : Google Scholar
|
|
12
|
Prietsch RF, Monte LG, da Silva FA, Beira
FT, Del Pino FA, Campos VF, Collares T, Pinto LS, Spanevello RM,
Gamaro GD and Braganhol E: Genistein induces apoptosis and
autophagy in human breast MCF-7 cells by modulating the expression
of proapoptotic factors and oxidative stress enzymes. Mol Cell
Biochem. 390:235–242. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guo R, Hu N, Kandadi MR and Ren J:
Facilitated ethanol metabolism promotes cardiomyocyte contractile
dysfunction through autophagy in murine hearts. Autophagy.
8:593–608. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao DJ, Wang ZV, Battiprolu PK, Jiang N,
Morales CR, Kong Y, Rothermel BA, Gillette TG and Hill JA: Histone
deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by
suppressing autophagy. Proc Natl Acad Sci USA. 108:4123–4128. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Borges JC, Silva JA Jr, Gomes MA, Lomez
ES, Leite KM, Araujo RC, Bader M, Pesquero JB and Pesquero JL:
Tonin in rat heart with experimental hypertrophy. Am J Physiol
Heart Circ Physiol. 284:H2263–H2268. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gowen M, Stroup GB, Dodds RA, James IE,
Votta BJ, Smith BR, Bhatnagar PK, Lago AM, Callahan JF, DelMar EG,
et al: Antagonizing the parathyroid calcium receptor stimulates
parathyroid hormone secretion and bone formation in osteopenic
rats. J Clin Invest. 105:1595–1604. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Silve C, Petrel C, Leroy C, Bruel H,
Mallet E, Rognan D and Ruat M: Delineating a Ca2+ binding pocket
within the venus flytrap module of the human calcium-sensing
receptor. J Biol Chem. 280:37917–37923. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pan W, Zhong Y, Cheng C, Liu B, Wang L, Li
A, Xiong L and Liu S: MiR-30-regulated autophagy mediates
angiotensin II-induced myocardial hypertrophy. PloS One.
8:e539502013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Abdulrahman BA, Khweek AA, Akhter A,
Caution K, Tazi M, Hassan H, Zhang Y, Rowland PD, Malhotra S,
Aeffner F, et al: Depletion of the ubiquitin-binding adaptor
molecule SQSTM1/p62 from macrophages harboring cftr ΔF508 mutation
improves the delivery of burkholderia cenocepacia to the autophagic
machinery. J Biol Chem. 288:2049–2058. 2013. View Article : Google Scholar
|
|
21
|
Kimura S, Fujita N, Noda T and Yoshimori
T: Monitoring autophagy in mammalian cultured cells through the
dynamics of LC3. Methods Enzymol. 452:1–12. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang WH, Lu FH, Zhao YJ, Wang LN, Tian Y,
Pan ZW, Lv YJ, Wang YL, Du LJ, Sun ZR, et al: Post-conditioning
protects rat cardiomyocytes via PKCepsilon-mediated calcium-sensing
receptors. Biochem Biophys Res Commun. 361:659–664. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang WH, Fu SB, Lu FH, Wu B, Gong DM, Pan
ZW, Lv YJ, Zhao YJ, Li QF, Wang R, et al: Involvement of
calcium-sensing receptor in ischemia/reperfusion-induced apoptosis
in rat cardiomyocytes. Biochem Biophys Res Commun. 347:872–881.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hunter JJ and Chien KR: Signaling pathways
for cardiac hypertrophy and failure. N Engl J Med. 341:1276–1283.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nemeth EF: The search for calcium receptor
antagonists (calcilytics). J Mol Endocrinol. 29:15–21. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Komuro I and Yazaki Y: Control of cardiac
gene expression by mechanical stress. Annu Rev Physiol. 55:55–75.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sadoshima J and Izumo S: The cellular and
molecular response of cardiac myocytes to mechanical stress. Annu
Rev Physiol. 59:551–571. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen C, Huo R, Tong Y, Sheng Y, Liu HB,
Gao X, Nakajima O, Yang BF and Dong DL: Systemic heme oxygenase-1
transgenic overexpression aggravates pressure overload-induced
cardiac hypertrophy in mice. Cell Physiol Biochem. 28:25–32. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Terman A and Brunk UT: Autophagy in
cardiac myocyte homeostasis, aging and pathology. Cardiovasc Res.
68:355–365. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rothermel BA and Hill JA: Autophagy in
load-induced heart disease. Circ Res. 103:1363–1369. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yan L, Vatner DE, Kim SJ, Ge H, Masurekar
M, Massover WH, Yang G, Matsui Y, Sadoshima J and Vatner SF:
Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci
USA. 102:13807–13812. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hein S, Arnon E, Kostin S, Schönburg M,
Elsässer A, Polyakova V, Bauer EP, Klövekorn WP and Schaper J:
Progression from compensated hypertrophy to failure in the
pressure-overloaded human heart: Structural deterioration and
compensatory mechanisms. Circulation. 107:984–991. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bursch W: The autophagosomallysosomal
compartment in programmed cell death. Cell Death Differ. 8:569–581.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cao X, Liu B, Cao W, Zhang W, Zhang F,
Zhao H, Meng R, Zhang L, Niu R, Hao X and Zhang B: Autophagy
inhibition enhances apigenin-induced apoptosis in human breast
cancer cells. Chin J Cancer Res. 25:212–222. 2013.PubMed/NCBI
|
|
35
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li MH, Zhang YJ, Yu YH, Yang SH, Iqbal J,
Mi QY, Li B, Wang ZM, Mao WX, Xie HG and Chen SL: Berberine
improves pressure overload-induced cardiac hypertrophy and
dysfunction through enhanced autophagy. Eur J Pharmacol. 728:67–76.
2014. View Article : Google Scholar : PubMed/NCBI
|