Introduction
The normal heart rhythm is particularly dependent
upon proper contractile function of cardiomyocytes. However, the
reactive oxygen species (ROS) generated under several pathological
conditions, including ischemia-reperfusion injury, may be one of
the major causes of heart muscle injury (1). ROS lead to an imbalanced
intracellular redox condition, which is essential for the induction
of apoptosis and impairment of ATP production. Due to the limited
regenerative capacity of cardiomyocytes, understanding the
mechanisms of oxidative stress-induced apoptosis may potentially
assist in developing novel treatment or preventative strategies for
ischemia-reperfusion injury of the heart.
Previous studies of microRNAs (miRs) have indicated
a a number of novel mechanisms of heart diseases. It has been
reported that miRs not only control arrhythmogenesis, but are also
significantly involved in the regulation of cardiomyocyte death
(2–4). Previously, miR-153 has been shown to
suppress cancer cell proliferation, invasion and migration
(5–8). Despite several targets, which have
been identified in cancer cells, the role of miR-153 in
cardiomyocytes remains to be fully elucidated.
As one of the major cascades of apoptosis, the
intrinsic apoptotic pathway is initiated from mitochondria and is
tightly controlled by B cell-lymphoma-2 (Bcl-2) family member
proteins. One member of the Bcl-2 family, myeloid cell leukemia-1
(Mcl-1), has been reported to exert critical functions in apoptosis
and mitochondrial homeostasis (9–13).
Previous studies have demonstrated that the loss of Mcl-1 is
essential in heart failure (10,14).
However, the mechanism by which Mcl-1 is regulated under oxidative
stress remains to be fully elucidated. A mechanism revealed by a
previous study showed that Mcl-1 functions to affect autophagy, a
major degradation process in the lysosome, which is critical for
cardiac homeostasis (10). A
number of studies have shown that autophagy has a beneficial effect
on the survival of cardiomyocyte under conditions of stress
(15–17). However, whether Mcl-1 is involved
in the regulation of pro-survival autophagy, which acts against
oxidative damage, remains to be elucidated.
The present study aimed to identify a novel
microRNA-associated mechanism in cardiac oxidative stress. A
potential binding site of miR-153 was identified in the Mcl-1
3′-untranslated region, suggesting that miR-153 may be involved in
this process. Therefore, multiple experimental methods were used to
confirm our hypothesis that miR-153 regulates cardiomyocytes
survival upon oxidative stimuli. The present study identified a
novel role of miR-153 in regulating apoptosis and autophagy, and
thus may provide certain clinical implications for the treatment of
oxidative stress associated heart syndrome.
Materials and methods
Cell culture
A primary culture of rat ventricular cardiomyocytes
was used, as described previously (18). Each time, 10 neonatal
Sprague-Dawley rats (within 2-days-old) were used for cell culture.
The neonatal rats were purchased from the Experimental Animal
Center of Southern Medical University (Guangzhou, China). Male and
female rats used to breed the neonatal rats were housed at a
constant temperature of 22°C with 50% humidity and 12 h light/dark
cycles. The rats were sacrificed with CO2. Briefly, the
hearts of neonatal rats were dissected and rinsed in cold
phosphate-buffered saline (PBS), followed by being cut into several
2×2 mm sections. The heart tissues were then digested with a series
of 0.25% trypsin solution. Following the completion of digestion,
the cell suspension was collected. To isolate the fibroblasts, the
suspension was plated in flasks for 2 h at 37°C. The cardiomyocytes
were then diluted (25,000 cells/ml) and plated elsewhere. The
cardiomyocytes were cultured in Dulbecco's modified Eagle's medium
(GE Healthcare Life Sciences HyClone Laboratories, Logan, UT, USE)
supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis,
MO, USA) and penicillin-streptomycin solution at 37°C.
H2O2 was used at the final concentration of
100 µM. The present study was approved by the Ethics
Committee of Southern Medical University (Guangzhou, China).
Transfection
A standard transfection procedure was used for
delivering miR-153 or miR-153 antisense inhibitor into the cells.
Lipofectamine 2000 tranfection reagent (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) was used, according to the
manufacturer's protocol for the kit. The Mcl-1 small interfering
(si)RNA, miR-153 and miR-153 antisense inhibitor were designed and
synthesized by Guangzhou RiboBio Co., Ltd. (Guangzhou, China). The
final transfection concentrations for siRNA and miR were 150
nmol/l.
MTT assay
To assess the extent of cell death, an MTT method
was used. Briefly, the cells were plated (25,000 cells/ml) in
96-well plates and treated, as indicated in each experiment. The
cells were treated with 100 µM H2O2
for 6, 12 and 24 h. For cells transfected with anti-miR-153 or
anti-miR-153 in combination with si-Mcl-1, cells were treated with
100 µM H2O2 for 24 h. Following
treatment, 20 µl MTT (5 mg/ml) solution was added to each
well, and the cells were allowed to incubate for another 4 h at
37°C. The culture medium was then discarded prior to visualization
with dimethyl sulfoxide. The absorbance value at 490 nm was
determined using a spectrophotometer (BioTek, Winooski, VT,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was isolated from the cardiomyocytes using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. Reverse transcription was
performed using a specific stem-loop primer obtained from Guangzhou
RiboBio Co., Ltd. SYBR green master mix (Promega Corp., Madison,
WI, USA) was used to amplify the cDNA (1:15; 5 µl) with
specific primers for miR-153 (Guangzhou RiboBio Co., Ltd.) on an
ABI7500 FAST Real-Time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The following cycles were used: 40 cycles
at 95°C for 2 min, 95°C for 30 sec and 58°C for 40 sec. U6 was used
as an internal control for normalization. The fold changes in
miR-153 in each experimental group were calculated by normalizing
to the negative control (NC) or control group using the
2−ΔΔCq method.
Western blot analysis
Western blot analysis was performed to detect the
expression levels of the proteins of interest. Briefly, the cells
were homogenized with SDS lysis buffer (Beyotime Institute of
Biotechnology, Beijing, China) and denatured with sample buffer
(Beyotime Institute of Biotechnology). The protein quantity was
determined using a bicinchoninic acid assay. Electrophoresis of 30
µg protein on an SDS-PAGE gel was performed, followed by
transfer onto the supporting media of a polyvinylidene fluoride
membrane. The membranes were blocked with 5% non-fat milk at room
temperature for 2 h. The proteins were identified by incubation
with specific primary antibodies overnight at 4°C, washed with PBS
containing Tween-20 three times, followed by
horseradish-peroxidase-conjugated secondary antibody specification
for 40 min at room temperature. The membranes were washed as
before. The bands were visualized using an ECL kit (Beyotime
Institute of Biotechnology). Antibodies for LC3 (cat. no. 2775;
1:500), cleaved caspase-3 (cat. no. 9664; 1:500) and cleaved
caspase-9 (cat. no. 9507; 1:1,000) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA), antibodies for
β-actin (cat. no. sc-130656; 1:1,000) and Mcl-1 (cat. no. sc-819;
1:1,000), and secondary antibody was horseradis peroxidase-linked
goat anti-rabbit immunoglobulin G (cat. no. sc-2301; 1:2,000) were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). Protein bands were quantified using Quantity One software
(v4.62; Bio-Rad Laboratories, Hercules, CA, USA).
Dual luciferase activity assay
A fragment of the Mcl-1 3′UTR was amplified by PCR,
and the short sequence was inserted into the 3′UTR of an pMIRGLO
construct, which expresses firefly luciferase. HEK293 cells
(20,000/ml) were co-transfected with 0.1 µg firefly
luciferase reporter containing the Mcl-1 3′UTR and miR-153 or its
antisense inhibitor, along with pRL-TK as an internal control using
Lipofectamine 2000 (Thermo Fisher Scientific, Inc.). The 3′-UTR
sequence was amplified from the cDNA using the primer: forward:
5′-GAGCTCTACAAGAGGGTGAAGGAGGA-3′ and reverse:
5′-AAGCTTCAGGCTTAAACTTGTGTTAAAC-3′. The amplification program was
as follows: 95°C for 10 min; 35 cycles at 95°C for 30 sec, 56°C for
30 sec and 72°C for 45 sec; followed by 72°C for 5 min. The empty
pMIRGLO plasmid and the PCR products were digested with
HindIII and SacI at 37°C for 1 h, followed by T4
ligation reaction at room temperature for 2 h. The ligation product
was then transformed into competent cells (Transgene Biotech,
Beijing, China) and the positive plasmid was verified by PCR. All
the restriction enzymes were purchase from Takara Bio., Inc.
(Dalian, China). The luciferase activity in each group was
normalized to that of the renilla luciferase expressed by the
pRL-TK plasmid. The whole procedure was performed using a
Dual-Luciferase Reporter Assay system (Promega Corp.) in accordance
with the manufacturer's protocol.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. Comparisons between two groups were performed using
Student's t-test; comparisons between three groups were
performed using one way analysis of variance followed by a
Bonferroni test. SPSS 19.0 software (IBM SPSS, Chicago, IL, USA)
was used for analysis. P<0.05 (two-tailed) was considered to
indicate a statistically significant difference.
Results
miR-153 is upregulated under apoptotic
stimuli induced by oxidative stress
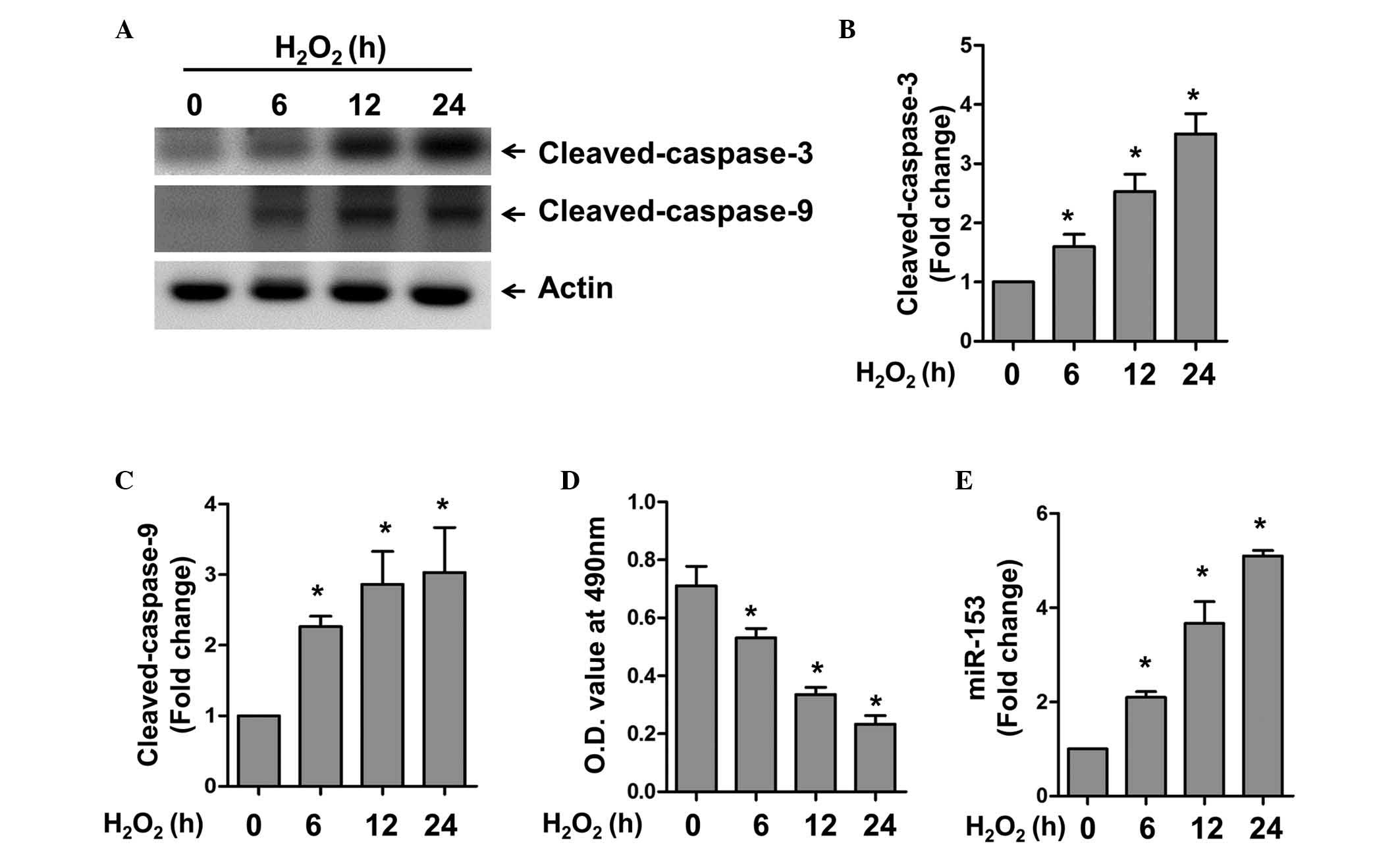
The present study first examined the expression
level of miR-153 under oxidative stress. The cardiomyocytes were
treated with H2O2 for different time periods
to induce apoptosis in the cardiomyocytes. Using western blot
analysis, H2O2 was found to induce caspase-3
and 9 activation in a time-dependent manner (Fig. 1A–C). Accordingly, a significant
decrease in cell viability was observed under these conditions, as
revealed by the MTT assay (Fig.
1D). RT-qPCR was used to detect the expression of miR-153 at
various time points. In accordance with the expression of the
apoptotic markers, caspase-3 and 9, miR-153 was also upregulated in
a time-dependent manner. These results confirmed the successful
establishment of an apoptotic model in the cardiomyocytes, and
indicated that miR-153 may be involved in oxidative stress-induced
apoptosis.
Inhibition of miR-153 reduces the
apoptosis of cardiomyocytes
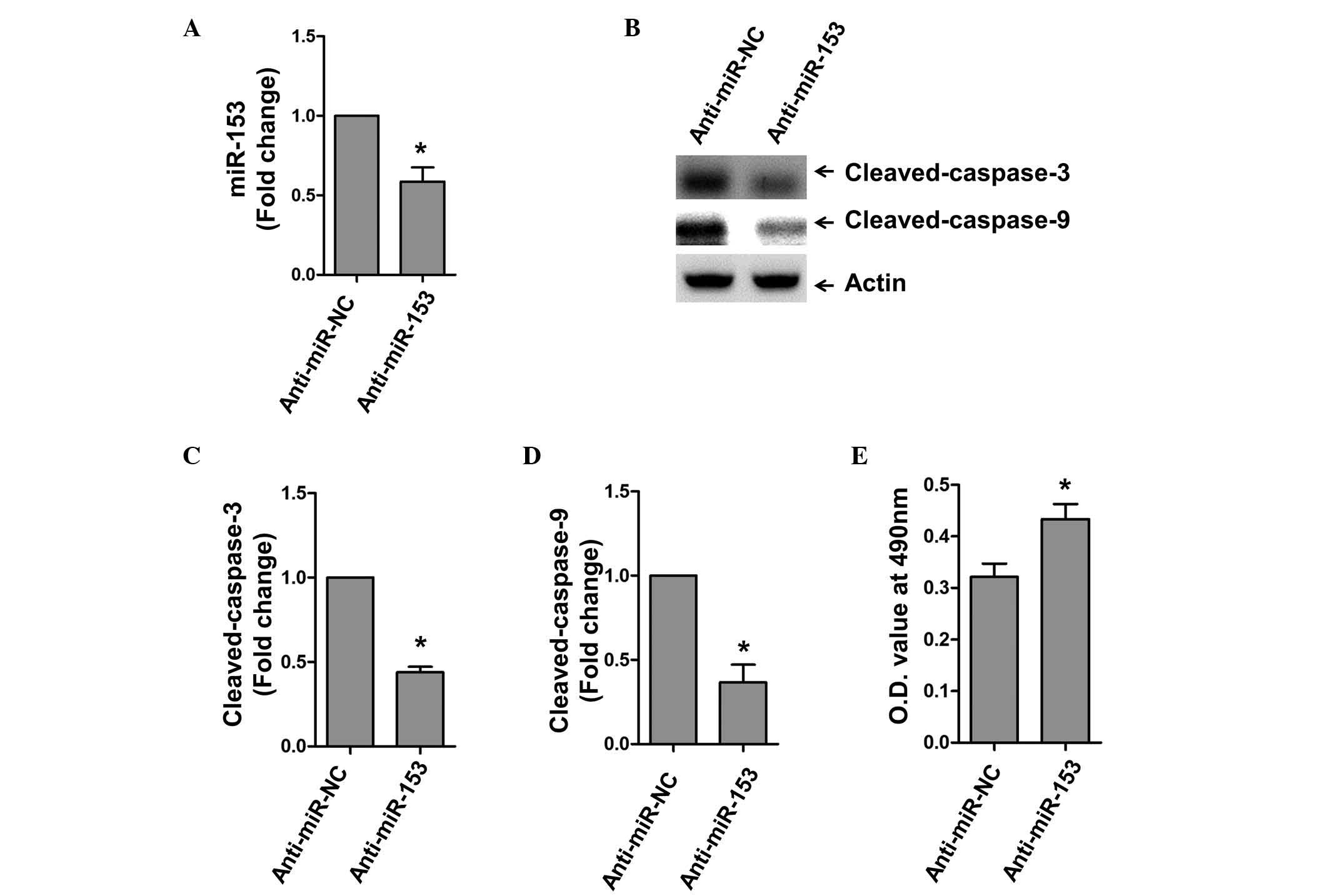
Following the above observation that miR-153 was
upregulated during oxidative stress, the present study subsequently
transfected cells with the antisense inhibitor of miR-153
(anti-miR-153) to manipulate its endogenous level. Anti-miR-153
significantly reduced the expression of miR-153 under the
H2O2 stimuli (Fig. 2A). The results of the western blot
analysis showed that the expression levels of activated caspase-3
and caspase-9 were significantly lower following miR-153 inhibition
(Fig. 2B–D). Consistently, the
inhibition of miR-153 by its antisense inhibitor increased the
survival of cardiomyocytes upon H2O2
stimulation (Fig. 2E).
Mcl-1 is a target of miR-153
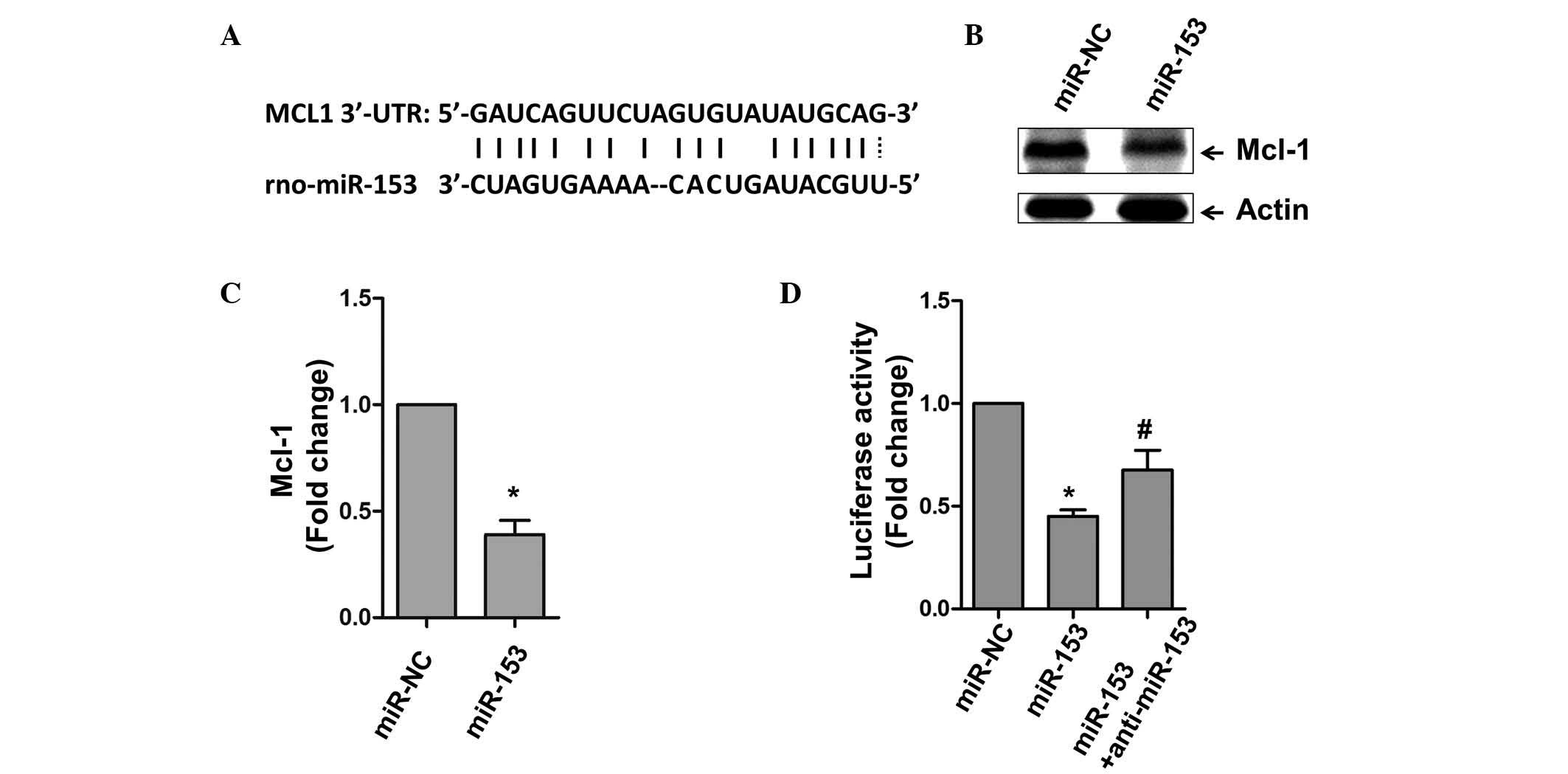
miRs commonly function to repress gene expression by
binding to the 3′UTR region of mRNA. Using bioinformatical
alignment, the present study identified a potential binding site
within the 3′UTR of Mcl-1 (Fig.
3A). Whether this predicted region is actually targeted by
miR-153 was then assessed. The cardiomyocytes were transfected with
miR-153, and it was found that the expression of Mcl-1 was
significantly inhibited, compared with the NC group of cells
(Fig. 3B and C). A luciferase
assay was also used to examine the association between Mcl-1 and
miR-153. As shown in Fig. 3D, the
overexpression of miR-153 markedly inhibited luciferase activity,
and anti-miR-153 rescued the inhibitory effect of miR-153 on
luciferase activity.
miR-153 inhibits Mcl-1-dependent
autophagy
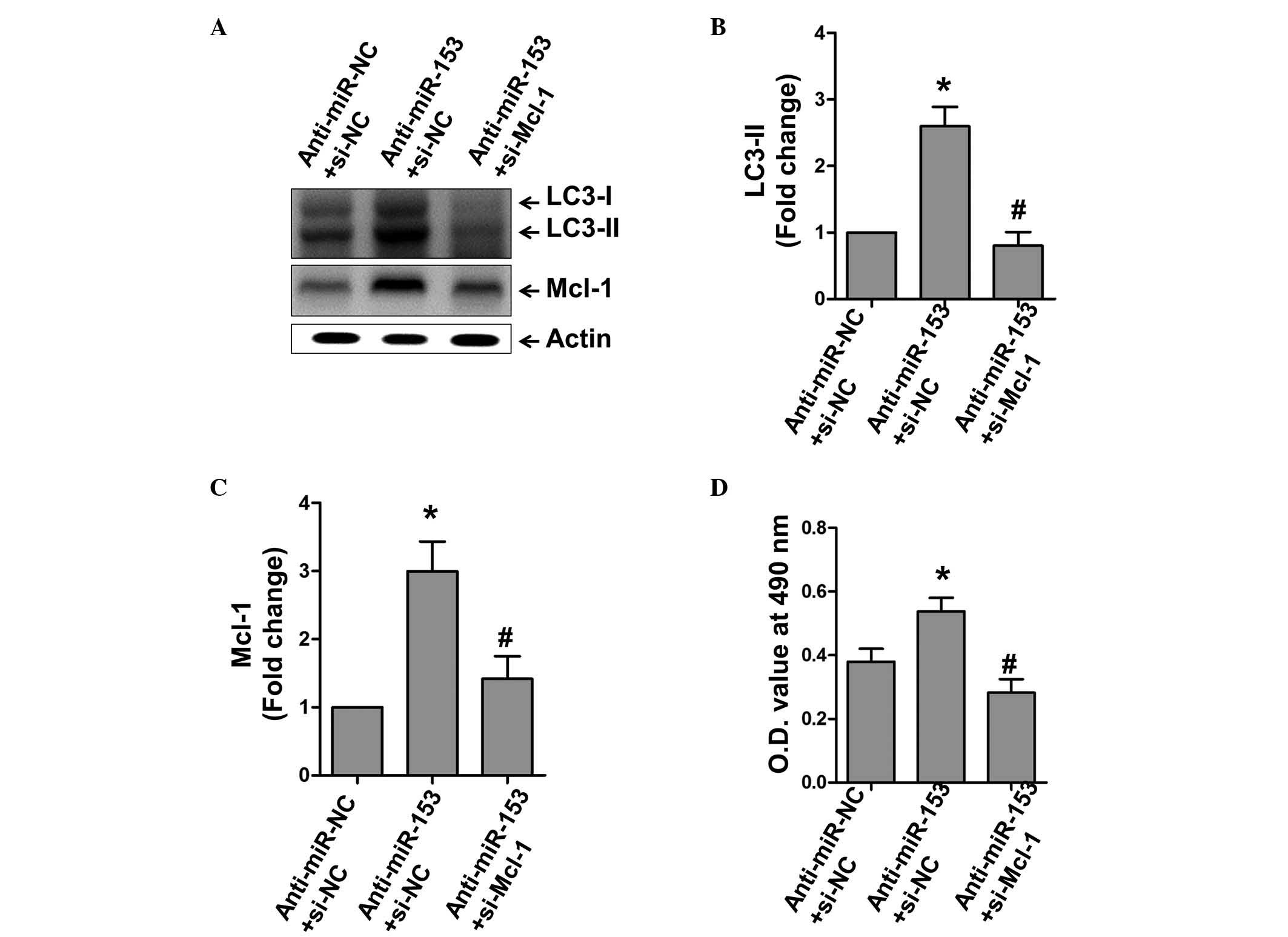
As Mcl-1 has been established as a target of
miR-153, and it has been previously shown to affect cardiac
autophagy, the present study investigated whether Mcl-1-dependent
autophagy is also under the control of miR-153 (10,19).
The inhibition of miR-153 under H2O2
stimulation markedly increased the expression of the autophagy
marker, LC3-II, whereas the inhibition of Mcl-1 inhibited this
effect (Fig. 4A–C). In particular,
the MTT assay showed that the inhibition of Mcl-1 inhibited the
protective effect of anti-miR-153, whereas inhibiting anti-miR-153
induced autophagy, suggesting that Mcl-1-dependent autophagy was
critically involved in the promotion of cell death by miR-153
(Fig. 4D).
Discussion
In the present study, it was demonstrated that
miR-153 was involved in oxidative stress-induced cell death in
cardiomyocytes. On investigating the molecular mechanism by which
miR-153 regulated cell survival, the present study identified Mcl-1
as its direct target. As a consequence, the cardiomyocytes
exhibited limited autophagic activity and enhanced apoptosis, which
may have accounted, at least partially, for the
H2O2-induced cardiomyocyte death. These
results may have important clinical implications for the prevention
and treatment of cardiac syndromes, including ischemia-reperfusion
injury.
miRs have emerged as a class of important regulators
in the heart. The interplay between miRs and several critical
regulators in the apoptotic machinery have attracted wide attention
in cardiac investigations. For example, a previous study by Wang
et al showed that miR-361 inhibits apoptosis by regulating
prohibitin during myocardial ischemia (20), and Xu et al reported that
the β-blocker, carvedilol, exerts a protective function on
cardiomyocytes against oxidative stress-induced apoptosis by
inducing the expression of miR-133 (21). These earlier findings suggest that
miRs may have a profound effect on cardimyocyte survival. miR-153
is located on chromosome 6 in the rat genome, and previous studies
have reported that miR-153 inhibits the growth and invasive
behavior of cancer cells. Bai et al reported its importance
in pancreatic ductal adenocarcinoma (22), and Zhou et al showed its
antitumor effect in ovary cancer cells (6). However, there are few reports on the
direct role of miR-153 on apoptosis, particularly in heart cells.
The present study provided the first evidence, to the best of our
knowledge, that miR-153 was increased upon apoptosis induced by
H2O2 in cardiomyocytes, and that modulation
in its level had a direct effect on cell apoptosis.
Apoptosis and autophagy are two essential biological
processes, which act reciprocally to control cell survival under
oxidative stress. It has been reported that basal autophagic
activity is required for heart cells to maintain homeostasis, and
that inhibition of this process by the deletion of its essential
gene, ATG5, leads to heart failure (23). It has also been demonstrated that
autophagy is required for cardimyocytes to confer their
anti-apooptotic effects (24).
Previous studies have shown that Mcl-1 is involved, not only in the
apoptotic machinery, but also in the quality control of
mitochondria by autophagy. Thomas et al reported that the
deletion of Mcl-1 in heart muscle results in impaired autophagy and
heart failure (10). The present
study identified a free-energy favorable binding site of miR-153
within the Mcl-1 3′UTR, suggesting that miR-153 may modulate cell
survival through the downstream effect of Mcl-1. Following
validation of Mcl-1 as a direct target of miR-153, the present
study investigated whether autophagy was also involved. Consistent
with previous studies, the increased expression of the autophagic
marker, LC3-II, was observed upon miR-153 inhibition, which was
reversed by Mcl-1 silencing. Thus, the present study is the first,
to the best of our knowledge, to provide a novel mechanism
underlying the regulation of cell apoptosis and autophagy in heart
cells by miR-153.
The way in which miRs are regulated under different
pathological conditions is an important issue and requires further
understanding of the behavior of cardiomyocytes. Previous studies
demonstrating the regulation of transcriptional factors of miRs may
provide insights into this issue. For example, serum response
factor transcriptionally regulates miR-1 in the heart, and p53
transcriptionally downregulates the expression of miR-499 to
promote apoptosis (25,26). It is conceivable that several
oxidative stress-specific transcriptional factors may regulate the
expression of miR-153. Further investigation is warranted to
investigate the upstream events of miR-153.
In conclusion, the present study demonstrated the
key role of miR-153 in oxidative stress-induced apoptosis. Mcl-1
was identified as a novel target, through which miR-153 regulated
essential programs, including apoptosis and autophagy. These
results may provide novel clues for understanding the control by
miRs of multiple effects under certain stresses. The results of the
present study may also guide the development of novel therapies for
cardiac syndromes.
References
|
1
|
Hafstad AD, Nabeebaccus AA and Shah AM:
Novel aspects of ROS signalling in heart failure. Basic Res
Cardiol. 108:3592013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang J and Martin JF: Macro advances in
microRNAs and myocardial regeneration. Curr Opin Cardiol.
29:207–213. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sala V, Bergerone S, Gatti S, Gallo S,
Ponzetto A, Ponzetto C and Crepaldi T: MicroRNAs in myocardial
ischemia: Identifying new targets and tools for treating heart
disease. New frontiers for miR-medicine. Cell Mol Life Sci.
71:1439–1452. 2014. View Article : Google Scholar
|
|
4
|
Port JD and Sucharov C: Role of microRNAs
in cardiovascular disease: Therapeutic challenges and potentials. J
Cardiovasc Pharmacol. 56:444–453. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xia W, Ma X, Li X, Dong H, Yi J, Zeng W
and Yang Z: miR-153 inhibits epithelial-to-mesenchymal transition
in hepatocellular carcinoma by targeting Snail. Oncol Rep.
34:655–662. 2015.PubMed/NCBI
|
|
6
|
Zhou J, Xie M, Shi Y, Luo B, Gong G, Li J,
Wang J, Zhao W, Zi Y, Wu X and Wen J: MicroRNA-153 functions as a
tumor suppressor by targeting SET7 and ZEB2 in ovarian cancer
cells. Oncol Rep. 34:111–120. 2015.PubMed/NCBI
|
|
7
|
Hua HW, Jiang F, Huang Q, Liao Z and Ding
G: MicroRNA-153 promotes Wnt/β-catenin activation in hepatocellular
carcinoma through suppression of WWOX. Oncotarget. 6:3840–3847.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shan N, Shen L, Wang J, He D and Duan C:
MiR-153 inhibits migration and invasion of human non-small-cell
lung cancer by targeting ADAM19. Biochem Biophys Res Commun.
456:385–391. 2015. View Article : Google Scholar
|
|
9
|
Carroll RG, Hollville E and Martin SJ:
Parkin sensitizes toward apoptosis induced by mitochondrial
depolarization through promoting degradation of Mcl-1. Cell Rep.
9:1538–1553. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thomas RL, Roberts DJ, Kubli DA, Lee Y,
Quinsay MN, Owens JB, Fischer KM, Sussman MA, Miyamoto S and
Gustafsson ÅB: Loss of MCL-1 leads to impaired autophagy and rapid
development of heart failure. Genes Dev. 27:1365–1377. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jokinen E and Koivunen JP: Bcl-xl and
Mcl-1 are the major determinants of the apoptotic response to dual
PI3K and MEK blockage. Int J Oncol. 47:1103–1110. 2015.PubMed/NCBI
|
|
12
|
Cerella C, Muller F, Gaigneaux A, Radogna
F, Viry E, Chateauvieux S, Dicato M and Diederich M: Early
downregulation of Mcl-1 regulates apoptosis triggered by cardiac
glycoside UNBS1450. Cell Death Dis. 6:e17822015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Varadarajan S, Poornima P, Milani M, Gowda
K, Amin S, Wang HG and Cohen GM: Maritoclax and dinaciclib inhibit
MCL-1 activity and induce apoptosis in both a MCL-1-dependent and
-independent manner. Oncotarget. 6:12668–12681. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang X, Bathina M, Lynch J, Koss B,
Calabrese C, Frase S, Schuetz JD, Rehg JE and Opferman JT: Deletion
of MCL-1 causes lethal cardiac failure and mitochondrial
dysfunction. Genes Dev. 27:1351–1364. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li L, Xu J, He L, Peng L, Zhong Q, Chen L
and Jiang Z: The role of autophagy in cardiac hypertrophy. Acta
Biochim Biophys Sin. 2016.
|
|
16
|
Jia Z, Liu Y, Su H, Li M, Zhang M, Zhu Y,
Li T, Fang Y and Liang S: Safflower extract inhibiting apoptosis by
inducing autophagy in myocardium derived H9C2 cell. Int J Clin Exp
Med. 8:20254–20262. 2015.
|
|
17
|
Schiattarella GG and Hill JA: Therapeutic
targeting of autophagy in cardiovascular disease. J Mol Cell
Cardiol. 2015.PubMed/NCBI
|
|
18
|
Wang K, Liu F, Zhou LY, Ding SL, Long B,
Liu CY, Sun T, Fan YY, Sun L and Li PF: miR-874 regulates
myocardial necrosis by targeting caspase-8. Cell Death Dis.
4:e7092013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu J, Liao X and Wong C: Downregulations
of B-cell lymphoma 2 and myeloid cell leukemia sequence 1 by
microRNA 153 induce apoptosis in a glioblastoma cell line
DBTRG-05MG. Int J Cancer. 126:1029–1035. 2010.
|
|
20
|
Wang K, Liu CY, Zhang XJ, Feng C, Zhou LY,
Zhao Y and Li PF: miR-361-regulated prohibitin inhibits
mitochondrial fission and apoptosis and protects heart from
ischemia injury. Cell Death Differ. 22:1058–1068. 2015. View Article : Google Scholar :
|
|
21
|
Xu C, Hu Y, Hou L, Ju J, Li X, Du N, Guan
X, Liu Z, Zhang T, Qin W, et al: β-Blocker carvedilol protects
cardiomyocytes against oxidative stress-induced apoptosis by
up-regulating miR-133 expression. J Mol Cell Cardiol. 75:111–121.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bai Z, Sun J, Wang X, Wang H, Pei H and
Zhang Z: MicroRNA-153 is a prognostic marker and inhibits cell
migration and invasion by targeting SNAI1 in human pancreatic
ductal adenocarcinoma. Oncol Rep. 34:595–602. 2015.PubMed/NCBI
|
|
23
|
Nakai A, Yamaguchi O, Takeda T, Higuchi Y,
Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et
al: The role of autophagy in cardiomyocytes in the basal state and
in response to hemodynamic stress. Nat Med. 13:619–624. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dutta D, Xu J, Kim JS, Dunn WA Jr and
Leeuwenburgh C: Upregulated autophagy protects cardiomyocytes from
oxidative stress-induced toxicity. Autophagy. 9:328–344. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang JX, Jiao JQ, Li Q, Long B, Wang K,
Liu JP, Li YR and Li PF: miR-499 regulates mitochondrial dynamics
by targeting calcineurin and dynamin-related protein-1. Nat Med.
17:71–78. 2011. View
Article : Google Scholar
|
|
26
|
Zhao Y, Samal E and Srivastava D: Serum
response factor regulates a muscle-specific microRNA that targets
Hand2 during cardiogenesis. Nature. 436:214–220. 2005. View Article : Google Scholar : PubMed/NCBI
|