Introduction
Reactive oxygen species (ROS), a product of normal
cellular metabolism, are usually managed effectively by the
cellular defense systems, thereby having minimal effect on cellular
health (1). However, under
situations of exaggerated stress or hypoxia, the cellular defenses
may be insufficient to overcome ROS overload (1). Of note, in the hypertrophied and
failing heart, elevated levels of ROS and oxidative stress in
cardiomyocytes are associated with maladaptive ventricular
remodeling and a progressive decline in cardiovascular function
(2,3). Oxidative stress has been clinically
shown to be relevant in the progression of heart failure,
myocardial infarction, cardiac ischemia/reperfusion injury and
atrial fibrillation (4,5). Excess ROS can cause various types of
cellular damage, including mitochondrial dysfunction and DNA
damage, and can ultimately lead to apoptosis, with apoptosis of
cardiomyocytes being critical in tissue damage and eventual heart
failure (6–8). Therefore, understanding the mechanism
underlying the effects of ROS and apoptosis are necessary for
further improvement in the diagnosis and therapy of heart
diseases.
MicroRNAs (miRNAs) are small, non-coding RNAs, which
inhibit the translation of target mRNAs through imperfect
sequence-specific binding to the 3′-untranslated region of target
mRNAs (9). Various studies have
demonstrated that the expression of miRNAs is tightly regulated in
a tissue-specific and a time-dependent manner (10,11).
By affecting protein translation, miRNAs have been identified as
potent regulators of a wide range of biological processes,
including cardiac morphogenesis, heart failure and arrhythmias
(12,13). It has been recognized that miRNAs
can be efficiently inhibited for prolonged periods using antisense
technologies, which has led to increased interest in the inhibition
of specific miRNAs as a feasible therapeutic option for selected
cardiovascular diseases (14).
According to these characteristics, miRNAs may represent an optimal
target for disease therapy.
In our previous study, it was demonstrated that
miRNA (miR)-423-5p is involved in congestive heart failure (CHF)
through the direct targeting of O-GlcNAc transferase (OGT) and the
induction of apoptosis in cardiomyocytes (15). Clinical experiments indicated that
miR-423-5p is associated with CHF and the expression levels of
prohormone brain natriuretic peptide (proBNP). Furthermore, the
expression of miR-423-5p significantly regulates the expression of
OGT and associated downstream targets, and induces apoptosis in
cardiomyocytes. However, the role of miR-423-5p during ROS-induced
apoptosis remains to be elucidated. The present study investigated
the expression of miR-423-5p in cardiomyocytes following exposure
to H2O2 for different durations and at
different concentrations. Furthermore, the role of miR-423-5p
during H2O2-induced apoptosis in
cardiomyocytes was also determined. The present results
demonstrated that miR-423-5p mediates
H2O2-induced apoptosis in cardiomyocytes.
Silencing of miR-423-5p significantly protects cardiomyocytes from
H2O2-induced apoptosis. It may provide a
novel therapeutic target for apoptosis-related heart diseases.
Materials and methods
Cell culture and treatment
The mouse cardiomyocytes used in the present study
were obtained from the Shanghai Cell Bank (Shanghai, China). The
cardiomyocytes were cultured in Dulbecco's modified Eagle's medium
containing 10% fetal bovine serumm(Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). All cells were maintained in a humidified
atmosphere containing 5% CO2 at 37°C. Cell transfection
was performed using FuGENE® HD transfection reagent
(Roche Diagnostics, Indianapolis, IN, USA), according to the
manufacturer's protocol. Briefly, cardiomyocytes were seeded into
6-well plates at a density of 2×105 cells/well and
cultured for 24 h to reach 70–80% confluence. Subsequently, 2
µg of plasmids encoding m-miR-423-5p and miR-Ctrl were
diluted in 100 µl serum-free medium, and 5 µl
FuGENE® HD transfection reagent was added to the tubes
containing the diluted DNA. The contents were mixed, and the
transfection complex was incubated for 15 min at room temperature,
followed by its addition to the 6-well plates. Medium alone was
used as a blank control. When the cardiomyocytes reached 70–80%
confluence in the 6-well plates, H2O2 was
added to the wells at different concentrations (0, 0.1, 0.2, 0.4
and 0.8 mM). At 12, 24 and 48 h following transfection and/or
H2O2 treatment, the cardiomyocytes were
collected for further experiments.
Vector construction
The miR-423-5p and miR-423-5p-mimic expression
plasmids were constructed, as previously (15). Briefy, each miRNA and plasmid
oligonucleotide were annealed at 90°C for 3 min, cooled to 37°C and
incubated for 1 h. The annealed double-stranded DNA
oligonucleotides were ligated between the HindIII and
SacI sites (Fermentas; Thermo Fisher Scientifc, Inc.) on the
pMIR-REPORT vector (Ambion Life Technologies; Thermo Fisher
Scientific, Inc.). The resulting recombinant plasmids were termed
pMIR-miR-423-5p, pMIR-miR-423-5p-mimic and pMIR-miR-control (Ctrl).
The three constructs were verified using DNA sequencing. The
plasmids, pMIR-miR-423-5p, pMIR-miR-423-5p-mimic and
pMIR-miR-control, were exacted and sent to the technology center of
Invitrogen Life Technologies at Shanghai for DNA sequencing. The
results were comprised with Jellyfish software (version 3.2) The
plasmids were extracted using EndoFree Plasmid Giga kits (Qiagen
GmbH, Hilden, Germany) from DH5α Escherichia coli
transformants (Genewiz, Suzhou, China) and stored at −20°C until
further use. The concentration was determined by measuring the
A260/A280 ratio using a Thermo ND 2000 spectrophotometer (Thermo
Fisher Scientific, Inc.).
Cell viability detection assay
A Cell Counting Kit-8 (CCK8) assay (Beyotime
Institute of Biotechnology, Shanghai, China) was used to detect
cell viability. The absorbance was determined at a wavelength of
450 nm using a plate reader. The same experiments were performed
three times.
RT-qPCR
Total RNA was extracted from each experimental group
using TRIzol® reagent (Invitrogen Life Technologies,
Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocol. The RNA concentration was assessed spectrophotometrically
at 260 nm (Thermo ND 2000; Thermo Fisher Scientific., Inc.).
Reverse transcription was performed on the isolated total RNA using
a Reverse Transcription kit (Takara Bio, Inc., Otsu, Japan), and
qPCR was performed using a Real Time PCR kit (Takara Bio, Inc).
Reverse transcription was performed using 1 µg total RNA in
2 µl water at 65°C for 5 min, 30°C for 10 min, 42°C for
10–30 min and 2°C for 3 min. The qPCR conditions were as follows:
Denaturation at 94°C for 2 min, amplification for 30 cycles at 94°C
for 0.5 min, annealing at 60°C for 0.5 min and extension at 72°C
for 1 min, followed by a terminal elongation step at 72°C for 10
min. The RT-qPCR analysis was performed on a Bio-Rad CFX96 thermal
cycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA). U6 was
amplified as an internal control and the Cq value of each PCR
product was calculated, from which the fold change was analyzed
(15). The m-miR-423-5p and m-U6
primers were supplied by Guangzhou RiboBio Co., Ltd. (Guangzhou,
China). The sequences were not supplied due to the rules of the
company.
26S proteasome activity assay
26S proteasome function was assayed, as described
previously (16). Briefly, the
cells were washed with phosphate-buffered saline, followed by
washing with buffer I, containing 50 mM Tris (pH 7.4), 2 mM DTT, 5
mM MgCl2 and 2 mM ATP), and pelleted by centrifugation
at 1,500 g for 3 min at 4°C. Glass beads and homogenization buffer
containing 50 mM Tris (pH 7.4), 1 mM DTT, 5 mM MgCl2, 2
mM ATP and 250 mM sucrose, were added and vortexed for 1 min. The
beads and cell debris were removed by centrifugation at 1,000 × g
for 5 min and 10,000 × g for 20 min. Protein concentration was
determined using a bicinchoninic acid assay protocol (Pierce
Biotechnology, Inc., Rockford, IL, USA). The protein from each
sample (100 µg) was diluted with buffer I to a final volume
of 1,000 µl, and the fluorogenic proteasome substrate,
SucLLVY-7-amido-4-methylcou-marin (chymotrypsin-like;
Sigma-Aldrich) was added to a final concentration of 80 µM
in 1% DMSO. To access 26S function, buffer I was replaced with
ATP-free buffer containing 20 mM HEPES (pH 7.8), 0.5 mM EDTA and
0.03%SDS (16). Cleavage activity
was monitored continuously by the detection of free
7-amido-4-methylcoumarin using a fluorescence plate reader (Gemini,
Molecular Devices LLC, Sunnyvale, CA, USA) at 380/460 nm and 37°C.
As controls for the drug experiments, 7-amido-4-methylcoumarin (2
µM) was incubated with the drugs in buffer I without cell
extracts, and measurements of proteasome function were corrected
when necessary.
Western blot analysis
Following transfection of the cardiomyocytes with
m-miR-423-5p and miR-Ctrl (48 h), total protein was collected. The
cells were lysed on ice for 30 min with radioimmunoprecipitation
assay lysis buffer containing 50 mM Tris-HCl (pH 7.4), 1% NP-40;
0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1
µg/ml each of aprotinin, leupeptin and pepstatin, 1 mM
Na3VO4 and 1 mM NaF. The proteins (20
µg) were separated by 10% SDS-polyacrylamide gel
electrophoresis and electronically transferred onto a
polyvinylidene difluoride membrane (EMD Millipore, Bedford, MA,
USA). Following blocking with 5% non-fat milk in Tris-buffered
saline containing 0.5% Tween-20 (TBST), the membranes were
incubated with the recommended dilutions of primary antibodies
against OGT (rabbit polyclonal; 1:800; cat. no. sc-32921; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) for 1 h at 37°C,
phosphorylated (p) adenosine monophosphate (AMP)-activated protein
kinase (p-AMPK; cat. no. 4188; 1:1,000), AMPK (cat. no. 2532;
1:1,000), p53 (cat. no. 2527; 1:1,200) and caspase 3 (cat. no.
9662; 1:600) for 1 h at 37°C (all purchased from Cell Signaling
Technology, Inc., Danvers, MA, USA) and GAPDH (cat. no. sc-25778;
1:5,000; Santa Cruz Biotechnology, Inc.). Following washing with
TBST, the membranes were incubated with peroxidase-conjugated
secondary antibodies against goat anti-mouse immunoglobulin G (cat.
no. ab97040; 1:5,000) or goat anti-rabbit (cat. no. ab191866;
1:5,000; each from Abcam, Cambridge, MA, USA) at 37°C for 1 h. The
peroxidase-labeled bands were visualized using an enhanced
chemiluminescence kit (Pierce Biotechnology, Inc.). The ratios of
OGT and p-AMPK to GAPDH were calculated using densitometry, and
values were normalized by dividing the ratios by the ratio in the
blank control.
Terminal deoxynucleotidyl
transferase-deoxyuridine triphosphate nick-end labeling (TUNEL)
assay
In order to detect apoptotic cells in the
cardiomyocytes, a TUNEL assay was performed using a DeadEndTM
Fluorometric TUNEL system (Promega Corporation, Madison, WI, USA),
according to the manufacturer's protocol. Cell nuclei with dark
green fuorescent staining were defined as TUNEL-positive nuclei,
which were visualized using a fluorescence microscope (DTX500;
Nikon Corporation, Tokyo, Japan). In order to quantify the
TUNEL-positive cells, the number of green fluorescent-positive
cells were counted in randomly selected fields (n=5) at
magnification, ×200. The cell nuclei were then counterstained with
4′,6-diamidino-2-phenylindole (Beyotime Institute of
Biotechnology).
Statistical analysis
Statistical comparisons of all results were analyzed
using one-way analysis of variance. Statistical analyses were
performed using SPSS version 19.0 (IBM SPSS, Armonk, NY, USA).
Values are expressed as the mean ± standard error of the mean.
P<0.05 was considered to indicate a statistically significant
difference.
Results
H2O2 decreases cell
viability in cardiomyocytes
The effect of H2O2 treatment
on cardiomyocyte viability was determined using a CCK8 assay. As
shown in Fig. 1A,
H2O2 treatment (0.4 mM) for 12 h
significantly reduced cell viability. Increasing the duration of
treatment with H2O2 to 24 and 48 h, markedly
inhibited the viability of the cardiomyocytes (Fig. 1A). Furthermore, the effect of
H2O2 concentration on cardiomyocyte viability
was determined. As the results in Fig.
1B show, treatment with 0.1 mM H2O2 for
24 h had no significant effect on cardiomyocytes viability.
However, treatment with 0.2, 0.4 and 0.8 mM
H2O2 for 24 h significantly inhibited
cardiomyocyte growth (Fig. 1B).
These results indicated that the effect of
H2O2 treatment on cell viability was time-
and concentration-dependent.
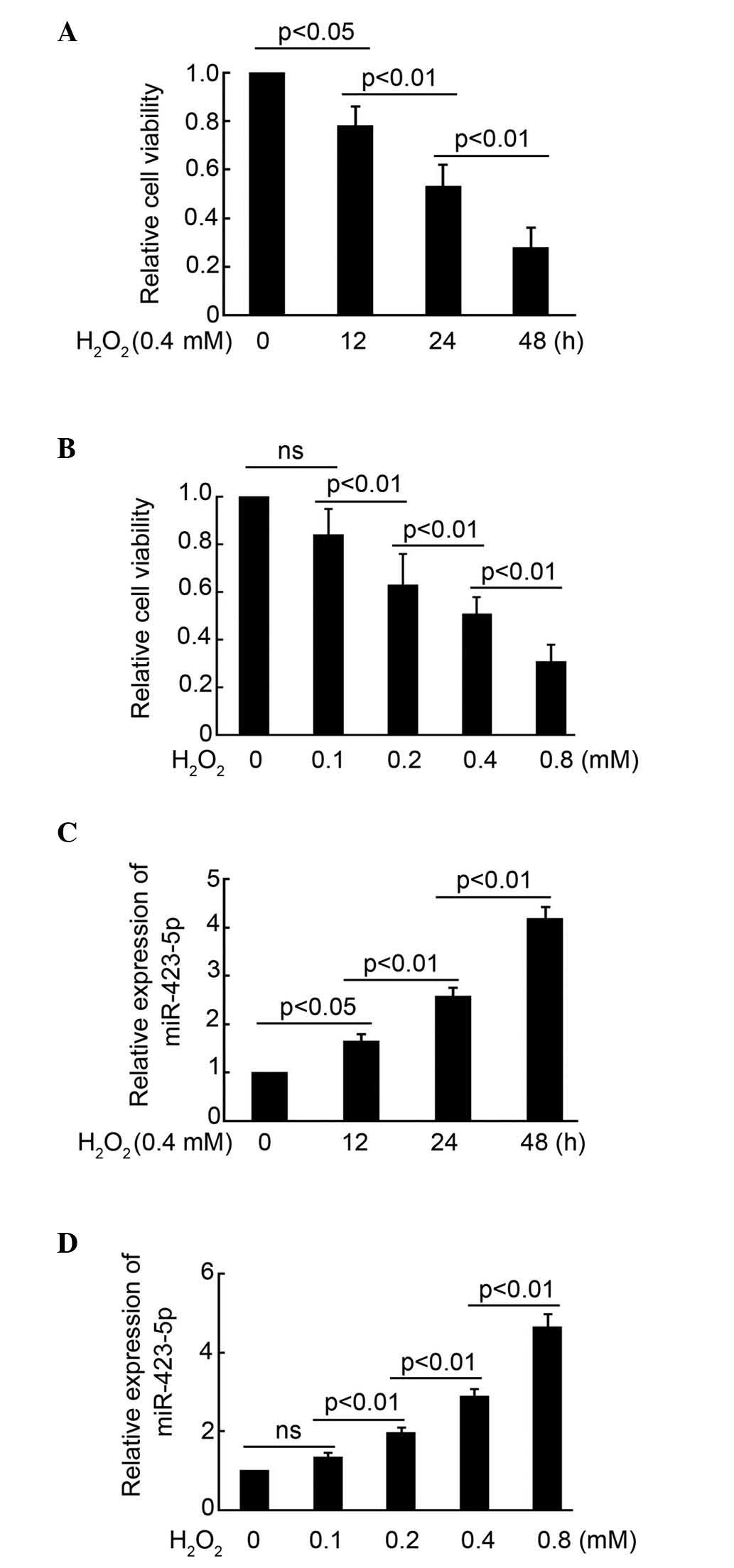 | Figure 1H2O2 decreases
cell viability and induces the expression of miR-423-5p in
cardiomyocytes. (A) Cardiomyocyte viability detection using a CCK8
assay following exposure to H2O2 (0.4 mM) for
0, 12, 24 and 48 h. (B) Cardiomyocyte viability detection using a
CCK8 assay following exposure to H2O2 (0,
0.1, 0.2, 0.4 and 0.8 mM) for 24 h. (C) RT-qPCR analysis of
m-miR-423-5p in cardiomyocytes following exposure to
H2O2 (0.4 mM) for 0, 12, 24 and 48 h. (D)
Real-time PCR of m-miR-423-5p in cardiomyocytes following exposure
to H2O2 (0, 0.1, 0.2, 0.4 and 0.8 mM) for 24
h. Data are presented as the mean ± standard error of the mean. The
same experiments were performed three times. miR, microRNA; CCK8,
Cell Counting Kit-8; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction; ns, no significant difference. |
H2O2 induces the
expression of miR-423-5p in cardiomyocytes
In the subsequent experiment, the expression of
miR-423-5p in cardiomyocytes following H2O2
treatment was examined using RT-q-PCR analysis. As shown in
Fig. 1C,
H2O2 treatment (0.4 mM) for 12 h
significantly induced the expression of miR-423-5p in the
cardiomyocytes. The expression of miR-423-5p in the cardiomyocytes
was higher following H2O2 treatment for 24
and 48 h, compared with that at 12 h. Treatment with 0.1 mM
H2O2 for 24 h had minimal effect on the
expression of miR-423-5p in the cardiomyocytes, compared with 0.2,
0.4 and 0.8 mM H2O2 treatment, which
significantly induced the expression of miR-423-5p in the
cardiomyocytes (Fig. 1D). Taken
together, these results indicated that H2O2
treatment significantly induced the expression of miR-423-5p in
cardiomyocytes, in a time- and concentration-dependent manner.
miR-423-5p mediates
H2O2-induced apoptosis in cardiomyocytes
To examine the effect of miR-423-5p on mediation of
H2O2-induced apoptosis in cardiomyocytes,
plasmids encoding miR-423-5p and miR-423-5p-mimic, which
neutralized the activity of miR-423-5p, were constructed. RT-qPCR
analysis demonstrated that transfection with the miR-423-5p plasmid
significantly induced the expression of miR-423-5p in the
cardiomyocytes (Fig. 2A). To
examine the effect of the miR-423-5p-mimic on the expression of
miR-423-5p, the cardiomyocytes were exposed to
H2O2 (0.4 mM) following miR-423-5p-mimic
transfection. After 48 h, the cells were collected for RT-qPCR
analysis. The results demonstrated the ability of the
miR-423-5p-mimic to inhibit the expression of miR-423-5p (Fig. 2B). Cardiomyocyte viability was
determined in the following treatment groups: Blank,
H2O2 (0.4 mM) alone,
H2O2 combined with miR-Ctrl transfection,
H2O2 combined with miR-423-5p-mimic
transfection, and miR-423-5p transfection without
H2O2 treatment. As shown in Fig. 2C, H2O2
treatment or transfection with miR-423-5p significantly decreased
cardiomyocyte viability (Fig. 2C).
No significant difference in cardiomyocyte viability was found
between the blank control group and the H2O2
combined with miR-423-5p-mimic transfection group (Fig. 2C). These results indicated that
cardiomyocyte viability was restored following inhibition of
miR-423-5p activity. A TUNEL assay was used to detect apoptotic
cardiomyocytes. It was found that H2O2
treatment or the overexpression of miR-423-5p significantly induced
cardiomyocyte apoptosis, compared with the miR-423-5p-mimic, which
markedly inhibited H2O2-induced apoptosis
(Fig. 2D and E). These results
demonstrated that miR-423-5p mediated
H2O2-induced apoptosis in the
cardiomyocytes.
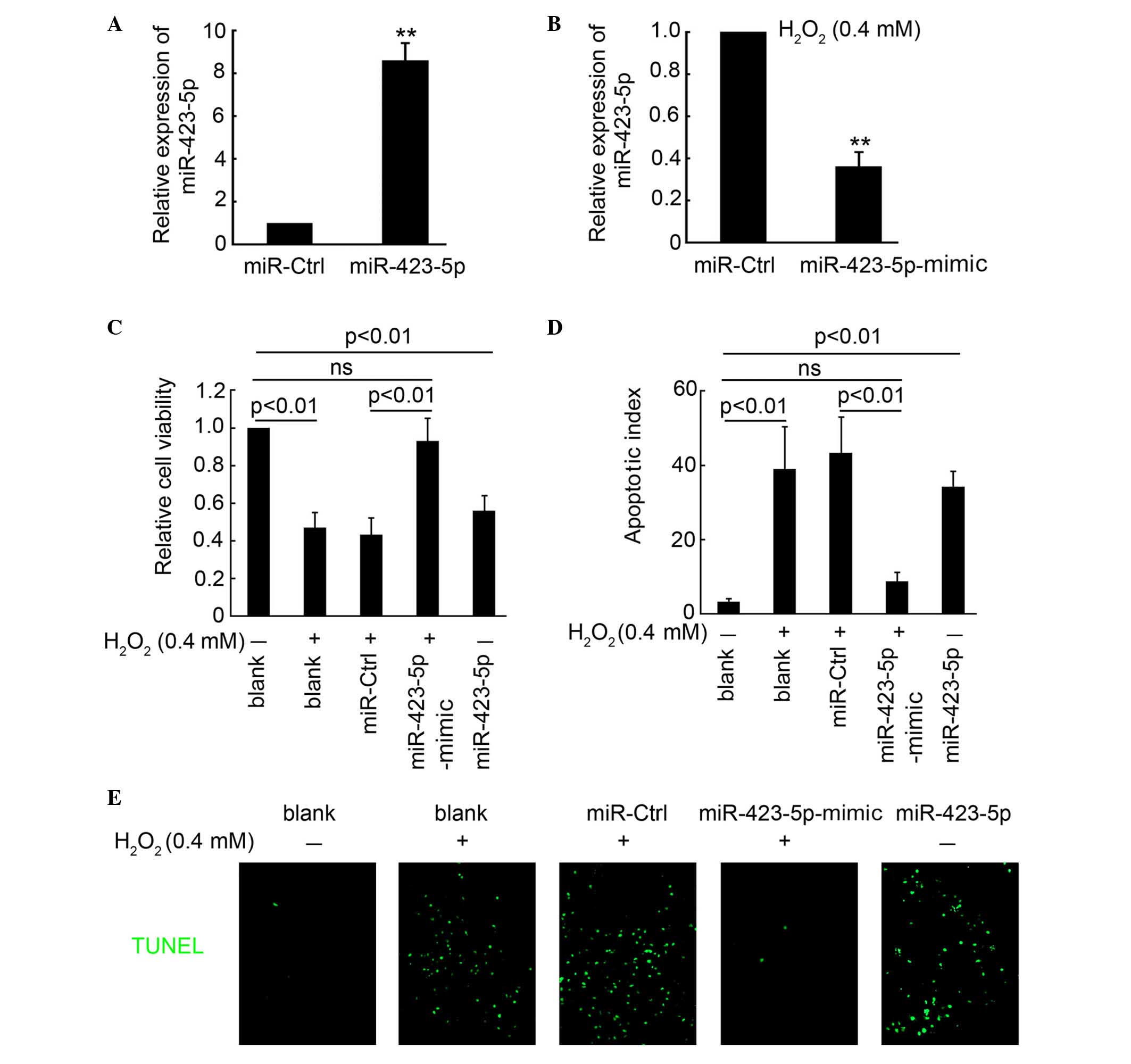 | Figure 2miR-423-5p mediates
H2O2-induced apoptosis in cardiomyocytes. (A)
RT-qPCR analysis of m-miR-423-5p in cardiomyocytes following
transfection with the m-miR-423-5p expression plasmid. (B) RT-qPCR
of m-miR-423-5p in cardiomyocytes following exposure to
H2O2 (0.4 mM) and transfection with miR-Ctrl
and m-miR-423-5p-mimic plasmids for 48 h. **P<0.01
compared with the miR-ctrl group. (C) Cardiomyocyte viability
detection using a CCK8 assay in the following treatment groups:
Blank, H2O2 (0.4 mM) treatment,
H2O2 combined with miR-Ctrl transfection,
H2O2 combined with miR-423-5p-mimic
transfection and miR-423-5p transfection without
H2O2 treatment. (D) Statistical analysis of
the apototic index of cardiomyocytes in the different treatment
groups (number of apoptotic cells / number of total cells), which
was determined by performing a TUNEL assay, according to the kit
instructions. (E) Positive cells are indicated in green; DAPI was
used to indicate all cells present. Data are presented as the mean
± standard error of the mean. The same experiments were performed
three times. Original magnification, ×200. miR, microRNA; Ctrl,
control; RT-qPCR, reverse transcription-quantitative polymerase
chain reaction; CCK8, Cell Counting Kit-8; TUNEL, terminal
deoxynucleotidyl transferase-deoxyuridine triphosphate nick-end
labeling; ns, no significant difference. |
miR-423-5p mediates
H2O2-regulated expression of OGT
In our previous study, it was demonstrated that OGT
is the direct target of miR-423-5p. Thus, in the present study, the
expression of OGT was examined. As shown in Fig. 3A and B, the expression of OGT was
significantly inhibited following exposure to
H2O2 for 12, 24 and 48 h. Further
investigation demonstrated that H2O2 markedly
decreased the expression of OGT in a concentration-dependent manner
(Fig. 3C and D). To examine the
role of miR-423-5p on mediation of the
H2O2-regulated expression of OGT, the
cardiomyocytes were treated in the following groups: Blank control,
H2O2 (0.4 mM) treatment,
H2O2 combined with miR-Ctrl transfection,
H2O2 combined with miR-423-5p-mimic
transfection, and miR-423-5p transfection without
H2O2 treatment. The results indicated that
treatment with H2O2 or transfection with
miR-423-5p significantly inhibited the expression of OGT. Of note,
the expression of OGT in cardiomyocytes was restored following
inhibition of miR-423-5p activity by the miR-423-5p-mimic. No
significant difference were observed between blank control group
and the H2O2 combined with miR-423-5p-mimic
transfection group. Collectively, miR-423-5p mediated the
H2O2-induced downregulation of OGT.
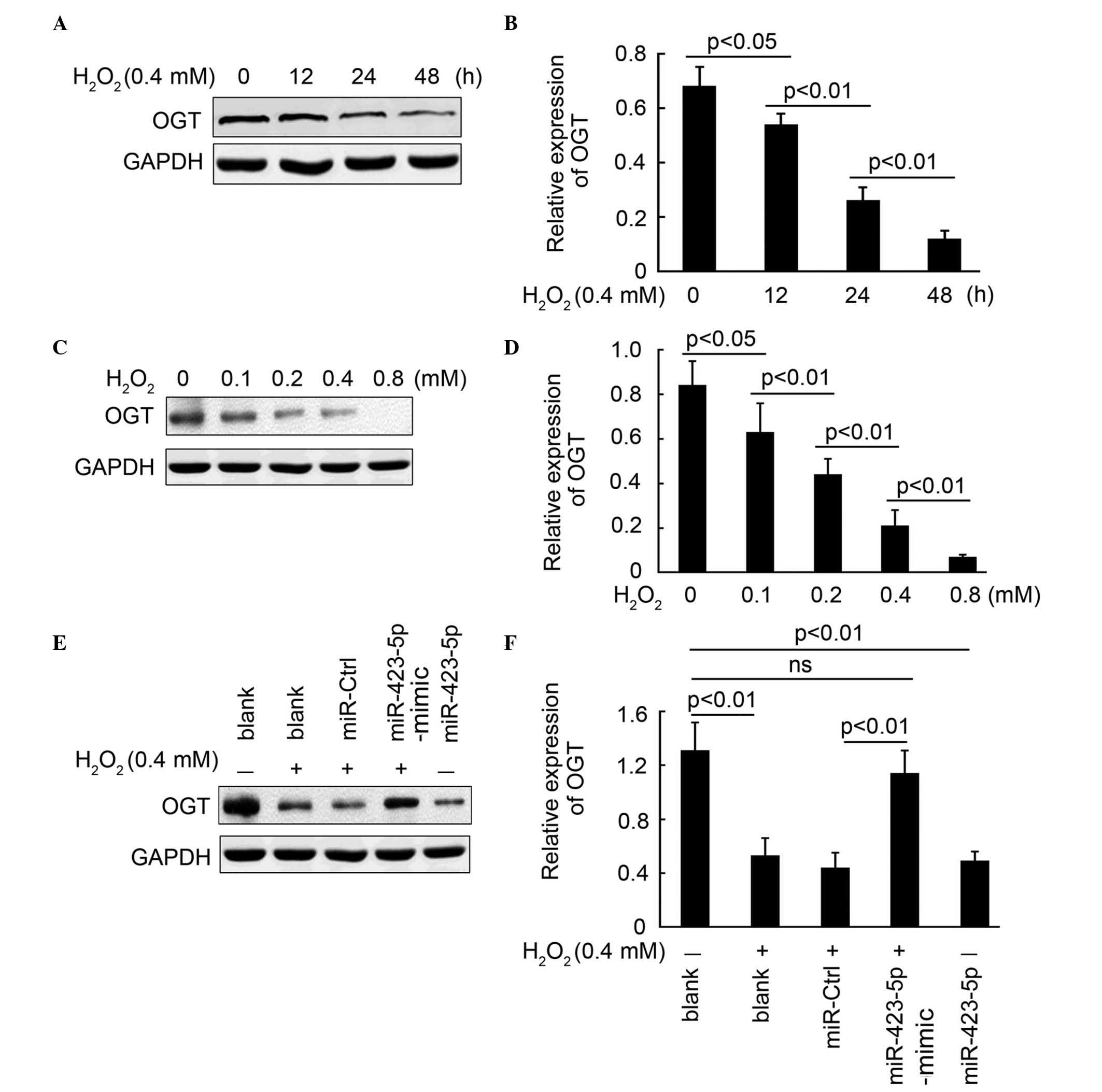 | Figure 3miR-423-5p mediates
H2O2-regulated expression of OGT. (A)
Expression of OGT in cardiomyocytes following exposure to
H2O2 (0.4 mM) for 0, 12, 24 and 48 h,
determined using western blot analysis and (B) quantified using
statistical analysis. GAPDH was used as the loading control. (C)
Expression of OGT in cardiomyocytes following exposure to
H2O2 (0, 0.1, 0.2, 0.4 and 0.8 mM) for 24 h,
determined using western blot analysis and (D) quantified using
statistical analysis. GAPDH was used as the loading control. (E)
Expression of OGT in cardiomyocytes in the following treatment
groups: Blank, H2O2 (0.4 mM) treatment alone,
H2O2 combined with miR-Ctrl transfection,
H2O2 combined with miR-423-5p-mimic
transfection and miR-423-5p transfection without
H2O2 treatment. Expression was determined
using western blot analysis and (F) quantified using statistical
analysis. GAPDH was used as the loading control. Data are presented
as the mean ± standard error of the mean. The same experiments were
performed three times. miR, microRNA; Ctrl, control; OGT, O-GlcNAc
transferase; ns, no significant difference. |
miR-423-5p mediates
H2O2-regulated expression of OGT downstream
targets
The activities of AMPK and 26S proteasome activity,
as the downstream targets of OGT, were also determined in the
present study. The phosphorylation of AMPK was inhibited following
H2O2 treatment or the overexpression
miR-423-5p through a reduction in OGT (Fig. 4A). Whereas, no significant change
in the expression of p-AMPK was observed between the blank control
group and the H2O2 combined with
miR-423-5p-mimic transfection group. According to the change in the
expression of p-AMPK, 26S proteasome activity was also increased
following H2O2 treatment or the
overexpression miR-423-5p (Fig.
4C). Transfection with the miR-423-5p-mimic also restored 26S
proteasome activity to a normal level (Fig. 4C). These results demonstrated that
miR-423-5p mediated the H2O2-regulated
expression of p-AMPK and activity of 26S proteasome, which are
downstream targets of OGT.
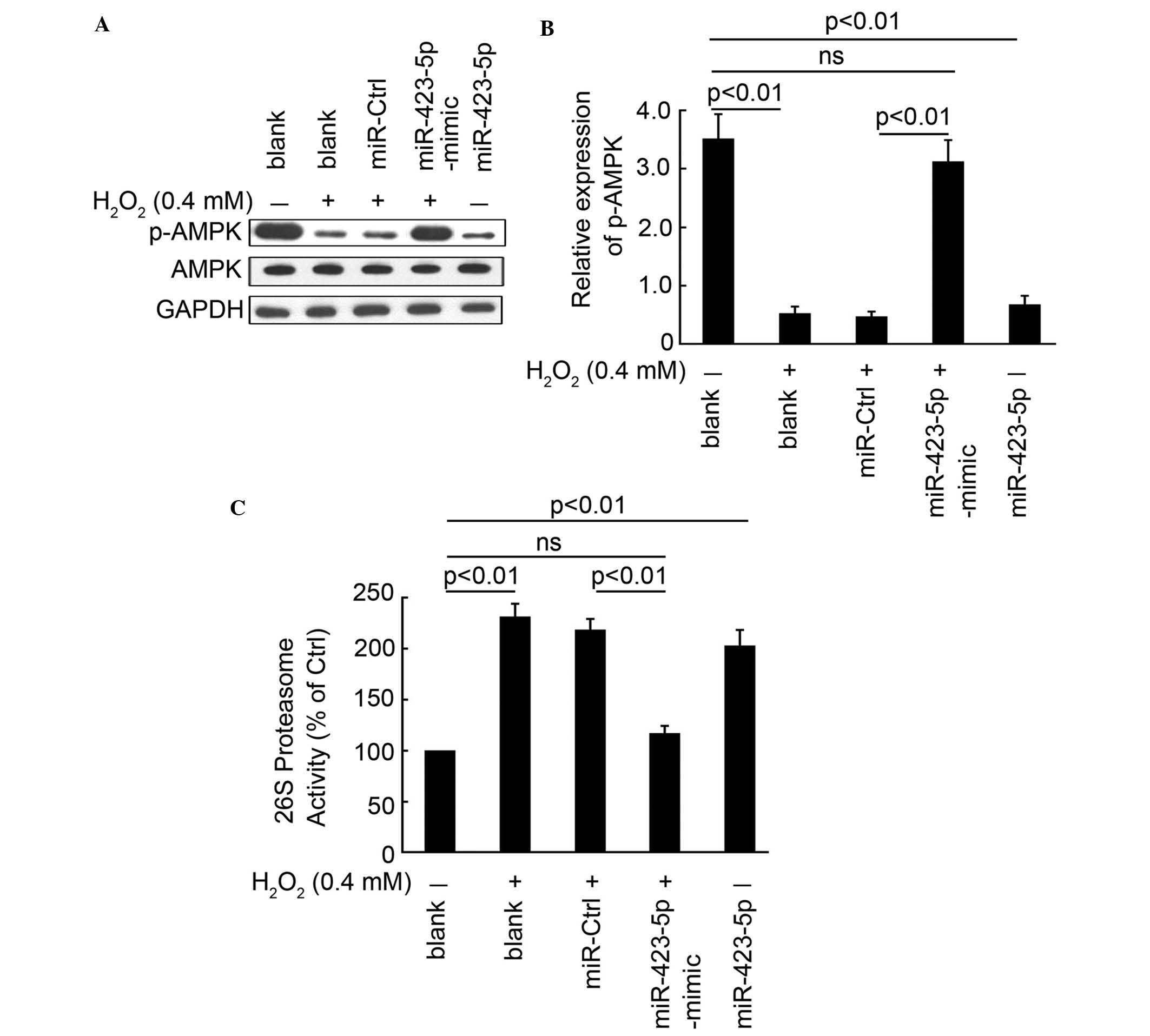 | Figure 4miR-423-5p mediates
H2O2-regulated expression of OGT downstream
targets. (A) Expression of AMPK and p-AMPK in cardiomyocytes in the
following treatment groups: Blank, H2O2 (0.4
mM) treatment alone, H2O2 combined with
miR-Ctrl transfection, H2O2 combined with
miR-423-5p-mimic transfection and miR-423-5p transfection without
H2O2 treatment. Expression was determined
using western blot analysis and was (B) quantified using
statistical analysis. GAPDH was used as the loading control. (C)
26S proteasome activity in cardiomyocytes in the following
treatment groups: Blank, H2O2 (0.4 mM)
treatment alone, H2O2 combined with miR-Ctrl
transfection, H2O2 combined with
miR-423-5p-mimic transfection and miR-423-5p transfection without
H2O2 treatment, determined according to the
kit instructions. Data are presented as the mean ± standard error
of the mean. The same experiments were performed in three times.
miR, microRNA; Ctrl, control; OGT, O-GlcNAc transferase; AMPK,
adenosine monophosphate-activated protein kinase; p-AMPK,
phosphorylated AMPK; ns, no significant difference. |
Discussion
In the present study, it was found that the
expression of miR-423-5p in cardiomyocytes was induced by
H2O2 in a time- and concentration-dependent
manner. The results indicated that the silencing of miR-423-5p by
transfection with miR-423-5p-mimic significantly inhibited
H2O2-induced apoptosis in cardiomyocytes.
Furthermore, the direct target of miR-423-5p, OGT, and downstream
targets, p-AMPK and 26S proteasome, were also demonstrated to be
involved in H2O2-induced apoptosis in
cardiomyocytes. Collectively, the results of the present study
demonstrated that miR-423-5p mediated
H2O2-induced apoptosis in the cardiomyocytes.
Silencing of miR-423-5p significantly protected the cardiomyocytes
from H2O2-induced apoptosis, and miR-423-5p
may provide a novel therapeutic target for apoptosis-associated
heart diseases.
As is already known, apoptosis is involved in the
pathological process of several cardiovascular diseases, including
heart failure, myocardial infarction, cardiac ischemia and
reperfusion injury, and atrial fibrillation (17–20).
Excess ROS can cause a variety of cellular damage, including
mitochondrial dysfunction and DNA damage, and ultimately leads to
apoptosis, with apoptosis of cardiomyocytes being critical in
tissue damage and eventually heart failure (6–8). The
administering of antioxidants, including vitamin C, have been
effective in preventing oxidative stress-mediated cardiovascular
dysfunction (21). Thus,
inhibiting the apoptosis of cardiomyocytes appears the most
effective clue to protecting the heart from injury. To examine the
correction between miR-423-5p and
H2O2-induced apoptosis in cardiomyocytes, the
expression of miR-423-5p was examined following exposure of the
cells to H2O2. It was found that miR-423-5p
was a sensor of H2O2-induced apoptosis.
In previous years, miR-423-5p has been widely
investigated as a potential biomarker and therapeutic target for
various heart diseases. miR-423-5p has been reported in array
studies to be upregulated in the failing myocardium in humans
(22). Tijsen et al
(23) showed that circulating
levels of miR-423-5p are increased in subjects with clinical heart
failure, defined by the Framingham criteria and elevated levels of
proBNP, and that the levels of miR-423-5p are associated with
proBNP and ejection fraction in this patient group. Our previous
study (15) demonstrated that
miR-423-5p corrects with CHF through the direct targeting of OGT
and inducing apoptosis in cardiomyocytes. Clinical experiments
indicated that miR-423-5p is associated with CHF and the expression
levels of proBNP. Furthermore, the expression of miR-423-5p
significantly regulated the expression levels of OGT and its
associated downstream targets, and induced apoptosis of the
cardiomyocytes. However, the role of miR-423-5p during ROS-induced
apoptosis remains to be fully elucidated. In the present study, it
was demonstrated that the expression of miR-423-5p in
cardiomyocytes was induced by H2O2 in a time-
and concentration-dependent manner. The silencing of miR-423-5p by
transfection with the miR-423-5p-mimic significantly inhibited
H2O2-induced apoptosis in cardiomyocytes.
These results demonstrated that miR-423-5p mediated
H2O2-induced apoptosis in cardiomyocytes, and
that silencing of miR-423-5p significantly protected the
cardiomyocytes from H2O2-induced
apoptosis.
In conclusion, the results of the present study
demonstrated that the expression of miR-423-5p was induced by
H2O2 in a time- and concentration-dependent
manner, and silencing of miR-423-5p significantly protected the
cardiomyocytes from H2O2-induced apoptosis.
These results indicated that miR-423-5p may be an effective
therapeutic target for the treatment of apoptosis-associated heart
diseases.
References
|
1
|
Khurana S, Hollingsworth A, Piche M,
Venkataraman K, Kumar A, Ross GM and Tai TC: Antiapoptotic actions
of methyl gallate on neonatal rat cardiac myocytes exposed to
H2O2. Oxid Med Cell Longev. 2014:6575122014.
View Article : Google Scholar
|
|
2
|
Dhalla AK, Hill MF and Singal PK: Role of
oxidative stress in transition of hypertrophy to heart failure. J
Am Coll Cardiol. 28:506–514. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hill MF and Singal PK: Right and left
myocardial antioxidant responses during heart failure subsequent to
myocardial infarction. Circulation. 96:2414–2420. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dhalla NS, Temsah RM and Netticadan T:
Role of oxidative stress in cardiovascular diseases. J Hypertens.
18:655–673. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maulik SK and Kumar S: Oxidative stress
and cardiac hypertrophy: A review. Toxicol Mech Methods.
22:359–366. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Peng YW, Buller CL and Charpie JR: Impact
of N-acetylcysteine on neonatal cardiomyocyte ischemia-reperfusion
injury. Pediatr Res. 70:61–66. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rodrigo R: Prevention of postoperative
atrial fibrillation: Novel and safe strategy based on the
modulation of the antioxidant system. Front Physiol. 3:932012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsutsui H, Kinugawa S and Matsushima S:
Oxidative stress and heart failure. Am J Physiol Heart Circ
Physiol. 301:H2181–H2190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cordes KR and Srivastava D: MicroRNA
regulation of cardiovascular development. Circ Res. 104:724–732.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu N and Olson EN: MicroRNA regulatory
networks in cardiovascular development. Dev Cell. 18:510–525. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ivey KN, Muth A, Arnold J, King FW, Yeh
RF, Fish JE, Hsiao EC, Schwartz RJ, Conklin BR, Bernstein HS and
Srivastava D: MicroRNA regulation of cell lineages in mouse and
human embryonic stem cells. Cell Stem Cell. 2:219–229. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao Y, Ransom JF, Li A, Vedantham V, von
Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ and
Srivastava D: Dysregulation of cardiogenesis, cardiac conduction
and cell cycle in mice lacking miRNA-1-2. Cell. 129:303–317. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
van Rooij E, Marshall WS and Olson EN:
Toward microRNA-based therapeutics for heart disease: The sense in
antisense. Circ Res. 103:919–928. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Luo P, He T, Jiang R and Li G:
MicroRNA-423-5p targets O-GlcNAc transferase to induce apoptosis in
cardiomyocytes. Mol Med Rep. 12:1163–1168. 2015.PubMed/NCBI
|
|
16
|
Fekete MR, McBride WH and Pajonk F:
Anthracyclines, proteasome activity and multi-drug-resistance. BMC
Cancer. 5:1142005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Al-Gubory KH, Fowler PA and Garrel C: The
roles of cellular reactive oxygen species, oxidative stress and
antioxidants in pregnancy outcomes. Int J Biochem Cell Biol.
42:1634–1650. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hauton D: Hypoxia in early pregnancy
induces cardiac dysfunction in adult offspring of Rattus
norvegicus, a non-hypoxia-adapted species. Comp Biochem Physiol A
Mol Integr Physiol. 163:278–285. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nanduri J, Makarenko V, Reddy VD, Yuan G,
Pawar A, Wang N, Khan SA, Zhang X, Kinsman B, Peng YJ, et al:
Epigenetic regulation of hypoxic sensing disrupts cardiorespiratory
homeostasis. Proc Natl Acad Sci USA. 109:2515–2520. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Patterson AJ and Zhang L: Hypoxia and
fetal heart development. Curr Mol Med. 10:653–666. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kane AD, Herrera EA, Camm EJ and Giussani
DA: Vitamin C prevents intrauterine programming of in vivo
cardiovascular dysfunction in the rat. Circ J. 77:2604–2611. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thum T, Galuppo P, Wolf C, Fiedler J,
Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J,
Haverich A, et al: MicroRNAs in the human heart: A clue to fetal
gene reprogramming in heart failure. Circulation. 116:258–267.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tijsen AJ, Creemers EE, Moerland PD, de
Windt LJ, van der Wal AC, Kok WE and Pinto YM: MiR423-5p as a
circulating biomarker for heart failure. Circ Res. 106:1035–1039.
2010. View Article : Google Scholar : PubMed/NCBI
|














