Introduction
Esophageal cancer is the eighth leading cause of
cancer globally with 456,000 new cases diagnosed in 2012 (1). It resulted in 345,000 mortalities in
1990 and ~400,000 in 2012 (1,2).
Rates of diagnosis vary widely among countries, with ~50% of all
cases occurring in China. It is ~3 times more common in men than in
women (1). The two predominant
sub-types of esophageal cancer are squamous cell carcinoma and
adenocarcinoma. Squamous cell carcinoma arises from the epithelial
cells that line the esophagus (3),
while adenocarcinoma arises from glandular cells present in the
lower third of the esophagus, often following a prior
transformation into an intestinal cell type as part of a condition
termed Barrett's esophagus) (1,4).
Surgical resection and adjuvant therapy are used to treat
esophageal cancer, however, patients often have a poor prognosis
with a 5-year survival rate of ~13–18% (5,6). The
high mortality rate for esophageal cancer is predominantly a result
of the advanced stage at diagnosis in the majority of cases, by the
time of diagnosis, 80% of esophageal cancers are no longer
localized to the esophagus. Thus, the development of effective
therapeutic strategies is required. Dietary modulation of signaling
pathways is a promising strategy for cancer prevention and
treatment. Vitamin Es, including tocopherols and tocotrienols,
exhibit natural antioxidant activity. Vitamin E succinate (VES) or
α-tocopheryl succinate, is obtained by esterification of
α-tocopherol and has been reported to inhibit growth and induce
apoptosis in a variety of types of cancer (7–9),
including prostate, breast, gastric, and colorectal cancer, in
addition to melanomas (10–13).
Previous studies have demonstrated that activation
of the phosphoinositide-3-kinase (PI3K), serine-threonine kinase
AKT (AKT) and mammalian target of rapamycin (mTOR) may be important
in cell proliferation and apoptosis (14). They are constitutively activated or
overexpressed in numerous types of cancer, and result in cancer
progression via stimulating cellular proliferation and suppressing
cell death signaling pathways (15). PI3K promotes tumor cell survival by
triggering the activation of downstream mediators of AKT (16). AKT exerts its effects via a diverse
array of effectors, which regulate key cellular processes,
including transcription, translation, apoptosis and cell cycle
progression (17). AKT directly
controls apoptosis by inducing phosphorylation and inactivation of
pro-apoptotic proteins, including Bcl-2-associated death receptor
(Bad) and caspase-9 (18–20). In addition, a major downstream
substrate of AKT is the serine/threonine kinase mTOR. mTOR is
directly activated by AKT via phosphorylated at Ser2448, and can be
indirectly activated by phosphorylation and inactivation of
tuberous sclerosis complex 2, also termed tuberin, by AKT. The
raptor-mTOR complex substrates, ribosomal protein S6 kinase β1
(p70S6K) and the eIF4E-binding protein 1 (4E-BP1), modulate
transcription and translation to selectively regulate downstream
proteins that control cell survival and death (21).
It has been reported that VES exerts its apoptotic
effect in cancer cells via multiple apoptotic signaling pathways.
VES regulates transforming growth factor-β (TGF-β) and Fas (CD95)
signaling pathways, thus, stimulating c-Jun N-terminal
kinase-induced apoptosis (22). In
addition, TGF-β-independent apoptotic mechanisms have also been
demonstrated (23). Reactive
oxygen species generated by mitocans mediate the formation of
mitochondrial outer membrane channels by activating
Bcl-2-associated X protein channels allowing translocation of
cytochrome c into the cytoplasm and activation of caspase-3
and -9, which is a key mechanism in VES-induced apoptosis (8). The present study demonstrated that
VES induced apoptosis in esophageal cancer cells via targeting the
PI3K/AKT signaling pathways and modulating the downstream effectors
Bad and caspase-9, in addition to mTOR. The results suggested that
VES in combination with AKT or mTOR inhibitors may be an effective
therapeutic strategy for esophageal cancer.
Materials and methods
Chemicals
VES was purchased from Sigma-Aldrich (St. Louis, MO,
USA). A PI3K inhibitor, LY294002, and AKT inhibitor, triciribine,
were purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA). mTOR inhibitor, rapamycin, was purchased from EMD Millipore
(Billerica, MA, USA).
Cell culture
The EC109 human esophageal squamous cell carcinoma
cell line was obtained from the Cell Bank of the Chinese Academy of
Science (Shanghai, China). EC109 cells were cultured in Dulbecco's
modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) containing 10% fetal bovine serum (Lonza Group,
Ltd., Basel, Switzerland) and 1% penicillin/streptomycin (complete
media) at 37°C in a humidified 5% CO2 incubator.
Western blotting
Cells were washed with phosphate-buffered saline and
lysed in lysis buffer (50 mM HEPES, pH 8.0; 1% Triton X-100; 1.5 mM
EDTA; 150 mM NaCl; 1 mM Na3VO4; 50 mM NaF; 1
mM MgCl2; 20 mM β-glycerophosphate; 10% glycerol; 1
µM pepstatin A; 1 mM phenylmethylsulphonyl fluoride; and 10
µg/ml aprotonin). Cell lysate was centrifuged at 10,000 × g
for 10 min and the supernatant was collected. Protein samples were
quantified using a bicinchoninic acid protein assay kit (Beyotime
Institute of Biotechnology, Haimen, China). Total protein samples
(20–50 µg) were separated by 12% SDS-PAGE and transferred to
nitrocellulose membranes (GE Healthcare Life Sciences, Chalfont,
UK). The membranes were blocked with 5% bovine serum albumin (Sigma
Aldrich) in Tris-buffered saline Tween 20 (TBST) for 1 h, and
incubated with specific primary antibodies overnight at 4°C.
Subsequently, the membranes were washed 3 times with TBST, followed
by incubation with horseradish peroxidase (HRP)-conjugated
secondary antibodies for 2 h at room temperature. Antibodies
against the following proteins were used: Rabbit polyclonal
anti-AKT (1:1,000; cat. no. 9272), Bad (1:1,000; cat. no. 9292),
casapse 9 (1:1,000; cat. no. 9502), phosphorylated (p)-mTOR
(Ser2448; 1:1,000; cat. no. 2971); rabbit monoclonal anti-p-AKT
(Ser473, 1:2,000, cat. no. 4060; and Thr308, 1:1,000, cat. no.
13038), p-Bad (Ser136; 1:1,000; cat. no. 4366), mTOR (1:1,000; cat.
no. 2983), p70S6K (1:1,000; cat. no. 2708) 4E-BP1 (1:1,000; cat.
no. 9644), p-4E-BP1 (Thr37/46; 1:1,000; cat. no. 2855) and GAPDH
(1:1,000; cat. no. 2118); mouse monoclonal anti-p-p70S6K (Thr389;
1:1,000, cat. no. 9206) (all obtained from Cell Signaling
Technology, Inc.); and goat polyclonal anti-p-caspase 9 (Ser196;
1:500; cat. no. sc11755; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA). HRP-conjugated goat anti-rabbit secondary antibody
(1:2,000; cat. no. 7074; Cell Signaling Technology, Inc.), horse
anti-mouse antibody (1:2,000; cat. no. 7076; Cell Signaling
Technology, Inc.) and donkey anti-goat antibody (1:5,000; cat. no.
sc2020; Santa Cruz Biotechnology, Inc.) were used. Enhanced
chemiluminescence-detecting reagent (GE Healthcare Life Sciences)
was used for development. The protein blots were quantified by
densitometry using QuantityOne software (version 4.5.0; Bio-Rad
Labroatories, Inc., Hercules, CA, USA) and the amounts were
expressed relative to the corresponding reference protein.
Cell survival assay
Cell survival was evaluated using the MTT assay.
Cells were seeded at a density of 1×104/well in a
96-well flat bottom plate. Cells were allowed to grow in a 37°C, 5%
CO2 incubator for 48 h, and then 20 µl of 5 mg/ml
MTT was added to each well for a further 4 h. Cells were washed by
phosphate-buffered saline and lysed by addition of 200 µl
dimethyl sulfoxide. Absorbance was detected at a wavelength of 490
nm using an enzyme-linked immunosorbent assay reader.
Quantification of apoptosis
Apoptosis was quantified using the Annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) assay
(Sigma-Aldrich) following the manufacturer's protocols. The Annexin
V-FITC/PI assay detects the amount of phosphatidylserine on the
outer surface of the plasma membrane, a biochemical alteration
observed in membranes of apoptotic cells, and the amount of PI, a
dye that easily enters dead cells or cells in the late stages of
apoptosis and binds DNA. Fluorescence was detected using a
FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ,
USA) by fluorescence-activated cell sorting (FACS) analysis, and
data were analyzed using CellQuest software (version 7.5.3; BD
Biosciences). Cells with phosphatidylserine on their surface were
considered to be apoptotic.
Statistical analysis
Data are presented as the mean ± standard error of
the mean and all the experiments were replicated at least 3 times.
SPSS software (version 11.0; SPSS, Inc., Chicago, IL, USA) was used
to perform statistical analysis. A Student's t-test was used to
compare treated and untreated cells. P<0.05 was considered to
indicate a statistically significant difference.
Results
VES inhibits the proliferation of
esophageal cancer cells
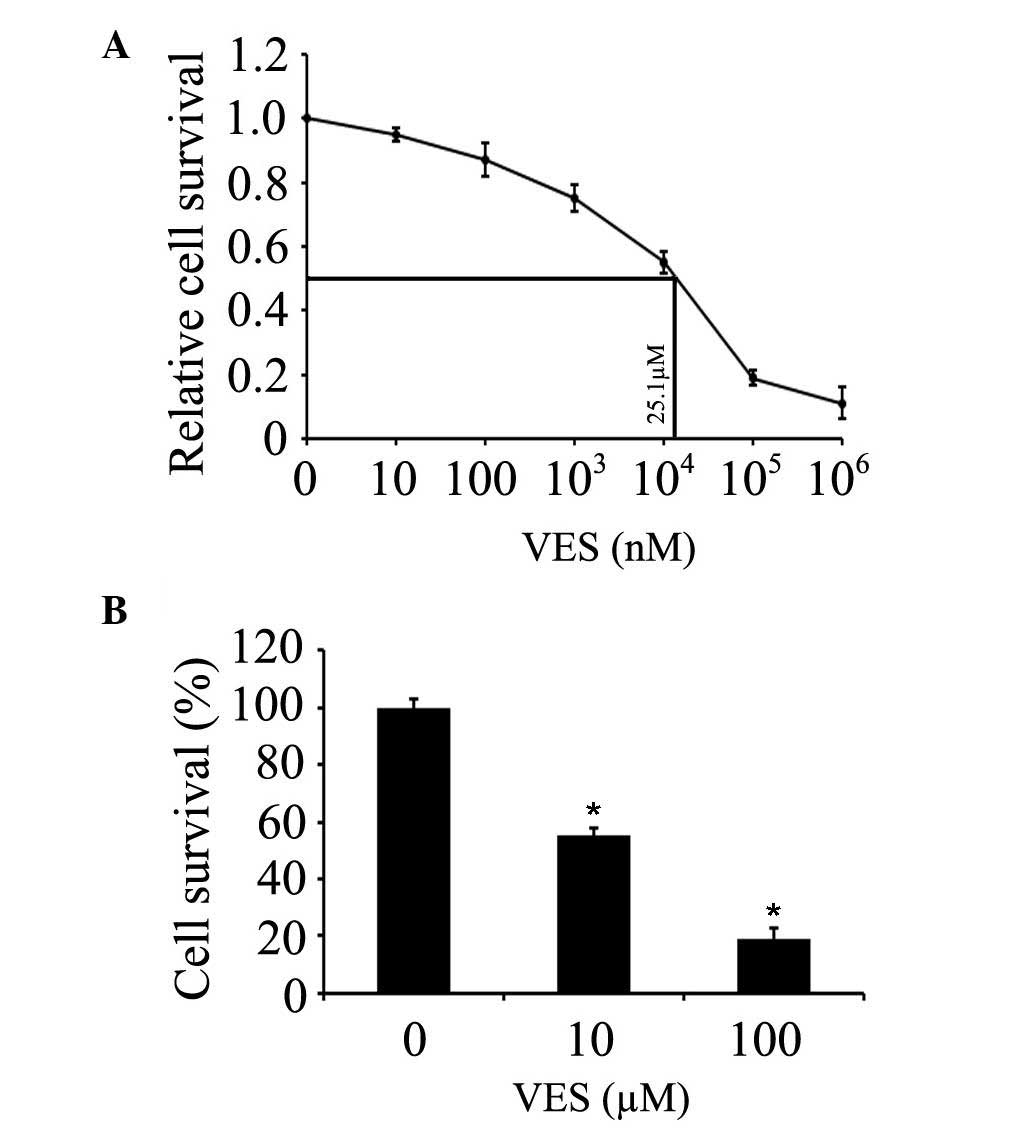
The effects of VES on the EC109 human esophageal
cancer cell line was detected in the present study. The
IC50 value of VES was determined by treating EC109 cells
with increasing concentrations of VES for 24 h. IC50
value was calculated to be 25.1 µM for EC109 cells at 24 h
(Fig. 1A). EC109 cells were
treated with 10–100 µM VES for 24 h, and the cell viability
was evaluated by MTT assay. The results demonstrated that the
growth was decreased by ~45 and ~81% following treatment with 10
and 100 µM VES in EC109 cells (Fig. 1B). The current study also
investigated whether the proliferation inhibition resulted from the
induction of apoptosis in the cells.
VES induces the apoptosis of esophageal
cancer cells
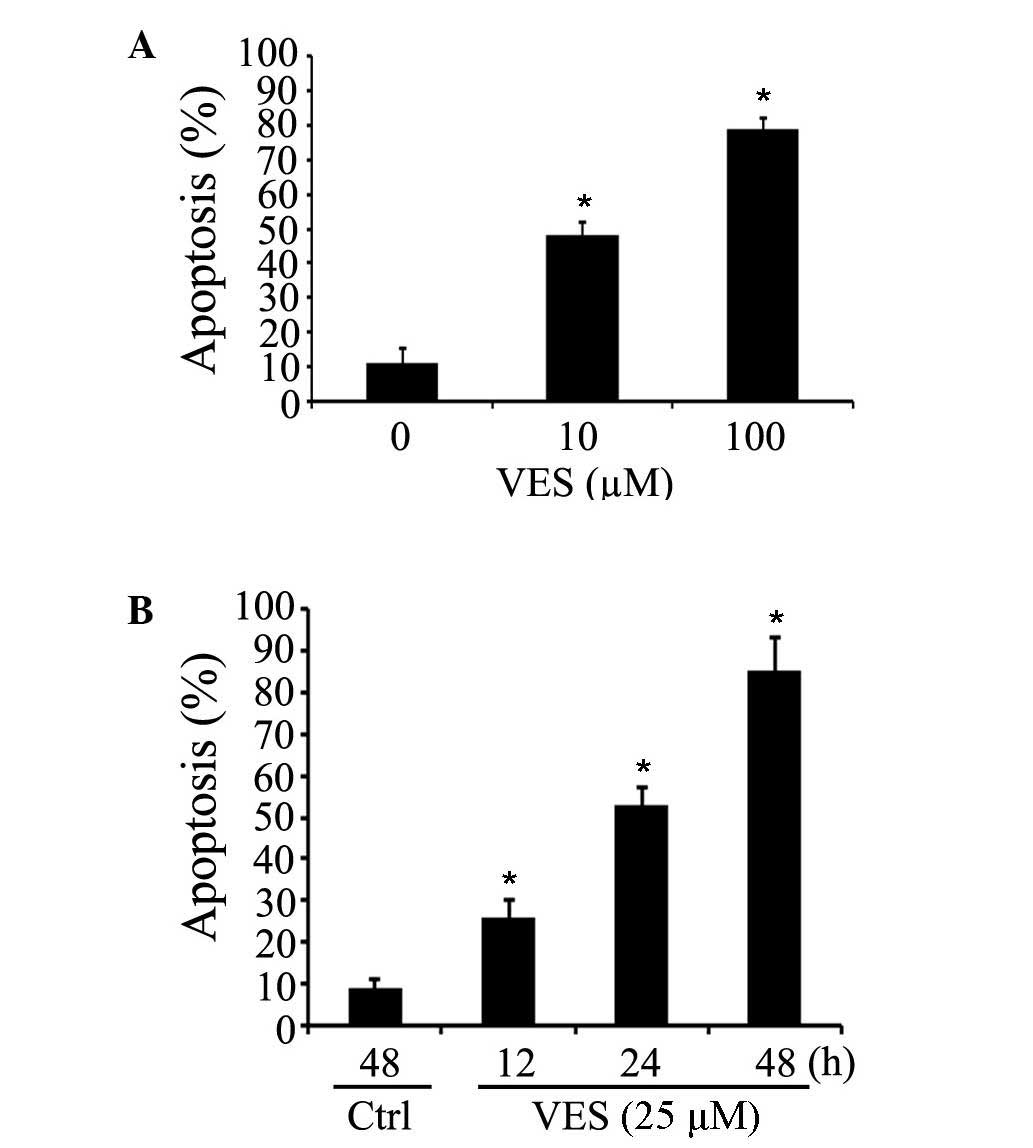
To investigate whether VES promotes apoptosis in
esophageal cancer cells, EC109 cells were treated with VES at
various concentrations (0, 10 or 100 µM; Fig. 2A) or for different periods of time
(12, 24 or 48 h; Fig. 2B).
Apoptosis was quantified by FACS analyses. Apoptosis in the EC109
cells was induced by 10 or 100 µM VES treatment for 24 h in
a dose-dependent manner (Fig. 2A).
Apoptosis of EC109 cells was induced by 25 µM VES for 12, 24
and 48 h in a time-dependent manner (Fig. 2B). These results suggest that VES
is a potent inducer of apoptosis in EC109 esophageal cancer
cells.
VES suppresses the active forms of AKT
and mTOR in esophageal cancer cells
EC109 cells that are not treated with VES express
high levels of p-AKT, and downstream substrates p-Bad and
p-caspase-9 (Fig. 3). EC109 cells
were treated with 25 µM VES for 12, 24 and 48 h, which
reduced levels of p-AKT (Ser473/Thr308), p-Bad (Ser136), and
p-caspase-9 (Ser196) in a time-dependent manner (Fig. 3A and C). It also downregulated
p-mTOR (Ser2448) and its substrates p-p70S6K (Thr389) and p-4E-BP1
(Thr37/46; Fig. 3B and C). These
data demonstrated that VES reduced the levels of active AKT, mTOR,
and their downstream effectors, promoting the activation of Bad and
caspase-9, which mediate cell apoptosis.
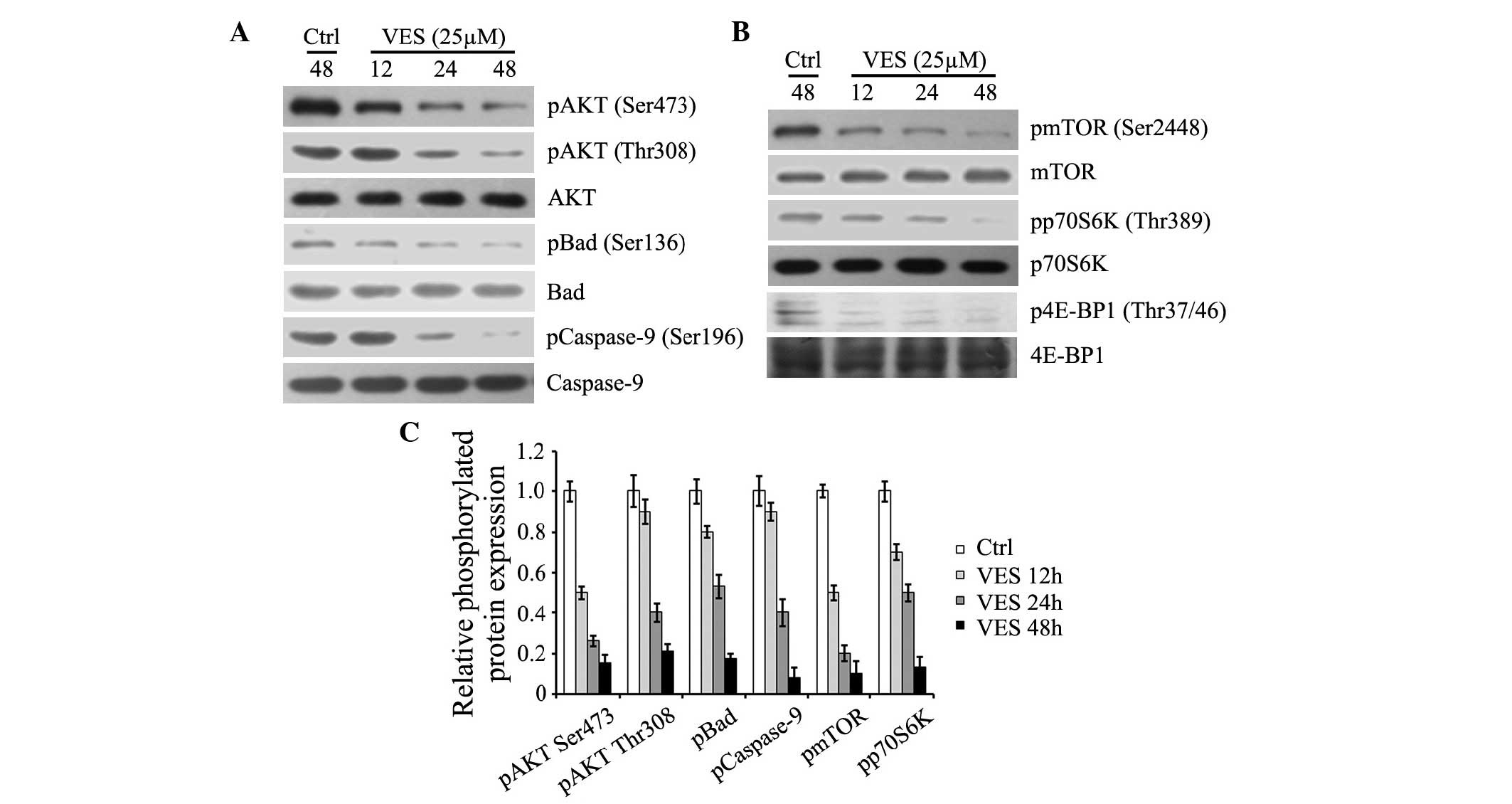 | Figure 3VES inhibits AKT and mTOR, in addition
to their downstream targets. EC-109 cells were treated with 25
µM VES for 12, 24 and 48 h. (A) Protein expression levels of
p-AKT (Ser473 and Thr308), p-Bad (Ser136), p-caspase-9 (Ser196),
and expression levels of total AKT, Bad and caspase-9 were detected
by western blotting. (B) Protein expression levels of p-mTOR
(Ser2448), p-p70S6K (Thr389), and p-4E-BP1 (Thr37/46), and
expression levels of total mTOR, p70S6 K and 4E-BP1 were determined
by western blotting. (C) Protein expression levels were quantified
and presented as the mean ± standard error of the mean. VES,
vitmain E succinate; AKT, serine-threonine kinase AKT; mTOR,
mammalian target of rapamycin; p, phosphorylated; Bad,
Bcl-2-associated death promoter; p70S6K, ribosomal protein S6
kinase β1; 4E-BP1, eIF4E-binding protein 1; Ctrl, control. |
PI3K inhibitor, LY294002 reduces
phosphorylation of AKT and mTOR in EC109 cells
To investigate the underlying mechanisms of
regulation of the levels of p-AKT and p-mTOR, EC109 cells were
treated with 2 µM PI3K inhibitor LY294002 for 12 h. LY294002
decreased the p-AKT and its substrates p-Bad (Ser136) and
p-caspase-9 (Ser196; Fig. 4A), in
addition to p-mTOR (Ser2448) and its substrate p-p70S6 K (Thr389)
(Fig. 4B). This indicates that
PI3K is a key contributor to regulations of p-AKT and p-mTOR.
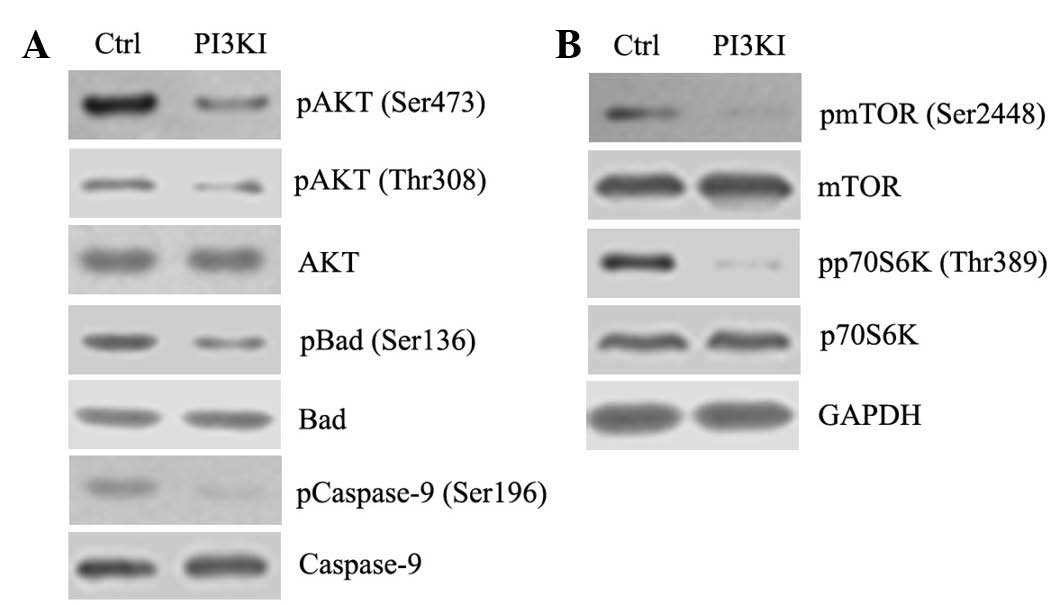 | Figure 4AKT and mTOR are downstream targets of
PI3K. EC109 cells were treated with 2 µM PI3K inhibitor
(PI3KI) LY294002 for 12 h. (A) Protein expression levels of p-AKT
(Ser473), p-Bad (Ser136) and p-caspase-9 (Ser196), and expression
levels of total AKT, Bad and caspase-9 were determined by western
blotting. (B) Protein expression levels of p-mTOR (Ser2448) and
p-p70S6K (Thr389), and total levels of mTOR and p70S6K were also
determined by western blotting. PI3K, phosphoinositide-3-kinase;
AKT, serine-threonine kinase AKT; mTOR, mammalian target of
rapamycin; p, phosphorylated; Bad, Bcl-2-associated death promoter;
p70S6K, ribosomal protein S6 kinase β1; Ctrl, control. |
VES cooperated with inhibitors of PI3K,
AKT and mTOR to induce apoptosis
To investigate the effects of inhibition of members
of PI3K signaling pathway on VES-induced apoptosis, EC109 cells
were treated with 15 µM VES plus 2 µM LY294002, 10
µM AKT inhibitor triciribine, and 50 nM mTOR inhibitor
rapamycin for 24 h. Single treatments with VES or inhibitors
induced apoptosis, whereas VES combined with each inhibitor
individually markedly increased the induction of apoptosis compared
with single treatments (Fig. 5A).
VES and the PI3K inhibitor decreased the expression levels of p-AKT
and p-mTOR, and the combination of VES + PI3K inhibitor
synergistically decreased expression levels of p-AKT and p-mTOR in
comparison with individual treatments or the control (Fig. 5B). AKT and mTOR inhibitors
increased VES downregulation of p-AKT and p-mTOR, respectively
(Fig. 5C). These data indicate
that VES inhibited the activation of AKT and mTOR, and may function
with the inhibitors of AKT and mTOR to promote esophageal cancer
cell apoptosis.
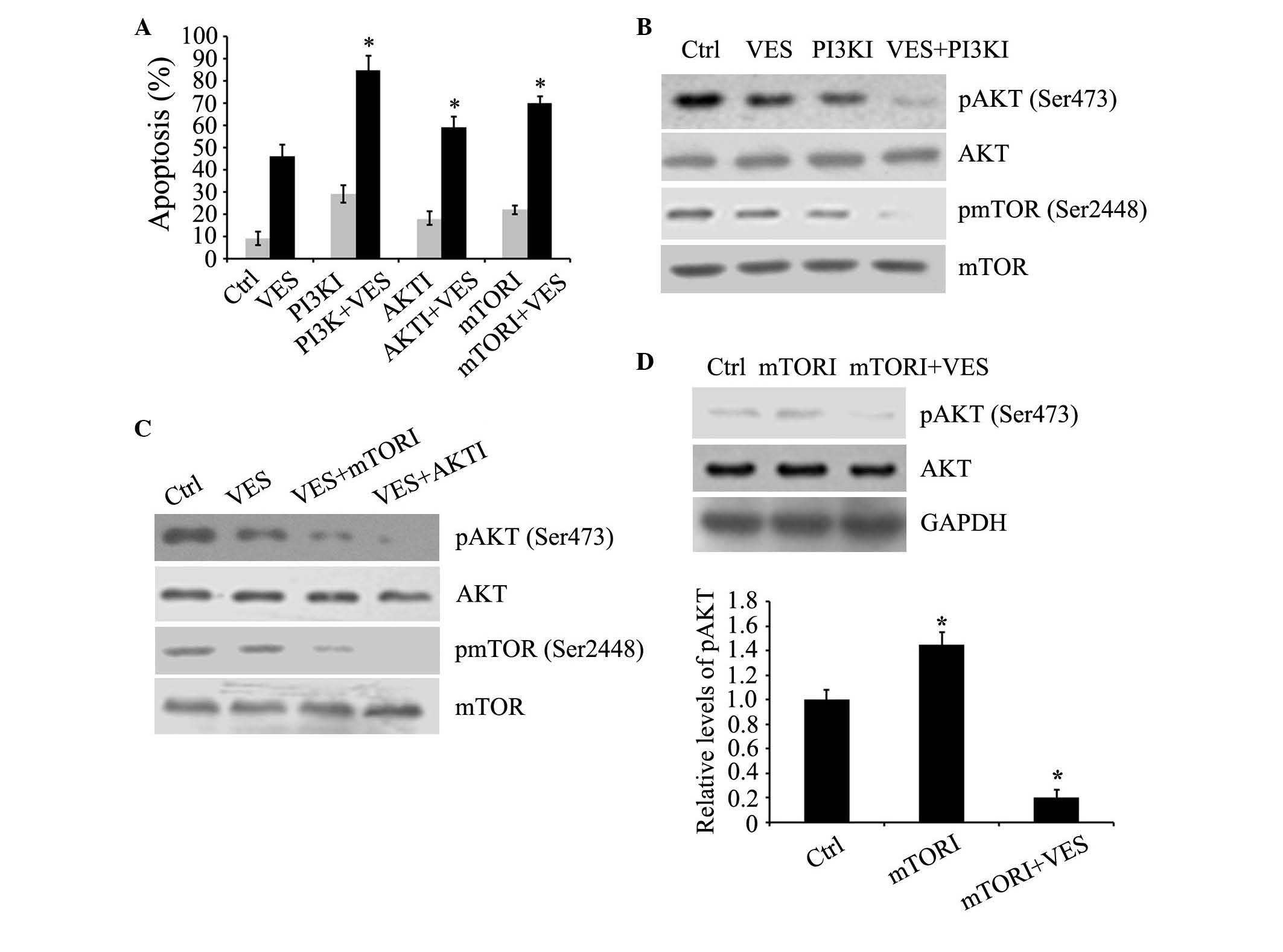 | Figure 5Inhibitors of PI3K, AKT and mTOR
enhance VES-induced apoptosis. (A) EC109 cells were cultured with
15 µM VES, plus 2 µM PI3KI, LY294002, 10 µM
AKTI, triciribine, and 50 nM mTORI, rapamycin for 24 h. Apoptosis
was determined by fluorescence-activated cell sorting analysis. (B
and C) Protein levels of p-AKT (Ser473) and p-mTOR (Ser2448), in
addition to expression levels of total AKT and mTOR were determined
by western blot analyses in EC109 cells treated with the three
inhibitors and VES. (D) EC109 cells were treated with 50 nM mTORI
rapamycin and 15 µM VES for 24 h. Protein expression levels
of p-AKT (Ser473), AKT and GAPDH were determined by western blot
analyses. Protein expression levels were quantified and presented
as the mean ± standard error of the mean. *P<0.05 vs.
ctrl group. PI3K, phosphoinositide-3-kinase; AKT, serine-threonine
kinase AKT; mTOR, mammalian target of rapamycin; VES, vitamin E
succinate; p, phosphorylated; PI3KI, PI3K inhibitor; AKTI, AKT
inhibitor; mTORI, mTOR inhibitor; Ctrl, control. |
VES blocked the increase of p-AKT induced
by the mTOR inhibitor
It has been indicated that the increase in
expression of p-AKT may be induced by the mTOR inhibitor rapamycin
via negative feedback regulation of insulin receptor substrate
(24,25). As expected, the mTOR inhibitor
induced an increase in expression of p-AKT, which attenuates its
anti-cancer efficacy. Notably, mTOR inhibitor treatment combined
with VES was able to suppress this counterproductive effect in AKT,
which is a pro-survival mediator (Fig.
5D).
Discussion
VES exhibits anti-tumor activity in colorectal,
breast, prostate, skin, and gastric cancer by inducing various
apoptotic signaling pathways. The results of the present study
demonstrated that VES inhibited cell proliferation and induced
apoptosis in EC109 esophageal cancer cells. To the best of our
knowledge, the present study is the first to indicate that VES
inhibited AKT-mediated anti-apoptotic events by suppressing
phosphorylation of Bad and caspase-9. In addition, VES suppressed
AKT and downregulated mTOR activity, resulting in reductions of
downstream effectors, p-p70S6K and p-4E-BP1. The present study
hypothesized that VES induced apoptosis by inhibiting PI3K/AKT
signaling pathways, which stimulated activation of Bad and
caspase-9. Furthermore, it was indicated that the side-effect of
the mTOR inhibitors, namely, activation of AKT, was prevented by
combination treatment with VES. These results demonstrated that VES
induced apoptosis via inhibition of the PI3K/AKT signaling pathways
and suppressed the mTOR inhibitor-mediated activation of AKT,
suggesting that combination treatment an mTOR inhibitor and VES may
improve clinical treatment outcome.
PI3K/AKT signaling is constitutively activated in
numerous types of human cancer. Activation of this signaling in
cancer cells has been associated with cancer cell viability, tumor
growth and drug resistance. Thus, PI3K/AKT signaling has attracted
growing attention as a potential target for cancer therapeutic
strategies. Downregulation of PI3K/AKT signaling in cancer cells
results in cellular apoptosis and sensitization to chemotherapy
(26,27). Various strategies using genetic and
pharmacologic inhibitors to inhibit anti-apoptotic proteins,
including X-linked inhibitor of apoptosis protein, survivin,
inhibitor of apoptosis proteins, matrix metalloproteinases, B-cell
lymphoma 2 (Bcl-2), nuclear factor-κB and AKT have been used to
sensitize cancer cells to chemotherapy and apoptosis (27,28).
α-Tocopherol ether-linked acetic acid (α-TEA) suppresses AKT and
contributes to α-TEA-induced apoptosis in prostate and ovarian
cancer cells (29,30). The present study demonstrated that
inhibition of PI3K/AKT signaling potentiates VES-induced apoptosis
in esophageal cancer cells.
Bad and caspase-9 are pro-apoptotic proteins. Bad is
a member of the Bcl-2 family, which accelerates apoptosis via the
formation of heterodimers with pro-survival factors, Bcl-2 and
B-cell lymphoma-extra large (Bcl-xL). Phosphorylation of Bad at
Ser112 and Ser136 blocks its binding with Bcl-2 or Bcl-xL,
promoting cell growth (31,32).
The phosphorylation of Bad has been reported to be enhanced by
extracellular signal-regulated kinase/ribosomal protein S6 kinase
α-1 and PI3K/AKT signaling pathways, respectively (32). The current study demonstrated that
VES suppressed phosphorylation of Bad at Ser136, suggesting that
downregulation of AKT activity mediated by VES results in
Bad-mediated apoptotic events. In addition, LY294002 inhibition of
PI3K reduced phosphorylation of Bad at Ser136, which suggested PI3K
was involved in the regulation of AKT in esophageal cancer cells.
Caspase-9 induces cell death via mitochondria-mediated initiation
of caspases (33). It has been
reported that AKT is involved in the inactivation of caspase-9 by
phosphorylating caspase-9 at Ser196 (19). Thus, caspase-9 is a target for AKT
to prevent cells from apoptosis. Thus, VES suppresses AKT activity
to downregulate phosphorylation of caspase-9 at Ser196, and then
contributes to mitochondria-dependent apoptosis.
Previous studies indicate that PI3K/AKT/mTOR augment
cancer cell growth and resistance to chemotherapeutics. The mTOR
inhibitor rapamycin exhibits marked growth inhibitory effects
against a broad range of types of human cancer (34,35).
The current study indicated that VES downregulated mTOR activity by
suppressing phosphorylation of mTOR and inhibited its downstream
effectors p70S6K and 4E-BP1. Furthermore, the results demonstrated
that VES augmented suppression of rapamycin on mTOR and promoted
apoptosis, and inhibited feedback activation of AKT induced by
rapamycin, providing a combination therapeutic strategy for
esophageal cancer using mTOR and VES.
In conclusion, the present study demonstrated that
VES targeted PI3K/AKT signaling pathways and induced apoptosis in
esophageal cancer cells. In addition, VES may work synergistically
with inhibitors of members of the PI3K/AKT signaling pathways in
order to more effectively control cancer cell survival and
growth.
Acknowledgments
The authors would like to thank Summus Biological
Technology Co., Ltd. (Harbin, China) for their technical
support.
References
|
1
|
Montgomery EA, Bosman FT, Brennan P and
Malekzadeh R: Oesophageal cancer. World Cancer Report 2014. Stewart
BW and Wild CP: International Agency for Research on Cancer; Lyon:
pp. 528–543. 2014
|
|
2
|
Lozano R, Naghavi M, Foreman K, Lim S,
Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et
al: Global and regional mortality from 235 causes of death for 20
age groups in 1990 and 2010: A systematic analysis for the Global
Burden of Disease Study 2010. Lancet. 380:2095–2128. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kelsen D, Daly JM, Kern SE, Levin B,
Tepper JE and Van Cutsem E: Gastrointestinal Oncology: Principles
and Practices. 2nd edition. Lippincott Williams & Wilkins;
Philadelphia, PA: pp. 42007
|
|
4
|
Schottenfeld D: Cancer Epidemiology and
Prevention. 3rd edition. Oxford University Press; Oxford: pp.
6972006
|
|
5
|
Ferri FF: Esophageal Tumors. Ferri's
Clinical Advisor. 2013, Mosby (Elsevier); Maryland Heights, MO: pp.
389–391. 2012
|
|
6
|
National Cancer Institute: Cancer
Statistics: SEER stat fact sheets, esophageal cancer. http://seer.cancer.gov/statfacts/html/esoph.html.
Accessed April 15, 2016.
|
|
7
|
Neuzil J, Tomasetti M, Mellick AS, Alleva
R, Salvatore BA, Birringer M and Fariss MW: Vitamin E analogues: A
new class of inducers of apoptosis with selective anti-cancer
effect. Curr Cancer Drug Targets. 4:355–372. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Neuzil J, Wang XF, Dong LF, Low P and
Ralph SJ: Molecular mechanism of 'mitocan'-induced apoptosis in
cancer cells epitomizes the multiple roles of reactive oxygen
species and Bcl-2 family proteins. FEBS Lett. 580:5125–5129. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang XF, Dong L, Zhao Y, Tomasetti M, Wu K
and Neuzil J: Vitamin E analogues as anticancer agents: Lessons
from studies with alpha-tocopheryl succinate. Mol Nutr Food Res.
50:675–685. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Malafa MP and Neitzel LT: Vitamin E
succinate promotes breast cancer tumor dormancy. J Surg Res.
93:163–170. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Malafa MP, Fokum FD, Andoh J, Neitzel LT,
Bandyopadhyay S, Zhan R, Iiizumi M, Furuta E, Horvath E and Watabe
K: Vitamin E succinate suppresses prostate tumor growth by inducing
apoptosis. Int J Cancer. 118:2441–2447. 2006. View Article : Google Scholar
|
|
12
|
Neuzil J: Vitamin E succinate and cancer
treatment: A vitamin E prototype for selective antitumour activity.
Br J Cancer. 89:1822–1826. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Quin J, Engle D, Litwiller A, Peralta E,
Grasch A, Boley T and Hazelrigg S: Vitamin E succinate decreases
lung cancer tumor growth in mice. J Surg Res. 127:139–143. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Annovazzi L, Mellai M, Caldera V, Valente
G, Tessitore L and Schiffer D: mTOR, S6 and AKT expression
inrelation to proliferation and apoptosis/autophagy in glioma.
Anticancer Res. 29:3087–3094. 2009.PubMed/NCBI
|
|
15
|
Falasca M: PI3K/Akt signaling pathway
specific inhibitors: A novel strategy to sensitize cancer cells to
anti-cancer drugs. Curr Pharm Des. 16:1410–1416. 2010. View Article : Google Scholar
|
|
16
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar
|
|
17
|
Chang F, Lee JT, Navolanic PM, Steelman
LS, Shelton JG, Blalock WL, Franklin RA and McCubrey JA:
Involvement of PI3K/Akt pathway in cell cycle progression,
apoptosis, and neoplastic transformation: A target for cancer
chemotherapy. Leukemia. 17:590–603. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Datta SR, Dudek H, Tao X, Masters S, Fu H,
Gotoh Y and Greenberg ME: Akt phosphorylation of BAD couples
survival signals to the cell-intrinsic death machinery. Cell.
91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cardone MH, Roy N, Stennicke HR, Salvesen
GS, Franke TF, Stanbridge E, Frisch S and Reed JC: Regulation of
cell death protease caspase-9 by phosphorylation. Science.
282:1318–1321. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mabuchi S, Ohmichi M, Kimura A, Hisamoto
K, Hayakawa J, Nishio Y, Adachi K, Takahashi K, Arimoto-Ishida E,
Nakatsuji Y, et al: Inhibition of phosphorylation of BAD and Raf-1
by Akt sensitizes human ovarian cancer cells to paclitaxel. J Biol
Chem. 277:33490–33500. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gibbons JJ, Abraham RT and Yu K: Mammalian
target of rapamycin: Discovery of rapamycin reveals a signaling
pathway important for normal and cancer cell growth. Semin Oncol.
36(Suppl 3): S3–S17. 2009. View Article : Google Scholar
|
|
22
|
Kline K, Yu W and Sanders BG: Vitamin E
and breast cancer. J Nutr. 134(Suppl 12): S3458–S3462. 2004.
|
|
23
|
Yu W, Sanders BG and Kline K:
RRR-alpha-tocopheryl succinate induction of DNA synthesis arrest of
human MDA-MB-435 cells involves TGF-beta-independent activation of
p21Waf1/Cip1. Nutr Cancer. 43:227–236. 2002. View Article : Google Scholar
|
|
24
|
Wan X, Harkavy B, Shen N, Grohar P and
Helman LJ: Rapamycin induces feedback activation of Akt signaling
through an IGF-1R-dependent mechanism. Oncogene. 26:1932–1940.
2007. View Article : Google Scholar
|
|
25
|
Jiang X, Sinnett-Smith J and Rozengurt E:
Carbachol induces p70S6K1 activation through an ERK-dependent but
Akt-independent pathway in human colonic epithelial cells. Biochem
Biophys Res Commun. 387:521–524. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Asanuma K, Moriai R, Yajima T, Yagihashi
A, Yamada M, Kobayashi D and Watanabe N: Survivin as a
radioresistance factor in pancreatic cancer. Jpn J Cancer Res.
91:1204–1209. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chawla-Sarkar M, Bae SI, Reu FJ, Jacobs
BS, Lindner DJ and Borden EC: Downregulation of Bcl-2, FLIP or IAPs
(XIAP and survivin) by siRNAs sensitizes resistant melanoma cells
to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 11:915–923.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tamm I, Wang Y, Sausville E, Scudiero DA,
Vigna N, Oltersdorf T and Reed JC: IAP-family protein survivin
inhibits caspase activity and apoptosis induced by Fas (CD95), Bax,
caspases, and anticancer drugs. Cancer Res. 58:5315–5320.
1998.PubMed/NCBI
|
|
29
|
Yu W, Shun MC, Anderson K, Chen H, Sanders
BG and Kline K: alpha-TEA inhibits survival and enhances death
pathways in cisplatin sensitive and resistant human ovarian cancer
cells. Apoptosis. 11:1813–1823. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shun MC, Yu W, Park SK, Sanders BG and
Kline K: Downregulation of epidermal growth factor receptor
expression contributes to alpha-TEA's proapoptotic effects in human
ovarian cancer cell lines. J Oncol. 2010:8245712010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Datta SR, Katsov A, Hu L, Petros A, Fesik
SW, Yaffe MB and Greenberg ME: 14-3-3 proteins and survival kinases
cooperate to inactivate BAD by BH3 domain phosphorylation. Mol
Cell. 6:41–51. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hayakawa J, Ohmichi M, Kurachi H, Kanda Y,
Hisamoto K, Nishio Y, Adachi K, Tasaka K, Kanzaki T and Murata Y:
Inhibition of BAD phosphorylation either at serine 112 via
extracellular signal-regulated protein kinase cascade or at serine
136 via Akt cascade sensitizes human ovarian cancer cells to
cisplatin. Cancer Res. 60:5988–5994. 2000.PubMed/NCBI
|
|
33
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chan S: Targeting the mammalian target of
rapamycin (mTOR): A new approach to treating cancer. Br J Cancer.
91:1420–1424. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vignot S, Faivre S, Aguirre D and Raymond
E: mTOR-targeted therapy of cancer with rapamycin derivatives. Ann
Oncol. 16:525–537. 2005. View Article : Google Scholar : PubMed/NCBI
|