Introduction
Osteoarthritis (OA) is a common disease, and its
incidence rate in people over 25 years old is 14%, with ~400,000
patients diagnosed with OA per year in the Catalonia region of
Spain (1). Clinically, local
intra-articular injection of DXM has an anti-inflammatory function,
relieving the symptoms of knee OA, such as pain and swelling.
However, long-term and repeated injections of DXM can disrupt the
metabolic balance in chondrocytes, leading to the catabolism in the
cartilage with increase of matrix metalloproteinases (MMPs), and
can accelerate cell apoptosis and death. This may result in a
reduction in chondrocyte number and further deterioration of
articular cartilage in OA. Therefore, a novel method to maintain
the survival of chondrocytes under DXM stimulation is required.
Autophagy, a self-protective mechanism, has been
suggested to maintain cellular homeostasis by removing protein
aggregates and damaged organelles by the fusion of autophagosomes
and lysosomes. Previous studies have demonstrated that autophagy is
a protective process preventing against chondrocyte apoptosis and
cartilage degeneration (2,3). In a mouse model, local injection of
rapamycin, an inducer of autophagy, has been demonstrated to impede
the progress of arthritis (2,4). It
is widely accepted that autophagy in mammalian cells can be
adaptively activated by different stresses (5–7),
therefore, it was hypothesized that DXM may be able to activate
autophagy in chondrocytes.
Previous studies have demonstrated that the ability
of joint tissues to restore the articular surface is reduced with
age; patients with intra-articular fractures of the knee >50
years have a greater risk of developing OA than younger patients
(8,9). Age also increases the risk of
post-traumatic OA in patients with anterior cruciate ligament tears
(9). With the aging of cartilage,
senescent chondrocytes may accumulate in the cartilage, leading to
the loss of ability to maintain and restore articular cartilage
effectively (10). A previous
study additionally observed a clear correlation between increasing
age and incidence of OA (11).
Aging is the largest risk factor for OA, and cell senescence serves
an important role in the occurrence and development of OA. However,
the exact mechanism involved in the effect of DXM on chondrocyte
senescence remains unclear.
A previous study demonstrated that autophagy is a
protective mechanism in human chondrocytes with mitochondrial
dysfunction (3). These
observations suggest that autophagy serves an important role to
protect chondrocytes from oxidative stress, and additionally
supported the theory that pharmacological interventions targeting
autophagy may prevent cartilage degradation (3). The effect of autophagy on chondrocyte
senescence remains to be fully elucidated. Therefore, the current
study aimed to culture rat chondrocytes treated with DXM, in order
to determine the effect of DXM on chondrocyte senescence and
autophagy and the association between these two events.
Materials and methods
All procedures involving Sprague-Dawley rats were
performed under the approval and guidance of the Animal Care and
Use Committee at Southern Medical University (Guangzhou,
China).
Rat knee chondrocyte culture
A total of 34 3-month-old Sprague-Dawley male rats,
ranging in weight between 300 and 340 g, were obtained from the
Laboratory Animal Center of Southern Medical University. These rats
were anesthetized by 10% chloral hydrate and sacrificed by cervical
dislocation, and the articular cartilage was separated from the
femoral condyles and tibial plateaus under a microscope. Cartilage
slices were incubated with trypsin (0.5 mg/ml) (Sigma-Aldrich;
Merck Millipore, Darmstadt, Germany) for 30 mins at 37°C.
Subsequent to removal of trypsin, the cartilage slices were
incubated with 0.1% collagenase, type II (Sigma-Aldrich; Merck
Millipore) in Dulbecco's modified Eagle's medium (DMEM; Life
Technologies; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
with 5% fetal calf serum (FCS; Life Technologies; Thermo Fisher
Scientific, Inc.) for 4 h at 37°C with shaking. The isolated
chondrocytes were recovered and plated in DMEM supplemented with
10% FCS and 1% penicillin/streptomycin. The chondrocytes were
incubated at 37°C in a humidified gas mixture containing 5%
CO2 balanced with air. In the current study, all cells
used were second-generation chondrocytes. DXM (Sigma-Aldrich; Merck
Millipore) was added to the chondrocytes at various concentrations
(0, 0.1, 1, 25 and 50 µg/ml).
3-MA and DXM were added to chondrocytes at various
concentrations (0 µg/ml, 10 mmol/l 3-MA, 10 mmol/l 3-MA + 25
µg/ml DXM and 25 µg/ml DXM).
Alcian blue staining
Chondrocytes were fixed with 4% paraformaldehyde for
30 min at 37°C. Chondrocytes were stained with 1% Alcian Blue 8GX
(Sigma-Aldrich; Merck Millipore) dissolved in glacial acetic acid
for 30 min at room temperature. The sections were counterstained
with 0.1% nuclear read dissolved in 5% aluminium sulfate for 20 sec
followed by routine dehydration. The stained sections and cells
were washed three times with phosphate-buffered saline (PBS) and
observed by light microscopy.
LysoTracker Red staining
Chondrocytes (3×105 cells/well) were
fixed with various concentrations of DXM for 4 days at 37°C in
24-well plates and rinsed with DMEM three times. LysoTracker Red
culture medium (66 mM; 1 ml) was added to each well and then the
cells were cultured for 30 min at 37°C. Subsequent to washing with
PBS three times, the chondrocytes were observed using an inverted
fluorescence microscope.
Monodansylcadaverine (MDC) staining
Monodansylcadaverine is a specific in vivo
marker for autophagic vacuoles. Chondrocytes (3×106
cells/well) were fixed at different concentrations of DXM for 4
days at 37°C in 6-well plates and rinsed with PBS three times.
Subsequently, chondrocytes were incubated at 37°C in 0.05 mM MDC
(Sigma-Aldrich; Merck Millipore) for 30 min. Following incubation,
chondrocytes were washed three times with PBS at 37°C and fixed for
30 min in 4% paraformaldehyde. Following fixation, chondrocytes
were washed four times with PBS and observed under a fluorescence
microscope. To observe the rate of autophagy, stained chondrocytes
were tested using flow cytometry.
Western blotting
Chondrocytes incubated with various concentrations
of DXM (0, 0.1, 1, 25 and 50 µg/ml) were washed with PBS.
Total proteins were isolated, and were mixed with a standard
protein solution from the Bicinchoninic Acid (BCA) assay kit to
produce the BCA working fluid. Each well was filled with the
working fluid (200 µl) for 15 min at room temperature. The
protein concentration was determined according to the measured
optical density values. According to the protein concentration of
each group (sample quantity, 30 µg; total volume, 20
µl), the total amount of protein in each group was
calculated. Subsequent to heating with loading buffer (Beyotime
Institute of Biotechnology, Haimen, China) and PBS at 100°C for 5
min, protein samples were prepared. Equal quantities (30 µg)
of protein from each sample were separated using 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and were transferred
onto polyvinylidene difluoride membranes (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Following blocking with 5% nonfat milk,
the membranes were incubated with the following rabbit anti-rat
primary antibodies overnight at 4°C: Polyclonal anti-beclin-1
(1:1,000; Abcam, Cambridge, MA, USA; cat. no. ab55878), mouse
monoclonal anti-p62 (1:1,000; Abcam; cat. no. ab56416), rabbit
polyclonal anti-LC3 (1:1,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA), mouse polyclonal anti-β-actin (1:1,000; Beyotime
Institute of Biotechnology; cat. no. AA128). Next, the membranes
were incubated with horseradish peroxidase-conjugated secondary
antibodies (goat anti-rabbit; 1:2,000; cat. no. A0208; Beyotime
Institute of Biotechnology) for 2 h at 37°C. The bands were
detected using ECL plus reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) on an enhanced chemiluminescence detection system
(PerkinElmer, Inc., Waltham, MA, USA). In addition, the intensity
of the bands was quantified using Alpha Ease FC software, version
4.0 (Alpha Innotech Corporation; ProteinSimple, Santa Clara, CA,
USA).
β-galactosidase (β-gal) staining
Chondrocytes were treated with 3-MA and DXM at
various concentrations (0 µg/ml, 10 mmol/l 3-MA, 10 mmol/l
3-MA + 25 µg/ml DXM and 25 µg/ml DXM) for 24, 48 and
72 h; then, chondrocytes were fixed using a Senescence
β-Galactosidase Staining kit for 15 min at room temperature.
Chondrocytes were stained with β-gal dye for 12 h and then washed
three times. The stained cells were observed using a phase-contrast
microscope to determine the percentage of positive cells out of the
total chondrocytes under magnification of ×200.
Red fluorescent protein (RFP) -green
fluorescent protein (GFP) -light chain 3 (LC3) assay
Prior to treatment with glucocorticoids,
chondrocytes were transfected with monomeric (m)RFP-GFP-LC3 when
the confluence was 50–70% using RFP-GFP-LC3 adenoviral vectors
(HanBio Technology Co., Ltd., Shanghai, China). The multiplicity of
infection was 100. The chondrocytes were incubated with the
adenovirus in DMEM with no serum for 2 h at 37°C. The transfected
chondrocytes were incubated with 10% DMEM supplemented with fetal
bovine serum overnight prior to glucocorticoid treatment to
eliminate the effect of starvation on an autophagic level.
Following treatment with different doses of glucocorticoids for 4
days, autophagosomes and autolysomes in chondrocytes were observed
under a confocal microscope (SP8; Leica Microsystems GmbH, Wetzlar,
Germany).
Statistical analysis
Statistical analyses were performed using SPSS
statistical software, version 16 (SPSS, Inc., Chicago, IL, USA).
One-way analysis of variance was used to analyze the differences
between groups. Tukey's significance test was used to detect
differences between two groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Chondrocyte identification
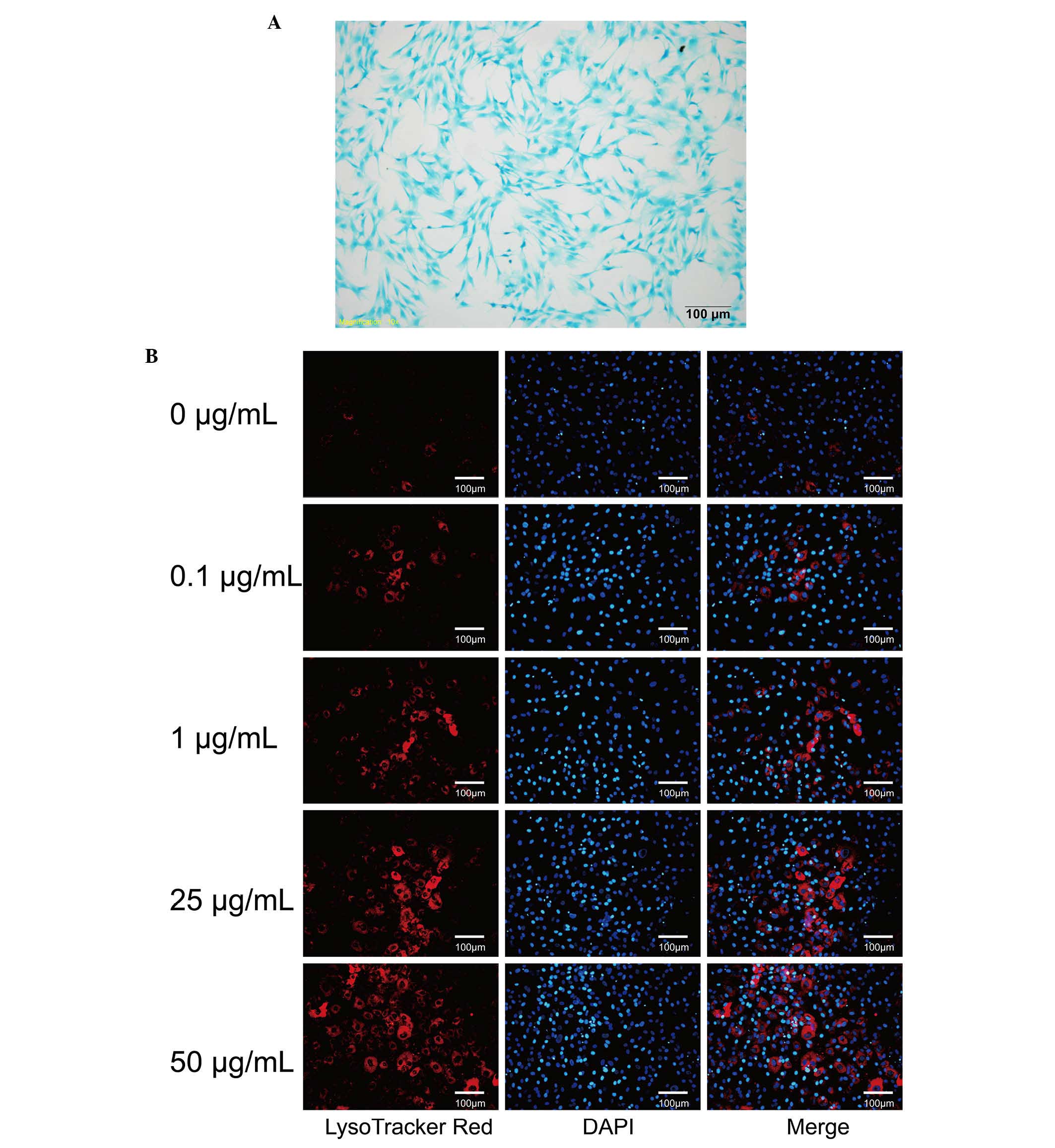
Chondrocytes were stained with toluidine blue. The
extracellular matrices of the chondrocytes were stained blue and
the nuclei were stained dark blue, which is consistent with typical
chondrocyte characteristics (Fig.
1A).
DXM promote chondrocyte autophagy
Chondrocytes were incubated with different
concentrations of DXM for 4 days and stained with LysoTracker and
MDC to observe autophagic vacuoles. Compared with the control
group, only a small number of chondrocytes displayed
LysoTracker-positive staining (Fig.
1B). It was observed that the intensity of LysoTracker-positive
cells increased markedly with increasing concentrations of DXM
(Fig. 1B). The majority of
chondrocytes were incubated with 50 µg/ml DXM for 4 days and
were positively stained with LysoTracker. Therefore, DXM is able to
promote autophagous vesicles in the chondrocytes.
The results of LysoTracker staining were similar to
that of MDC staining. In the control group, only certain cells were
stained positive for MDC (Fig. 2A)
and the intensity of MDC staining in the cells was markedly
increased with increasing concentrations of DXM. The MDC-stained
chondrocytes were observed by flow cytometry, and it was
demonstrated that the cells that were incubated with the different
concentrations of DXM had higher incidences of autophagy compared
with the control group after 4 days (P<0.05). In the 50
µg/ml group, autophagy occurred with an average incidence of
56.74%.
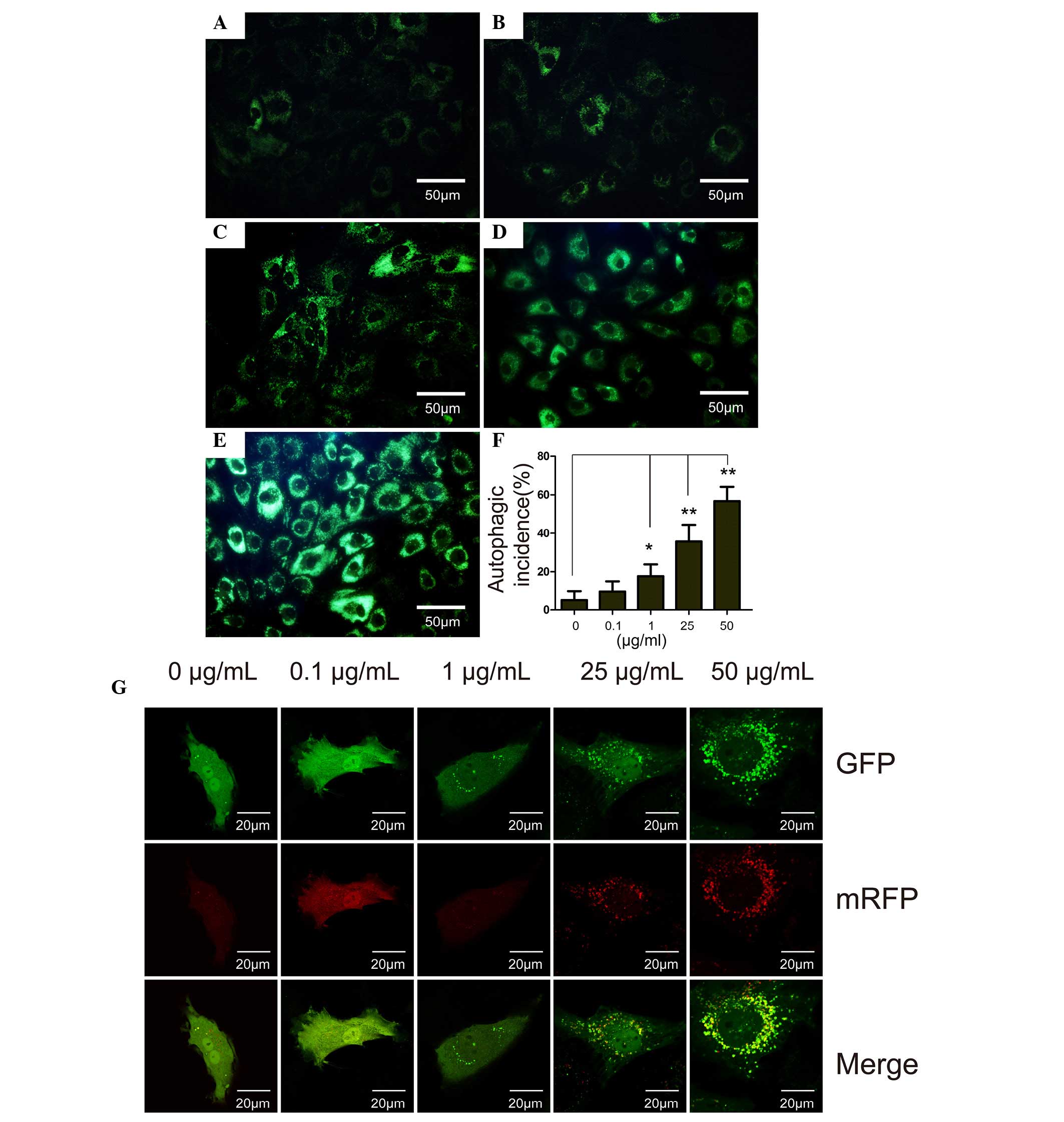 | Figure 2The MDC staining, flow cytometry
analysis and RFP-GFP-LC3 assay of chondrocytes treated with DXM.
MDC staining of chondrcytes treated with different concentrations
of DXM: (A) 0 µg/ml, (B) 0.1 µg/ml, (C) 1
µg/ml, (D) 25 µg/ml and (E) 50 µg/ml. Scale
bar, 50 µm. (F) Autophagic incidence analyzed by flow
cytometry. Mean ± standard deviation. n=3. *P<0.05
vs. control; **P<0.01 vs. control. (G) Prior to DXM
treatment, chondrocytes were transfected with RFP-GFP-LC3
adenoviral vectors. A dose-dependent increase in the number of
autophagosomes formed was observed in the DXM-treated chondrocytes.
MDC, monodansylcadaverine; GFP, green fluorescent protein; RFP, red
fluorescent protein; DXM, dexamethasone; LC3, light chain 3. |
RFP-GFP-LC3 assay
Autophagy is a recycling process including the
maturation of autophagosomes, fusion of autophagosomes and
lysosomes, autolysosome formation and degradation. The total
process is termed autophagic flux. In vitro, autophagic flux
can be determined by the transfection of an adenovirus harboring
mRFP-GFP-LC3. Subsequent to transfection, the autophagosomes in
cells are presented as yellow dots (the combination of red and
green fluorescence), and the autolysosomes are presented as red
dots (the extinction of GFP in the acid environment of lysosomes).
Following culture for 4 days, the yellow autophagosomes were
increased in the cytoplasm of chondrocytes with increasing DXM dose
(Fig. 2G), indicating the
stimulatory role of DXM on autophagic flux in chondrocytes.
DXM upregulates autophagy-associated
proteins
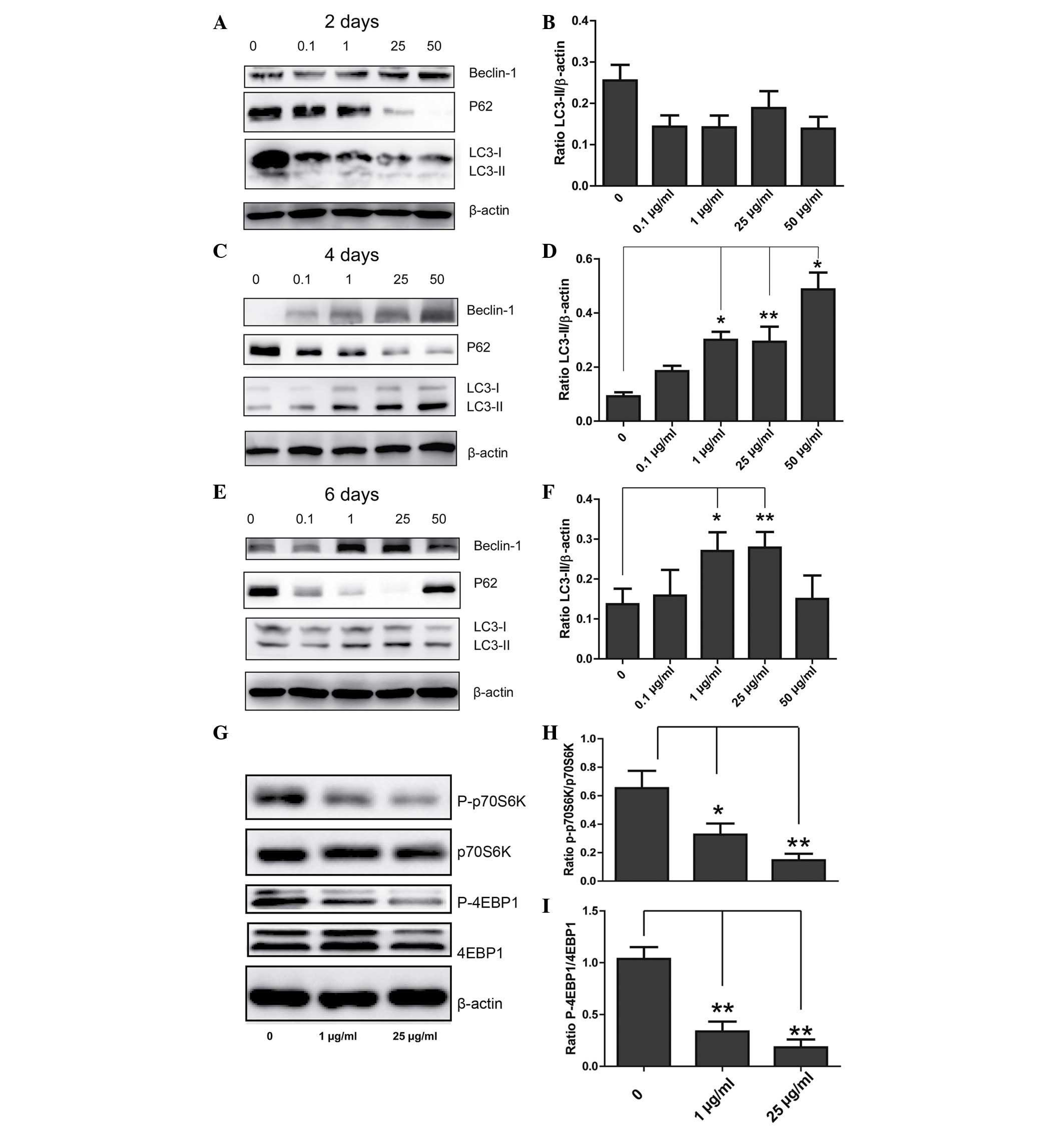
To determine the role of DXM in regulating
chondrocyte autophagy, western blotting was conducted in order to
observe the expression levels of LC3, beclin-1 and P62. When
chondrocytes were incubated with different concentrations of DXM
for 2 days, no significant differences in the expression levels of
LC3-II/β-actin in the different groups were observed. However,
after 4 days, the expression levels of LC3-II/β-actin were
significantly increased with increasing concentrations of DXM, and
after 6 days, the expression levels of LC3-II/β-actin were reduced
at the highest concentration of DXM (Fig. 3). Changes in the expression levels
of P62 and beclin-1 were also similar, however, notably, the
expression levels of P62 reduced with increasing concentrations of
DXM after 2 days.
DXM inhibits the mechanistic target of
rapamycin (mTOR) signaling pathway
The mechanism of activating autophagy is
predominantly associated with mTOR-dependent and mTOR-independent
pathways, with p70S6K and 4EBP1 involved as downstream effectors in
mTOR-dependent pathways. In the 1 and 25 µg/ml treatment
groups, the expression levels of P-p70S6K and 4EBP1 were reduced
when compared with the control group, and the difference was
identified to be statistically significant (Fig. 3G), indicating that DXM inhibits
autophagy through an mTOR-dependent pathway.
The role of DXM and autophagy in
chondrocyte senescence
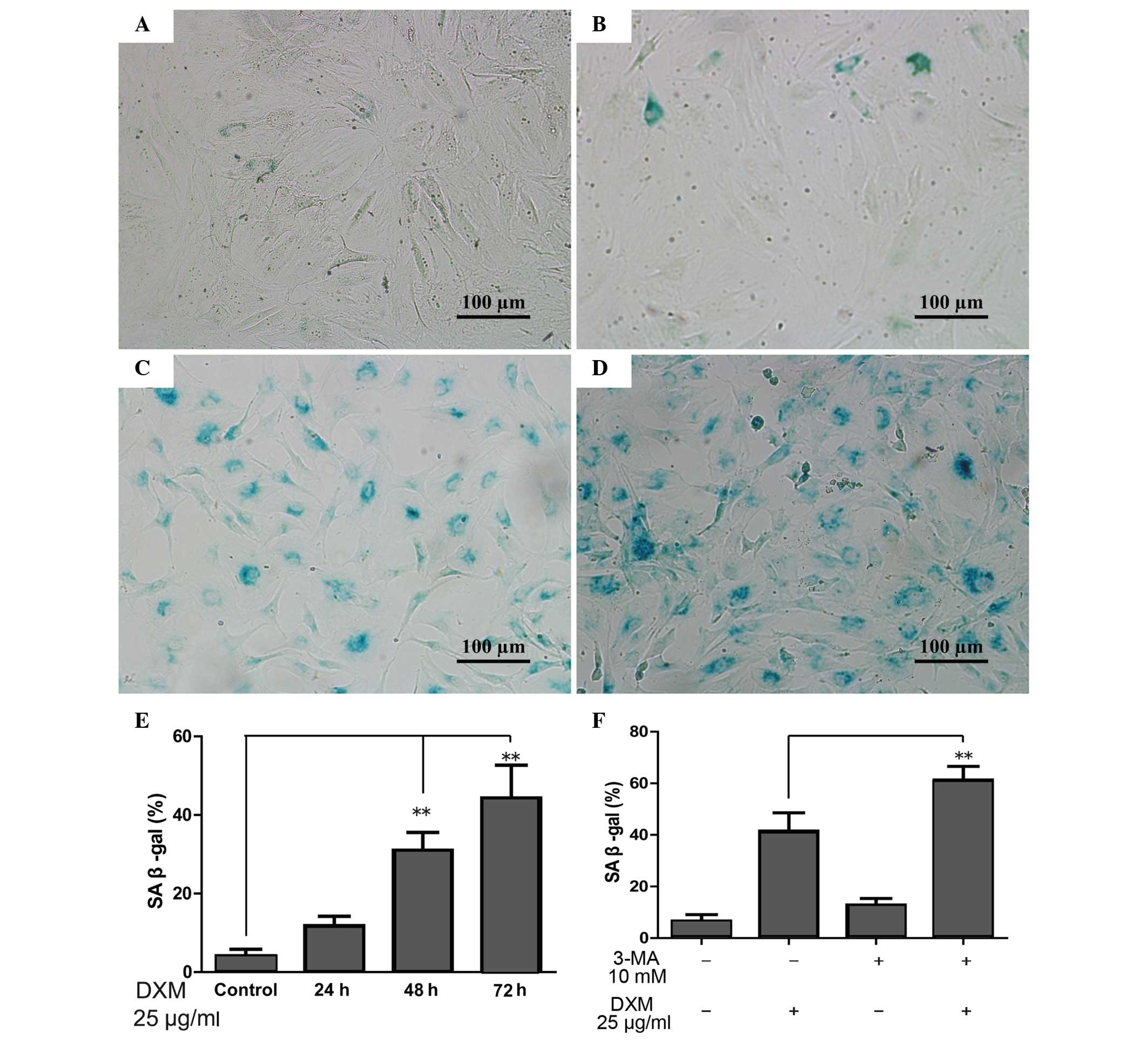
When the cells become senescent, β-gal-positive
staining occurs. The proportion of β-gal-positive chondrocytes
significantly increased with time upon treatment with 25
µg/ml glucocorticoid, and was greatest following treatment
for 72 h (Fig. 4A–E). However, the
proportion of β-gal-positive chondrocytes increased significantly
when 3-MA, which can inhibit autophagy, was added in comparison
with the DXM group alone after 72 h. Furthermore, 3-MA alone was
not able to increase the proportion of β-gal-positive chondrocytes
(Fig. 4F).
Discussion
In the current study, it was investigated whether
DXM is able to inhibit chondrocyte autophagy through an
mTOR-dependent pathway and induce senescence. Senescence activated
by DXM was observed to be inhibited by autophagy, and it was
identified that chondrocyte senescence increased with the
inhibition of autophagy.
DXM is widely used to relieve a variety of symptoms
caused by OA, however long-term and repeated treatment often
results in complications (12,13).
DXM can lead to chondrocyte apoptosis, growth inhibition, and a
reduction in biological activity (13,14).
Apoptosis may reduce the number of chondrocytes and reduce the
extracellular matrix, including proteoglycan and collagen type II,
thus it is suggested that chondrocyte apoptosis is an important
factor in the pathogenesis of OA (15). While autophagy is a programmed cell
death program similar to apoptosis, the effects of glucocorticoid
treatment on chondrocyte autophagy remain to be investigated.
Autophagy is not only similar to apoptosis but also
has a protective effect on chondrocytes in OA (2,3,16).
Autophagy has gained research focus due to its potential in
regulating the aging process. A previous study has reported that
the activity of autophagy was reduced in senescent tissues
(17). When cells encounter
certain stress situations or bacterial invasion, double membranous
vesicles called autophagosomes are formed. These autophagosomes
either fuse with endosomes or lysosomes, leading to the formation
of autolysosomes. The process begins with the formation of a double
membranous phagophore that elongates into an autophagosome, during
which cellular material becomes enclosed (18). In this way, autophagy can transport
larger organelles or pathogens. In vitro, autophagy can
inhibit chondrocyte apoptosis and prevent degradation of the
extracellular matrix induced by interleukin 1β (19). In vivo, it remains unclear
whether intraperitoneal injection of rapamycin or local
intra-articular injection of rapamycin can promote chondrocyte
autophagy and reduce the progress of degeneration of articular
cartilage (2,4). In the current study, it was
identified that DXM significantly activates chondrocyte autophagy,
thus it was hypothesized that autophagy may be compensatory to the
chondrocyte damage induced by DXM.
mTOR is one of the components of mTOR complex 1
(mTORC1) and serves as a key switch in regulating autophagy, thus
the autophagic signal pathway can be divided into mTOR-dependent
and mTOR-independent pathways (20). Commonly, the stimulation of
autophagy begins with the inhibition of mTOR, and the most
important of two downstream targets of mTORC1 are p70S6K and 4EBP1.
It was identified that DXM can reduce p70S6K and 4EBP1
phosphorylation and activity, and thus, DXM activates autophagy in
chondrocytes through an mTOR-dependent pathway. Similarly,
glucosamine, which is the current treatment for OA, also activates
autophagy through an mTOR-dependent pathway (21). These results demonstrate that the
predominant signal pathway of autophagy activation in chondrocytes
is mTOR-dependent.
DXM can reduce cell growth and inhibit cell
activity. Poulsen et al (22) identified that DXM can reduce tendon
cell growth and induce senescence. However, whether DXM can induce
chondrocyte senescence remains to be determined. When cell-cycle
progression is arrested, the cell can be in a static state or
become senescent. When the cell senesces, cell proliferation stops,
however its volume loss continues to increase, resulting in aging
cells being larger than normal cells (23). In the current study, it was
identified that DXM can promote chondrocyte senescence and that
this effect is more marked as time progresses. Chondrocyte
senescence has been considered to be a correlative factor for OA,
thus it is suggested that the long-term use of
glucocorticoid-induced senescence may be an explanation for
chondrocytes undergoing degeneration and necrosis.
The association between autophagy and senescence
remains controversial. Kamalakannan et al (24) identified that autophagy can prevent
mononuclear cell senescence caused by heat shock protein, however
it induces bronchial epithelial cells to undergo senescence caused
by oxidative stress (25). For
chondrocytes, senescence was activated when 3-MA was used to
inhibit autophagy, which indicates that glucocorticoid-induced
autophagy may be a compensatory protective effect against
senescence.
In conclusion, the current study identified that DXM
can inhibit chondrocyte autophagy through an mTOR-dependent pathway
and induce senescence. Autophagy may therefore serve as a
protective process against senescence.
Acknowledgments
The present study was supported by a grant from the
WenZhou Science and Technology Research Project (grant no.
Y20140582).
References
|
1
|
Reyes C, Garcia-Gil M, Elorza JM,
Mendez-Boo L, Hermosilla E, Javaid MK, Cooper C, Diez-Perez A,
Arden NK, Bolibar B, et al: Socio-economic status and the risk of
developing hand, hip or knee osteoarthritis: A region-wide
ecological study. Osteoarthritis Cartilage. 23:1323–1329. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Takayama K, Kawakami Y, Kobayashi M, Greco
N, Cummins JH, Matsushita T, Kuroda R, Kurosaka M, Fu FH and Huard
J: Local intra-articular injection of rapamycin delays articular
cartilage degeneration in a murine model of osteoarthritis.
Arthritis Res Ther. 16:4822014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
López de Figueroa P, Lotz MK, Blanco FJ
and Caramés B: Autophagy activation and protection from
mitochondrial dysfunction in human chondrocytes. Arthritis
Rheumatol. 67:966–976. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Caramés B, Hasegawa A, Taniguchi N, Miyaki
S, Blanco FJ and Lotz M: Autophagy activation by rapamycin reduces
severity of experimental osteoarthritis. Ann Rheum Dis. 71:575–581.
2012. View Article : Google Scholar :
|
|
5
|
Zhang M, Zhang J, Lu L, Qiu ZY, Zhang X,
Yu SB, Wu YP and Wang MQ: Enhancement of chondrocyte autophagy is
an early response in the degenerative cartilage of the
temporomandibular joint to biomechanical dental stimulation.
Apoptosis. 18:423–434. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Martinet W, De Meyer GR, Herman AG and
Kockx MM: Amino acid deprivation induces both apoptosis and
autophagy in murine C2C12 muscle cells. Biotechnol Lett.
27:1157–1163. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park EY and Park JB: High glucose-induced
oxidative stress promotes autophagy through mitochondrial damage in
rat notochordal cells. Int Orthop. 37:2507–2514. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stevens DG, Beharry R, McKee MD, Waddell
JP and Schemitsch EH: The long-term functional outcome of
operatively treated tibial plateau fractures. J Orthop Trauma.
15:312–320. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Martin JA, Brown TD, Heiner AD and
Buckwalter JA: Chondrocyte senescence, joint loading and
osteoarthritis. Clin Orthop Relat Res. (427 Suppl): S96–S103. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Martin JA and Buckwalter JA: Telomere
erosion and senescence in human articular cartilage chondrocytes. J
Gerontol A Biol Sci Med Sci. 56:B172–B179. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Buckwalter JA, Martin J and Mankin HJ:
Synovial joint degeneration and the syndrome of osteoarthritis.
Instr Course Lect. 49:481–489. 2000.PubMed/NCBI
|
|
12
|
Tu Y, Xue H, Francis W, Davies AP,
Pallister I, Kanamarlapudi V and Xia Z: Lactoferrin inhibits
dexamethasone-induced chondrocyte impairment from osteoarthritic
cartilage through up-regulation of extracellular signal-regulated
kinase 1/2 and suppression of FASL, FAS, and Caspase 3. Biochem
Biophys Res Commun. 441:249–255. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zaman F, Chrysis D, Huntjens K, Chagin A,
Takigawa M, Fadeel B and Sävendahl L: Dexamethasone differentially
regulates Bcl-2 family proteins in human proliferative
chondrocytes: Role of pro-apoptotic Bid. Toxicol Lett. 224:196–200.
2014. View Article : Google Scholar
|
|
14
|
Hong D, Chen HX, Yu HQ, Wang C, Deng HT,
Lian QQ and Ge RS: Quantitative proteomic analysis of
dexamethasone-induced effects on osteoblast differentiation,
proliferation, and apoptosis in MC3T3-E1 cells using SILAC.
Osteoporos Int. 22:2175–2186. 2011. View Article : Google Scholar
|
|
15
|
Aigner T, Hemmel M, Neureiter D, Gebhard
PM, Zeiler G, Kirchner T and McKenna L: Apoptotic cell death is not
a widespread phenomenon in normal aging and osteoarthritis human
articular knee cartilage: A study of proliferation, programmed cell
death (apoptosis), and viability of chondrocytes in normal and
osteoarthritic human knee cartilage. Arthritis Rheum. 44:1304–1312.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Caramés B, Olmer M, Kiosses WB and Lotz
MK: The relationship of autophagy defects and cartilage damage
during joint aging in a mouse model. Arthritis Rheumatol.
67:1568–1576. 2015. View Article : Google Scholar :
|
|
17
|
Wu X, Won H and Rubinsztein DC: Autophagy
and mammalian development. Biochem Soc Trans. 41:1489–1494. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sasaki H, Takayama K, Matsushita T, Ishida
K, Kubo S, Matsumoto T, Fujita N, Oka S, Kurosaka M and Kuroda R:
Autophagy modulates osteoarthritis-related gene expressions in
human chondrocytes. Arthritis Rheum. 64:1920–1928. 2012. View Article : Google Scholar
|
|
20
|
Alayev A and Holz MK: mTOR signaling for
biological control and cancer. J Cell Physiol. 228:1658–1664. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Caramés B, Kiosses WB, Akasaki Y, Brinson
DC, Eap W, Koziol J and Lotz MK: Glucosamine activates autophagy in
vitro and in vivo. Arthritis Rheum. 65:1843–1852. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Poulsen RC, Watts AC, Murphy RJ, Snelling
SJ, Carr AJ and Hulley PA: Glucocorticoids induce senescence in
primary human tenocytes by inhibition of sirtuin 1 and activation
of the p53/p21 pathway: In vivo and in vitro evidence. Ann Rheum
Dis. 73:1405–1413. 2014. View Article : Google Scholar :
|
|
23
|
Muller M: Cellular senescence: Molecular
mechanisms, in vivo, significance and redox considerations.
Antioxid Redox Signal. 11:59–98. 2009. View Article : Google Scholar
|
|
24
|
Kamalakannan V, Shiny A, Babu S and
Narayanan RB: Autophagy protects monocytes from wolbachia heat
shock protein 60-induced apoptosis and senescence. PLoS Negl Trop
Dis. 9:e00036752015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ito S, Araya J, Kurita Y, Kobayashi K,
Takasaka N, Yoshida M, Hara H, Minagawa S, Wakui H, Fujii S, et al:
PARK2-mediated mitophagy is involved in regulation of HBEC
senescence in COPD pathogenesis. Autophagy. 11:547–559. 2015.
View Article : Google Scholar : PubMed/NCBI
|