Introduction
Kabuki syndrome (KS), first reported by Japanese
researchers Kuroki et al (1) and Niikawa et al (2), is a rare genetic disorder
characterized by intellectual disability and multiple congenital
anomalies (3). The incidence of KS
is officially 1 in 32,000 individuals (4). The cardinal diagnostic manifestations
of KS include characteristic facial features, mild-to-moderate
mental retardation, dermatoglyphic abnormalities, skeletal
anomalies, and postnatal growth deficiencies (5). In addition, speech and language
deficits are commonly present in patients with KS (6). Etiologically, KS was attributed
originally to loss-of-function mutations in the gene encoding
lysine methyltransferase 2D (KMT2D), namely KMT2D (formerly
MLL2), with an autosomal dominant inheritance pattern
(7). More recently, the KS
phenotype was identified in boys with a loss-of-function mutation
in KDMA6 on the X chromosome, the product of which is
important for KMT2D function (8).
Currently, no effective treatments for KS are available (9). Treatments are required that are
beneficial for KS patients with developmental disability or
cognitive limitations; group therapy may be an option (10). The present study reports the case
of a young Chinese girl with unclear speech (nasal and muffled
pronunciation) and possible mental retardation, who was diagnosed
with KS based on DNA sequencing analysis of KMT2D.
Case report
Written informed consent was obtained from the
patient's family. A 4-year-old Chinese girl was admitted to the
Central South University Xiangya School of Medicine Affiliated
Haikou Hospital (Haikou, China) on March 8th, 2015 with unclear
speech (nasal and muffled pronunciation), which her parents
indicated had been present for >2 years, and possible mental
retardation. Mental retardation was suspected due to the weak
expressive language ability of the patient compared with healthy
peers.
The patient was a first-born child with no siblings,
born full-term via uncomplicated vaginal delivery when her mother
was 24 years old. There was no history of consanguineous marriage
in the family, and no family history of genetic disorders. The
patient's father was employed as a road construction engineer and
her mother had previously worked with cosmetics. During the first
trimester, prior to awareness of the pregnancy, the patient's
mother reportedly had a febrile illness and received oral cold
medication at a local clinic (no records available). The patient's
mother did not consume alcohol, smoke, or take any other drugs or
medications during the pregnancy.
At 1 year of age, the patient was diagnosed with
right congenital hip dysplasia, and was treated successfully with
external bracing. The patient's speech was delayed; she was only
able to say one word by 1 year of age. Upon admission to the
hospital at 4 years of age, the patient was able to speak in 5- and
6-word sentences. Her speech was unclear, with muffled
pronunciation and a nasal tone. No seizures, meningitis, or other
severe illnesses were reported. In addition, no problems with
hearing or difficulties with understanding and following
instructions were observed. The patient was able to recognize
simple colors and the numbers 1–5, and could color with a crayon.
However, she was unable to read, write, or perform simple addition.
Her motor and social skills were observed to be normal.
A physical examination revealed that the patient was
of normal height (~100 cm) and weight (~16 kg). However, she
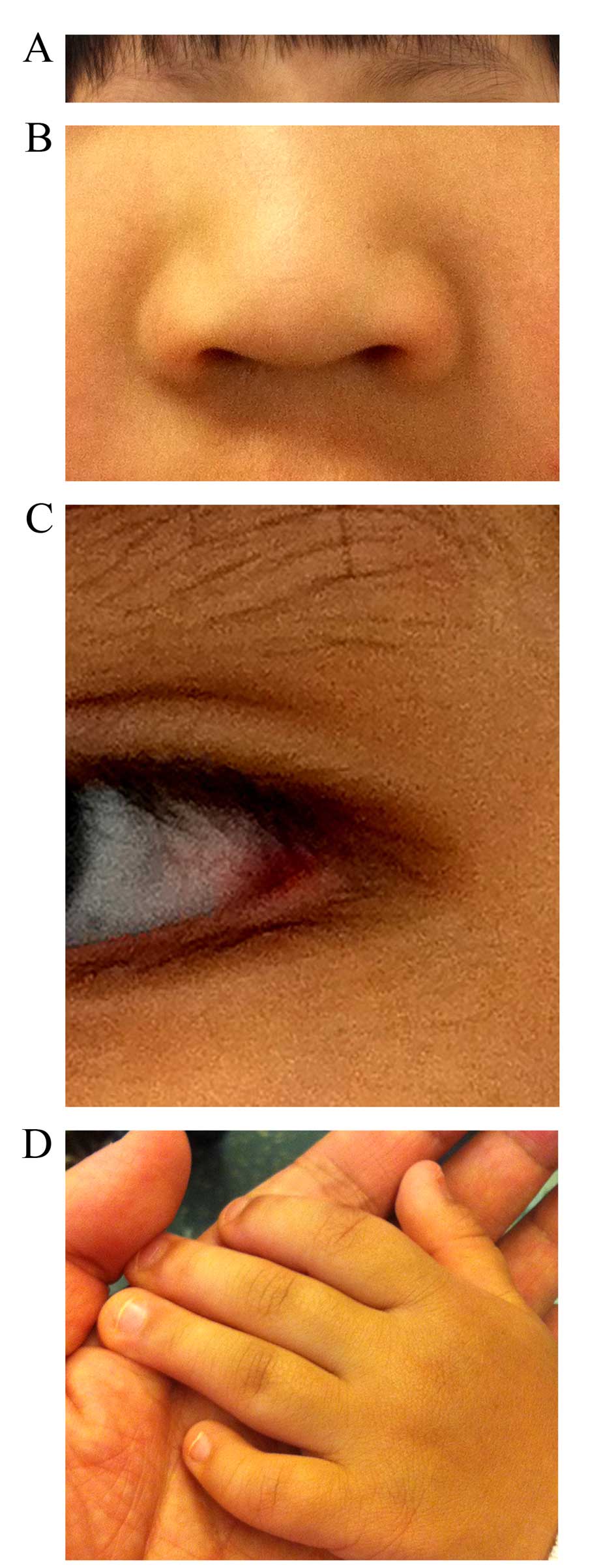
presented with abnormal facial features, including slanted eyes and
sparse eyebrows (Fig. 1A), a broad
nose and depressed nasal tip (Fig.
1B), and eversion of the lateral parts of the lower eyelids
(Fig. 1C). In addition, she had a
high palate, but without a cleft. Her fifth digits were short with
clinodactyly (Fig. 1D), but she
had normal palmar creases. The results of heart and respiratory
system examinations were normal, and there was no
hepatosplenomegaly. Neurologically, the patient exhibited normal
physiological reflexes and did not exhibit any pathological
reflexes. Her muscle strength appeared to be normal.
Blood analysis revealed normal complete blood count,
electrolyte measurements, blood lipid levels and renal and liver
functional markers, but high blood lactic acid levels (2.8 mmol/l,
August 2013; 3.4 mmol/l, January 2015; normal range, 0.7–2.1
mmol/l). In addition, the patient's blood ammonia levels were
normal and a urine mucopolysaccharide test was weakly positive [5
µl (+), 15 µl (+++) and 25 µl (+++)]. Gas
chromatography-mass spectrometry analysis of the patient's urine
and blood revealed no evidence of any amino acid or aliphatic acid
metabolic disorders. Furthermore, G-banding karyotyping
demonstrated that the patient had macroscopically normal
chromosomes (46, XX). Chest X-rays, retinal examinations, and brain
magnetic resonance imaging results were also normal. Notably,
X-rays of the fingers revealed the presence of three phalanges in
the short fifth digits of the two hands.
Due to the clinical manifestations and urine
mucopolysaccharide test results, it was suspected initially that
the patient may be suffering from mucopolysaccharidosis type III
(MPS III). MPS III and KS are characterized by atypical facial
features together with developmental and/or speech delays.
Specifically, MPS III is characterized by an early-onset
developmental and/or speech delay subsequent to an initial period
of normal development (11). The
developmental delays evolve into a progressive cognitive decline,
behavioral abnormalities and severe hyperactivity that does not
respond to treatment with stimulants (11). The somatic features of MPS III,
however, are relatively mild, including a dolichocephalic skull
shape with a short forehead, prominent eyebrows, an everted and
thick lower lip, and an upturned upper lip with a protruding
philtrum (11). The patient in the
present study exhibited abnormal facial features and delayed
speech, but displayed no overt behavioral abnormalities,
hyperactivity, or MPS III-characteristic facial features, and her
urine MPS test was only slightly abnormal. Therefore, a diagnosis
of MPS III was excluded. Autism was also considered due to the
patient's delayed speech development. Autism is a
neurodevelopmental disorder that is characterized by abnormalities
in reciprocal social and communicative behaviors, and an inflexible
adherence to routinized patterns of thought and behavior (12). The patient did not display any
difficulties in understanding or following instructions given by
her parents or preschool teacher. In addition, the patient gave
reliable responses and maintained eye contact when spoken to.
Furthermore, sensory processing difficulties are ubiquitous, albeit
highly variable, among people with autism. The patient of the
present study presented no evidence of sensory hyper- or
hypo-sensitivity, or any motor coordination delays, further
indicating that she did not have autism.
Following the exclusion of MPS III and autism, KS
was considered as a possible diagnosis. Various clinical
characteristics of the patient supported a diagnosis of KS,
including abnormal facial features (Fig. 1A–C), short fifth digits with
clinodactyly (Fig. 1D), skeletal
anomalies, delayed language development and unclear speech. DNA
sequence analysis of KMT2D, the gene primarily associated
with KS, was performed. A peripheral blood sample was first
obtained from the sample, and DNA was extracted from blood with a
QIAamp Blood DNA Mini kit (catalog no. 51106; Qiagen GmbH, Hilden,
Germany). High-throughput sequencing of KMT2D was performed
using the TruSight One Sequencing Panel (Illumina, San Diego, CA,
USA). The sequence was analyzed in ANNOVAR software (annovar.openbioinformatics.org/en/latest/)
(13), and various databases,
including 1,000 genomes (www.1000genomes.org/), Exome Sequencing Project 6500
(evs.gs.washington.edu/EVS/), Single
Nucleotide Polymorphisms (www.ncbi.nlm.nih.gov/SNP/) and Human Gene Mutation
Data (www.hgmd.cf.ac.uk/ac/index.php), were used for
screening and annotation of gene variants in accordance with the
American College of Medical Genetics and Genomics guidelines
(14). The DNA sequence analysis
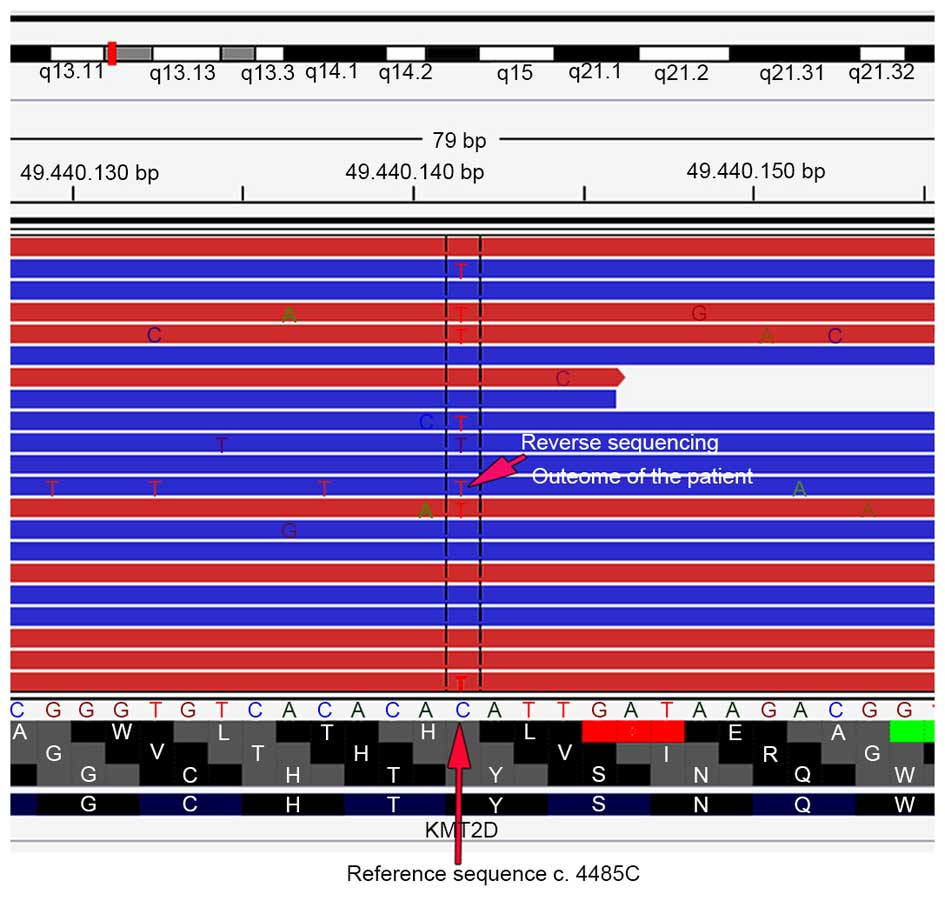
revealed a mutation in KMT2D (Fig. 2), which confirmed the diagnosis of
KS.
Subsequently, the patient's parents underwent
polymerase chain reaction (PCR) genetic testing together with their
daughter with the following KMT2D-Exon 16 primers, designed
using Primer Premier software version 5.0 (Premier Biosoft
International, Palo Alto, CA, USA): Forward,
5′-TATGATGTTGAGTATGTGACAGAGG-3′ and reverse,
5′-AATCCTAGCAGTGAAGAGACCAT-3′. The primers were used to amplify the
KMT2D gene using an ABI 9700 PCR instrument (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). Exon
16 was amplified in a reaction solution containing 1 µl 10X
PCR buffer, 0.35 µl dNTPs (10 mmol/l), 0.07 µl
FastStart Taq DNA Polymerase (5 U/µl; Roche Diagnostics,
Basel, Switzerland), 1 µl genomic DNA (100 ng/µl), 1
µl each primer (3.2 µmol/l) and 5.6 µl
ddH2O. The cycling conditions were as follows: 94°C for
12 min, followed by 35 cycles of 94°C for 30 sec, 55°C for 30 sec
and 72°C for 30 sec, and a final elongation step at 72°C for 10
min, as previously described (5,15).
An ABI 3500 Sequencer (Applied Biosystems; Thermo Fisher
Scientific, Inc.) was then used to sequence the PCR products by the
Sanger method, as previously described (15), and the resultant sequences were
compared with known sequences of KS-associated genes (NCBI
sequences, NM_003482.3 and NG_027827.1). The patient was found to
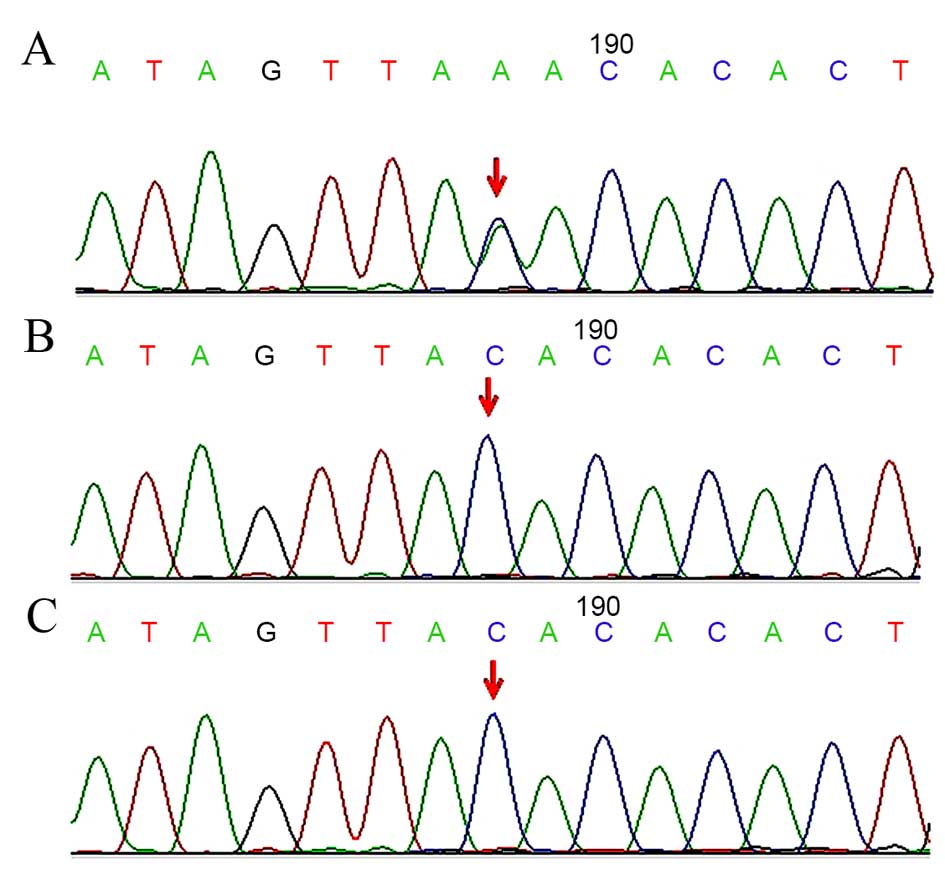
harbor a nonsense mutation in exon 16 of the KMT2D gene
(c.4485C>A, Tyr1495Ter; Fig.
3A); however, neither of her parents carried this mutation
(Fig. 3B and C). Based on the
clinical manifestations of the patient and the identification of a
de novo nonsense mutation of KMT2D, a diagnosis of KS
was established.
There is currently no cure for KS, and the only
treatments available are supportive and psychological therapies
(9). However, early diagnosis of
the syndrome is essential for optimal management. There are several
features that should be monitored, including the patient's height,
weight, head circumference, vision and hearing (9). The patient described in the present
report was referred immediately for speech therapy with the aim of
improving her speech quality.
The patient's prognosis is unclear; therefore, a
long-term follow-up plan was implemented with the intention of
providing potentially beneficial supportive and psychological
therapies as required. At a follow-up in May 2015, following 10
months of speech training, the patient's speech remained unclear.
Following her diagnosis in our hospital, she has not been subjected
to any further tests, has not exhibited any new manifestations of
pathology and has not been administered any other treatments beyond
speech therapy. The current case underscores the importance of
genetic testing in enabling a clear differentiated diagnosis of
KS.
Discussion
The present study reports a new case of the rare
genetic syndrome KS, which is characterized primarily by
intellectual deficiencies, multiple congenital malformations, and
specific craniofacial abnormalities (16). More specifically, KS is
characterized by the following five, mainly external, structural
features: i) A dysmorphic face, including eversion of the lower
lateral eyelids and arched eyebrows with sparse lateral eyebrows;
ii) dermatoglyphic abnormalities, with increased digital ulnar
loops and hypothenar loop patterns, absence of digital triradius,
and the presence of fingertip pads in 93% of patients; iii)
skeletal anomalies, with brachydactyly and spinal deformities with
or without sagittal cleft vertebrae in 92% of patients; iv)
mild-to-moderate mental retardation in 92% of patients; and v)
postnatal growth deficiencies in 83% of patients (17). Patients with KS frequently exhibit
internal malformations of the heart, kidneys, gastrointestinal
system, skeletal system and/or eyes (18). Certain patients present with
immunological defects and increased susceptibility to respiratory
infections. Speech and language deficits are common in KS, with the
most prominent deficit being dysarthria, characterized by imprecise
pronunciation of consonants, a harsh vocal quality, hypernasality,
reduced speech rate and stress, and a distorted pitch (6).
KS has an incidence rate of 1 in 32,000 individuals;
however, this figure is likely to be underestimated due to missed
diagnosis and misdiagnosis (4).
The vast majority of reported KS cases are sporadic (18). Diagnosis of KS can be challenging
due to the spectrum of clinical, radiological and biological
factors associated with KS, various complications, and incomplete
penetrance in certain cases (16).
Additionally, numerous other conditions present with features
similar to those observed in KS, including global developmental
delays, fetal alcohol syndrome, autism spectrum disorder and Down
syndrome (19). The majority of
patients diagnosed with KS are diagnosed based primarily on the
presentation of archetypical features. Currently, although genetic
testing of families is important for genetic counseling,
confirmatory genetic testing is performed only in a minority of
cases.
Mutations in KMT2D are present in 55–80% of
KS-diagnosed individuals subjected to genetic testing (9); KMD6A mutations have been
observed in the remaining KS cases in which genetic testing was
performed, although there may be other as yet unidentified genetic
mutations that lead to KS (20). A
number of different KMT2D mutations have been reported to
date, including missense mutations, nonsense mutations, frameshift
mutations, splice site mutations, and indel mutations (21). The majority of these result in a
truncated protein product. In the present study, a novel nonsense
mutation in exon 16 of KMT2D (c.4485C>A, Tyr1495Ter) was
identified. This mutation is a loss-of-function mutation in which
codon 1495 is replaced with a termination codon, resulting in
truncation of the encoded KMT2D protein product. KMT2D is a histone
methyltransferase, comprised of 5,537 amino acid residues, that
serves an important role in regulating gene transcription. KMT2D
methylates lysine 4 of histone 3; this methylation serves as a
marker of gene activation (22).
Numerous de novo KMT2D mutations have been
identified in sporadic KS cases (20). Among KS cases that are not
sporadic, autosomal dominant inheritance of KS due to
parent-to-child transmission of KMT2D mutations has
been observed (23). In the
present study, the patient's parents were not found to carry
KMT2D mutations, which indicates that the patient harbored a
de novo genetic mutation.
Although the molecular mechanisms underlying the KS
phenotype development remain to be elucidated, the clinical
manifestations of KS are generally presumed to be due to the loss
of KMT2D functionality. This view is supported by the fact that the
other gene in which mutations have been demonstrated to cause KS,
KDM6A, encodes a histone demethylase that interacts with
KMT2D (24). The KMT2D
mutation found in the present case (c.4485C>A) appears to be
novel in that it was not listed among the hundreds of previously
identified KS-causing mutations (5) represented in the Leiden Open
Variation Database (www.lovd.nl/),
the PubMed database of the National Center for Biotechnology
Information (www.ncbi.nlm.nih.gov/), the European Molecular Biology
Laboratory (www.embl.org/), or the Human Gene
Mutation Database (www.hgmd.org/).
In conclusion, clinicians should be aware that there
are a variety of clinical manifestations of KS. In the present
study, the patient had abnormal facial features and unclear speech;
however, her overall presentation was not clearly indicative of KS.
Genetic testing should be conducted to confirm KS diagnosis.
References
|
1
|
Kuroki Y, Suzuki Y, Chyo H, Hata A and
Matsui I: A new malformation syndrome of long palpebral fissures,
large ears, depressed nasal tip, and skeletal anomalies associated
with postnatal dwarfism and mental retardation. J Pediatr.
99:570–573. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Niikawa N, Matsuura N, Fukushima Y, Ohsawa
T and Kajii T: Kabuki make-up syndrome: A syndrome of mental
retardation, unusual facies, large and protruding ears, and
postnatal growth deficiency. J Pediatr. 99:565–569. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Verhagen JM, Oostdijk W, Terwisscha van
Scheltinga CE, Schalij-Delfos NE and van Bever Y: An unusual
presentation of Kabuki syndrome: Clinical overlap with CHARGE
syndrome. Eur J Med Genet. 57:510–512. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Niikawa N, Kuroki Y, Kajii T, Matsuura N,
Ishikiriyama S, Tonoki H, Ishikawa N, Yamada Y, Fujita M, Umemoto
H, et al: Kabuki make-up (Niikawa-Kuroki) syndrome: A study of 62
patients. Am J Med Genet. 31:565–589. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu S, Hong X, Shen C, Shi Q, Wang J,
Xiong F and Qiu Z: Kabuki syndrome: A Chinese case series and
systematic review of the spectrum of mutations. BMC Med Genet.
16:262015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Morgan AT, Mei C, Da Costa A, Fifer J,
Lederer D, Benoit V, McMillin MJ, Buckingham KJ, Bamshad MJ, Pope K
and White SM: Speech and language in a genotyped cohort of
individuals with Kabuki syndrome. Am J Med Genet A. 167:1483–1492.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hannibal MC, Buckingham KJ, Ng SB, Ming
JE, Beck AE, McMillin MJ, Gildersleeve HI, Bigham AW, Tabor HK,
Mefford HC, et al: Spectrum of MLL2 (ALR) mutations in 110 cases of
Kabuki syndrome. Am J Med Genet A. 155A:1511–1516. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bjornsson HT, Benjamin JS, Zhang L,
Weissman J, Gerber EE, Chen YC, Vaurio RG, Potter MC, Hansen KD and
Dietz HC: Histone deacetylase inhibition rescues structural and
functional brain deficits in a mouse model of Kabuki syndrome. Sci
Transl Med. 6:256ra1352014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Adam MP, Hudgins L and Hannibal M: Kabuki
Syndrome. GeneReviews(R). Pagon RA, Adam MP, Ardinger HH, Wallace
SE, Amemiya A, Bean LJH, Bird TD, Dolan CR, Fong CT, Smith RJH and
Stephens K: University of Washington, Seattle University of
Washington; Seattle: All rights reserved., Seattle (WA). 1993
|
|
10
|
Kasdon BD and Fox JE: Kabuki syndrome:
Diagnostic and treatment considerations. Ment Health Fam Med.
9:171–179. 2012.
|
|
11
|
Wijburg FA, Wegrzyn G, Burton BK and
Tylki-Szymanska A: Mucopolysaccharidosis type III (Sanfilippo
syndrome) and misdiagnosis of idiopathic developmental delay,
attention deficit/hyperactivity disorder or autism spectrum
disorder. Acta Paediatr. 102:462–470. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gaigg SB: The interplay between emotion
and cognition in autism spectrum disorder: Implications for
developmental theory. Front Integr Neurosci. 6:1132012. View Article : Google Scholar
|
|
13
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Y, Bögershausen N, Alanay Y, Simsek
Kiper PO, Plume N, Keupp K, Pohl E, Pawlik B, Rachwalski M, Milz E,
et al: A mutation screen in patients with Kabuki syndrome. Hum
Genet. 130:715–724. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Arnaud M, Barat-Houari M, Gatinois V,
Sanchez E, Lyonnet S, Touitou I and Geneviève D: Kabuki syndrome:
Update and review. Arch Pediatr. 22:653–660. 2015.In French.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin JL, Lee WI, Huang JL, Chen PK, Chan
KC, Lo LJ, You YJ, Shih YF, Tseng TY and Wu MC: Immunologic
assessment and KMT2D mutation detection in Kabuki syndrome. Clin
Genet. 88:255–260. 2015. View Article : Google Scholar
|
|
18
|
Banka S, Veeramachaneni R, Reardon W,
Howard E, Bunstone S, Ragge N, Parker MJ, Crow YJ, Kerr B, Kingston
H, et al: How genetically heterogeneous is Kabuki syndrome?: MLL2
testing in 116 patients, review and analyses of mutation and
phenotypic spectrum. Eur J Hum Genet. 20:381–388. 2012. View Article : Google Scholar :
|
|
19
|
Crane B, Alpert PT, Cyrkiel D and Jauregui
A: Kabuki syndrome: A challenge for the primary care provider. J Am
Assoc Nurse Pract. 25:522–526. 2013.PubMed/NCBI
|
|
20
|
Micale L, Augello B, Fusco C, Selicorni A,
Loviglio MN, Silengo MC, Reymond A, Gumiero B, Zucchetti F,
D'Addetta EV, et al: Mutation spectrum of MLL2 in a cohort of
Kabuki syndrome patients. Orphanet J Rare Dis. 6:382011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim SJ, Cho SY, Maeng SH, Sohn YB, Kim SJ,
Ki CS and Jin DK: A novel MLL2 gene mutation in a Korean patient
with Kabuki syndrome. Korean J Pediatr. 56:355–358. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo C, Chen LH, Huang Y, Chang CC, Wang P,
Pirozzi CJ, Qin X, Bao X, Greer PK, McLendon RE, et al: KMT2D
maintains neoplastic cell proliferation and global histone H3
lysine 4 monomethylation. Oncotarget. 4:2144–2153. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ng SB, Bigham AW, Buckingham KJ, Hannibal
MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM,
Mefford HC, et al: Exome sequencing identifies MLL2 mutations as a
cause of Kabuki syndrome. Nat Genet. 42:790–793. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lederer D, Grisart B, Digilio MC, Benoit
V, Crespin M, Ghariani SC, Maystadt I, Dallapiccola B and
Verellen-Dumoulin C: Deletion of KDM6A, a histone demethylase
interacting with MLL2, in three patients with Kabuki syndrome. Am J
Hum Genet. 90:119–124. 2012. View Article : Google Scholar :
|