Introduction
Aging is a complex physiological process involving
the progressive, irreversible accumulation of oxidative damage
sustained by cellular components during aerobic respiration
(1,2). Accelerated aging and death in
senescence-accelerated mice (SAM) is attributed to hyperoxidation
(3). Mitochondria, the primary
source of reactive oxygen species (ROS) and oxidative damage,
produce ROS during cellular respiration, which leads to impaired
physiological function, increased incidence of disease and reduced
lifespan (4). During respiration,
mitochondria produce the primary cellular energy component ATP,
which regulates cellular metabolism and modulates cell apoptosis
(5). Aging bodies are
characterized by abnormal energy metabolism, including decreased
mitochondrial ATP generation, increased ROS generation and
subsequent damage or disease (6).
Neurodegenerative diseases may arise from increased mitochondrial
dysfunction, oxidative stress and apoptosis (7). Cardiac function is particularly
susceptible to ROS production, mitochondrial DNA damage and
respiratory chain impairment (8).
Mitochondrial control of cellular respiration is associated with
cardiac function insufficiency and remodeling of the prostrated
heart (9). ATP is essential for
myocardial contraction and electrophysiology and maintaining normal
cardiac function during myocardial aging (10), while declining cardiac function is
marked by severe mitochondrial dysfunction and ATP deficiency
(6,11,12).
Antioxidants reduce oxidative damage arising from
abnormal mitochondrial metabolism (13). The oxidative stress theory of aging
describes antioxidants as ROS scavengers that enable the prevention
and repair of oxidative damage, which may reduce age-associated
illness (4). Melatonin, which is
synthesized from tryptophan primarily in the pineal gland, is a
free radical scavenger, and is the primary anti-oxidative defense
against reactive hydroxyl radicals (14,15).
As a pleiotropic molecule, melatonin exerts multiple beneficial
effects on age-related physiological functions, including metabolic
sensing, modulation and proliferation of mitochondria, the
antioxidative protection of biomolecules and anti-inflammatory
actions (16). In vitro and
in vivo studies have demonstrated that melatonin effectively
counteracts oxidative stress and oxidative damage (17–19).
Melatonin demonstrates obvious anti-aging effects, including
protecting the mitochondria of young and aged rats (20,21),
resisting mitochondrial dysfunction in SAM mice (22), and normalizing the energy status of
heart mitochondria and increasing ATP levels in SAM mice with
mitochondrial oxidative stress (2). Loss of melatonin production has long
been associated with aging (23).
Young hamsters exhibited an 8-fold increase in pineal melatonin
levels in the dark phase of a 24-h period, while aging hamsters
demonstrated no increase in nocturnal melatonin production,
indicating a marked reduction in pineal biosynthetic activity
associated with aging (24).
Similarly, melatonin levels in young, middle-aged and aged female
rats sacrificed during light and dark periods revealed low
melatonin levels in the pineal glands of all animals sacrificed
during light periods (25). By
contrast, marked increases (12-fold) in the melatonin levels of
young animals compared with older rats (6-fold) were observed
during dark periods, which indicates a notable reduction in pineal
melatonin production with age (25). Loss of melatonin is suggested to be
an indicator of ageing (23).
However, although melatonin may improve temporal organization in
advanced age and demonstrates beneficial effects on sleep and
age-related diseases, its protective effects against age-related
decline are not fully understood (26).
In the present study, the authors hypothesized that
melatonin may function in resistance to myocardial aging by
protecting the mitochondria, which could be demonstrated by
measuring cellular energy stores in aged rats. An accelerated aging
model was established by treating rats with D-galactose as
described previously (27). In
China, D-galactose-induced aging is a common method used to
generate animal models of aging and in studies of age-associated
pharmacotherapy (27). D-galactose
forms advanced glycation end products (AGEs) in vivo, and a
comparison of D-galactose-aged and AGEs-aged mice demonstrated that
both models resembled aged mice, suggesting that advanced glycation
may be part of the D-galactose aging mechanism (28). The aim of the present study was to
investigate the anti-aging effects of melatonin on the myocardial
mitochondria of D-galactose aged rats, and to explore associated
mechanisms.
Materials and methods
Animals
A total of 30 healthy male Sprague-Dawley (SD) rats
(age, 3 months; weight, 300–350 g) were purchased from the
Laboratory Animal Center of the Academy of Military Medical
Sciences (Beijing, China). All investigations were performed in
accordance with the Guide for the Care and Use of Laboratory
Animals of the Institutional Review Board of Chinese People's
Liberation Army (PLA) General Hospital (identification no.
2008-X1-70). Rats had ad libitum access to a commercial rat
chow diet and tap water. The animals were housed four per cage, and
maintained at 22±2°C and 50–60% relative humidity, under a 12-h
light/dark cycle. The Institutional Review Board of Chinese PLA
General Hospital reviewed and approved the study.
Grouping
All SD rats were randomly divided into 3 groups of
10 rats as follows: i) An accelerated aging group (D group) in
which 3-month-old rats were subject to accelerated ageing with
daily subcutaneous injections of 125 mg/kg D-galactose (Beijing
MENGYIMEI Biology Co., Ltd., Beijing China) for 6 weeks; ii) a
melatonin-treated group (M group) in which 3-month old rats were
aged with D-galactose administration (125 mg/kg/day) and treated
with 10 mg/kg/day melatonin (Shanghai Hengyuan Biological
Technology Co., Ltd., Shanghai, China) for 6 weeks; and iii) a
control group (C group) in which 3-month-old rats were treated with
equal volume of normal saline via daily subcutaneous and
intraperitoneal injections for 6 weeks.
Establishing the accelerated aging rat
model
A total of 10 three-month-old rats were subject to
accelerated ageing by administration of D-galactose (125 mg/kg/day
diluted in normal saline) delivered daily at 3:00 p.m. by
subcutaneous injection for 6 weeks, as described previously
(27,28). This method is based on the
metabolic disturbance theory of aging, which involves inducing
galactose overload via a high galactose diet or injection of
galactose, in order to promote subacute aging similar to natural
aging effects whereby D-galactose forms AGEs in vivo.
Successful and sufficient aging of rats in the present study was
characterized by the development of torpescence, sluggishness, grey
hair or trichomadesis of rats in the accelerated aging group (D
group) compared to the untreated control rats.
Materials and sample preparation
Rats in all three groups were sacrificed at the end
of the 6-week treatment period, when they were 4.5 months old.
Briefly, rats were anaesthetized with urethane via intraperitoneal
injection, and the hearts were removed and subsequently flushed
with saline as described previously (29). The free walls of the left
ventricles were then excised immediately. For each sample, a
portion of myocardial tissue was first placed into 1 mol/l
HClO4, then transferred into freezing tubes and
preserved in liquid nitrogen until required for evaluation. The
left ventricular myocardium was weighed precisely, and 100 mg
myocardial tissue was divided into pieces using eye scissors and
placed into a pre-cooled tissue grinder, to which 1 mol/l
HClO4 at 4°C was added at the ratio of 1 ml/100 mg. To
obtain homogenate, pieces of pre-cut tissue were homogenized in an
ice bath for 2 min using a CAT Scientific X120 Handheld Homogenizer
Drive (3000 r/min motor-driven homogenizer; PolyScience, Niles, IL,
USA). The homogenized tissues were centrifuged for 10 min at 4°C
and 6099 × g. For each sample, 800 µl supernatant was
transferred into a fresh 1.5 ml Eppendorf tube, to which 2 mol/l
KH2PO4 and 5 mol/l KOH were added. Samples
where then vigorously shaken on a vibrator, the pH was adjusted to
7.0, cooled to 4°C and then centrifuged for 10 min at 4°C and 6099
× g. For each sample, 200 µl of the supernatant was
transferred into a 0.5 ml Eppendorf tube for testing by high
performance liquid chromatography (HPLC). The remaining section of
myocardial tissue was subjected to freezing with liquid nitrogen to
extract myocardial mitochondria proteins, in order to observe
differences in the amounts of total mitochondrial proteins among
groups, and to measure alterations in the protein expression levels
of Bax, Bcl-2 in the mitochondria and cytochrome c (cyt-c) in the
cytoplasm.
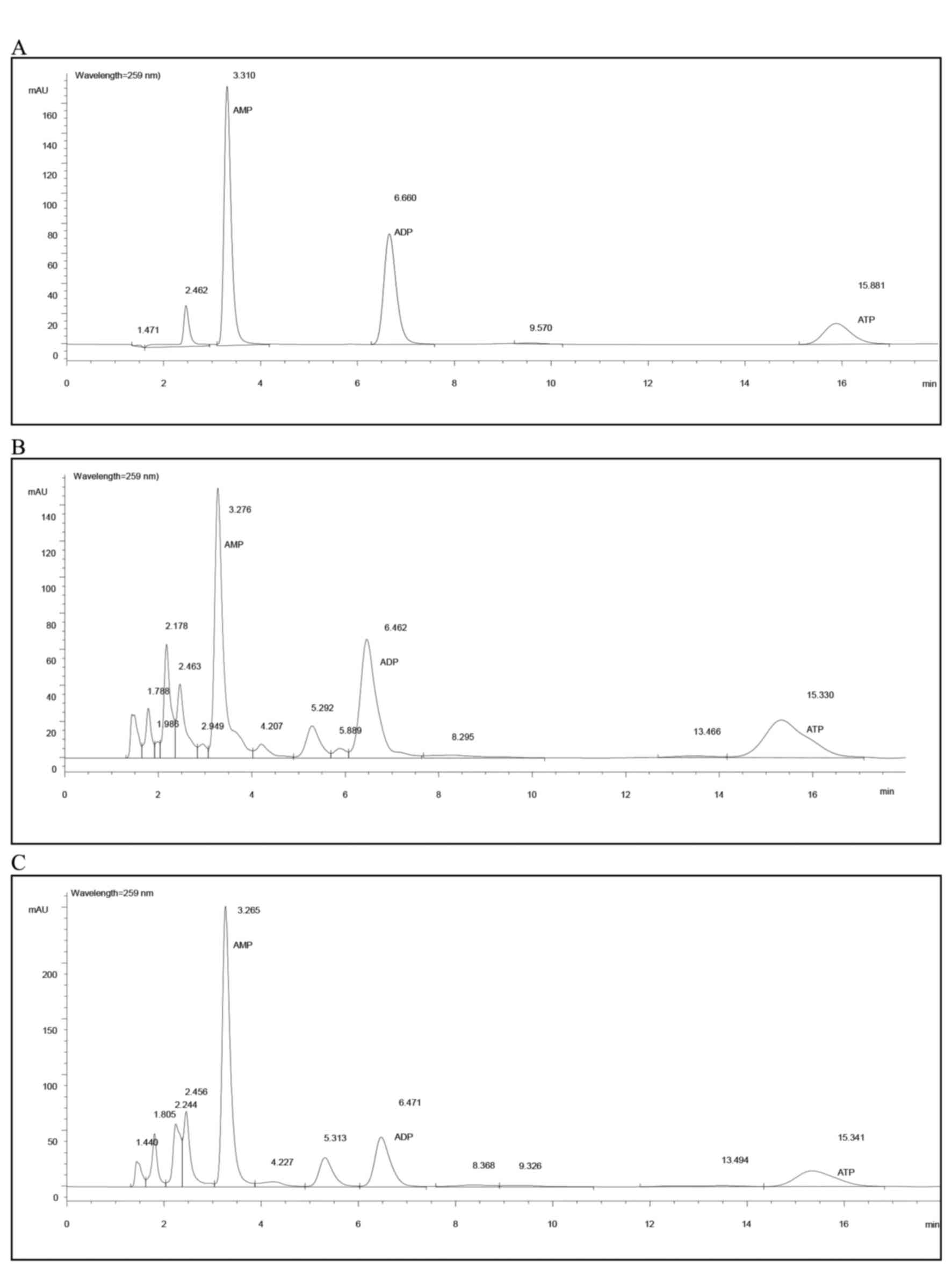
HPLC evaluation
An Agilent 1100 Series HPLC system (Agilent
Technologies, Inc., Santa Clara, CA, USA) was used. The
chromatographic analysis for the determination of ATP, ADP and AMP
was performed using a Zorbax, C (18) column (150×4.6 mm; 5 µm; Agilent
Technologies, Inc.). The mobile phase consisted of phosphate buffer
(solvent A: 6 mmol/l Na2HPO4 and 44 mmol/l
NaH2PO4, pH 5.8) and methanol (solvent B),
76/24, comprising the ion pair reagent A 5 mmol/l. For each sample,
20 µl was added to the sample injector of the HPLC instrument to
measure the levels of ATP, ADP and AMP at a detection wavelength of
259 nm. The solvent gradient elution conditions were 100% A, 0% B
for 0 min; 95% A, 5% B for 2 min; 80% A, 20% B for 4 min; 75% A,
25% B for 5 min; and 100% A, 0% B for 6 min at a flow-rate of 1.0
ml/min, and the column was maintained at a temperature of 25°C
throughout the study. Levels of ATP, ADP and AMP in rat myocardial
tissue were detected using the external standard method (30), and the total adenylic acid number
(TAN) was calculated using the following formula: TAN = ATP + ADP +
AMP. The energy state of myocytes was indicated by the energy
charge (EC), using the computational formula EC = (ATP + 1/2
ADP)/TAN, as described previously (31).
Mitochondria extraction
Briefly, 100 mg of myocardial tissue was weighed and
the mitochondria were extracted using a mitochondria/cytosol
fractionation kit (cat no. ab65320; Abcam, Cambridge, MA, USA).
Tissues were divided into 0.5 cm3 fragments with
scissors and placed into a small volume glass homogenizer as
described previously (32). The
total volume of the tissue fragments was estimated, and 1.5 ml
pre-cooled mitochondria/cytosol fractionation buffer (Mito-Cyto
Buffer; Mitochondria Isolation kit; Applygen Technologies Inc.,
Beijing, China) was added. The tissues were placed into a grinder
and ground 20 times. The tissue homogenate was then transferred to
a centrifuge tube and centrifuged at 27 × g for 5 min at
4°C. Cell nuclei, membrane fragments and non-disintegrated cells
were pelleted. The supernatant liquid was collected, transferred to
a new centrifuge tube and centrifuged at 27 × g for 5 min at
4°C. The precipitate was discarded, and the supernatant liquid was
transferred to a new centrifuge tube, and centrifuged at 6099 ×
g for 10 min at 4°C. The supernatant fluid containing
cytoplasmic components was then transferred to a new centrifuge
tube, leaving behind the mitochondrial pellet. To wash the
mitochondrial pellet, liquid and fragments adhering to the inner
wall of the tube were first removed and 0.2 ml Mito-Cyto Buffer was
added to re-suspend the mitochondria precipitate, which was
centrifuged at 6099 × g for 10 min at 4°C. The supernatant
was discarded and the mitochondria precipitate was re-suspended
with Mito-Cyto Buffer (50 µl/100 mg tissue). After detecting
protein concentration using the Bradford method (33), unused mitochondria precipitates
were preserved immediately at −70°C. The concentration of samples
was determined using a Bradford Protein assay kit (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) according to the
manufacturer's instructions.
Bax, Bcl-2, and cyt-c protein levels
in mitochondria and cytoplasm
Samples (50 µg) were loaded onto 10% SDS-PAGE gels
with 8 µl prestained and 4 µl illuminable protein molecular weight
markers. Electrophoresis was performed and proteins were
transferred to polyvinyldene difluoride (PVDF) membranes. Membranes
were blocked with 5% skimmed milk for 1 h at room temperature, and
incubated overnight at 4°C with the following primary antibodies
diluted in 5% skimmed milk: Rabbit anti-Bax (1:1,500; catalog no.
sc-6236), mouse anti-Bcl-2 (1:1,000; catalog no. sc-7382), goat
anti-cyt-c (1:2,000; catalog no. sc-8385) and rabbit anti-GAPDH
(1:2,000; catalog no. sc-25778), obtained from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Membranes were washed and
incubated with horseradish peroxidase-conjugated goat anti-rabbit
IgG (1:2,000; catalog no. sc-2004), goat anti-mouse IgG (1:2,000;
catalog no. sc-2005) or donkey anti-goat IgG (1:2,000; catalog no.
sc-2033) secondary antibodies, obtained from Santa Cruz
Biotechnology, Inc., for 1 h at 37°C. Proteins were detected using
an Enhanced Chemiluminescence reagent (Bio-Rad Laboratories, Inc.)
by adding Detection Reagent 2 (1:1 concentration) to each 6×9 cm
PVDF membrane, allowing liquid to cover the film surface, and
incubating at room temperature for 1 min. The reaction liquid was
drained and PVDF film was isolated with preservative film and
placed in an exposure box for compressed exposure in the darkroom
before photos were taken. Gel images were analyzed using Quantity
One software version 4.62 (Bio-Rad Laboratories Inc.), and the
results were presented as integral absorbance values.
Statistical analysis
Data are presented as mean ± standard deviation.
Differences among the three treatment groups were tested using a
one-way analysis of variance (ANOVA). The least significant
difference post-hoc comparisons test was performed when the
corresponding one-way ANOVA test reached statistical significance.
Differences were considered to be statistically significant when
P<0.05. All statistical analyses were performed using SPSS
(version, 15.0; SPSS Inc., Chicago, IL, USA).
Results
Comparison of adenylic acid content
and EC in rat myocardial tissues
No significant differences in AMP and ADP levels
among groups were observed (Table
IA). The ATP level in the melatonin-treated group (group M) was
significantly higher than those of the control (group C) and
accelerated-aging (group D) group (0.068 vs. 0.052 and 0.058
µmol/g; P=0.002 and P=0.045, respectively). The TAN level of the
melatonin-treated group (M) was also higher when compared with the
control and accelerated-aging group (0.160 vs. 0.133 and 0.142
µmol/g, respectively; Table IA),
but only the difference between the melatonin-treated group and
control group reached statistical significance (P=0.011). The
energy charge (EC) of the melatonin-treated group was increased,
however this did not reach statistical significance when compared
with the control and accelerated-ageing groups (0.570 vs. 0.529 and
0.561 µmol/g; Table IA and
Fig. 1).
 | Table I.Comparison of adenylic acid levels
and cyt-c, Bax and Bcl-2 protein expression among the treatment
groups. |
Table I.
Comparison of adenylic acid levels
and cyt-c, Bax and Bcl-2 protein expression among the treatment
groups.
| A, Adenylic acid
content of myocardial tissues |
|---|
|
|---|
| Adenylic
acid/EC | Group C
(µmol/g) | Group M
(µmol/g) | Group D
(µmol/g) |
|---|
| AMP | 0.044±0.005 | 0.044±0.011 | 0.041±0.009 |
| ADP | 0.032±0.007 | 0.037±0.006 | 0.035±0.003 |
| ATP | 0.052±0.009 |
0.068±0.012a |
0.058±0.008b |
| TAN | 0.133±0.027 |
0.160±0.020a | 0.142±0.017 |
| EC | 0.529±0.056 | 0.570±0.049 | 0.561±0.028 |
|
| B, Cyt-c protein
expression in myocardial mitochondria and cytoplasm |
|
| Fraction | Group C (IOD) | Group M (IOD) | Group D (IOD) |
|
| Cytoplasmic | 2.192±0.164 | 2.228±0.392 |
2.892±0.609a,b |
| Mitochondria | 1.067±0.209 | 0.999±0.226 | 1.055±0.288 |
|
Cytoplasmic/mitochondria | 2.134±0.488 | 2.272±0.359 |
2.838±0.635a,b |
|
| C, Protein
expression in myocardial mitochondria |
|
|
|
|
| Protein | Group C (IOD) | Group M (IOD) | Group D (IOD) |
|
| Bax | 0.809±0.178 | 0.674±0.099 | 0.687±0.158 |
| Bcl-2 |
1.217±0.218 |
0.959±0.166a |
0.780±0.194a,b |
| Bcl-2/Bax |
1.540±0.293 | 1.435±0.251 |
1.162±0.265a,b |
Comparison of Bax, Bcl-2 and cyt-c
protein levels in myocardial tissue mitochondria and cytoplasm
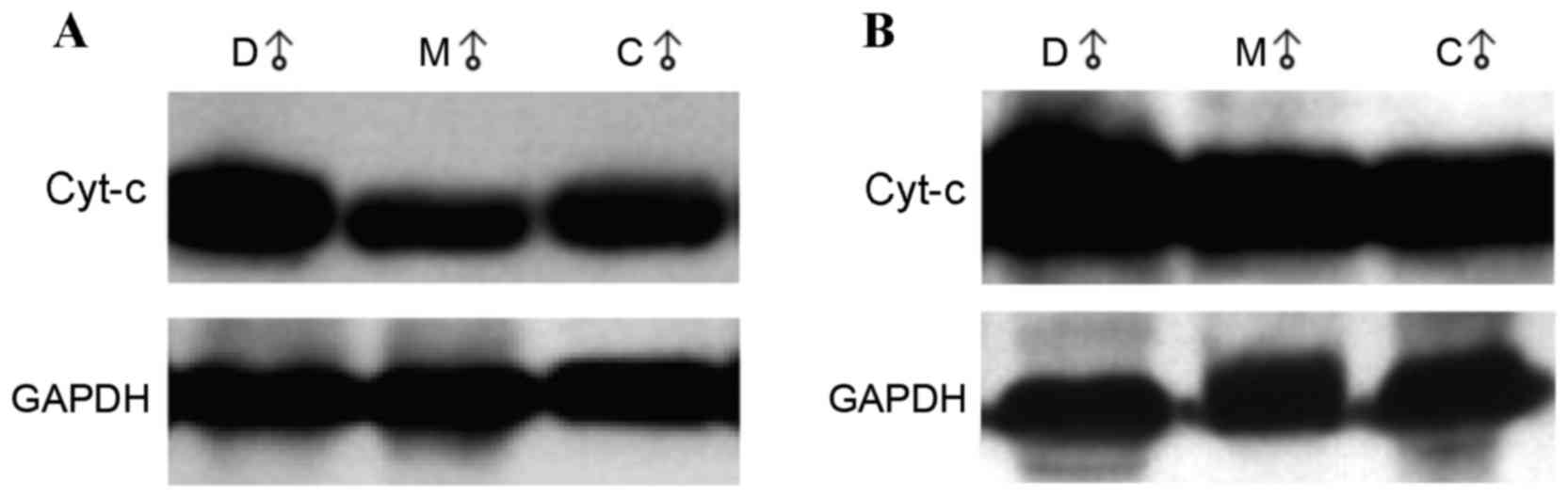
Cyt-c protein expression in the cytoplasm was
significantly higher in the accelerated-aging group compared with
the control and melatonin-treated groups (2.892 vs. 2.192 and
2.228; P=0.001 and P=0.002, respectively; Table IB). The representative western blot
images are shown in Fig. 2A. No
significant differences in the protein expression levels of cyt-c
in mitochondria were observed among groups (Table IB). The representative western blot
images are shown in Fig. 2B. The
cytoplasmic/mitochondrial ratio of cyt-c expression was
significantly higher in the accelerated aging group compared with
that of the control and melatonin-treated groups (2.838 vs. 2.134
and 2.272; P=0.004 and P=0.019, respectively; Table IB).
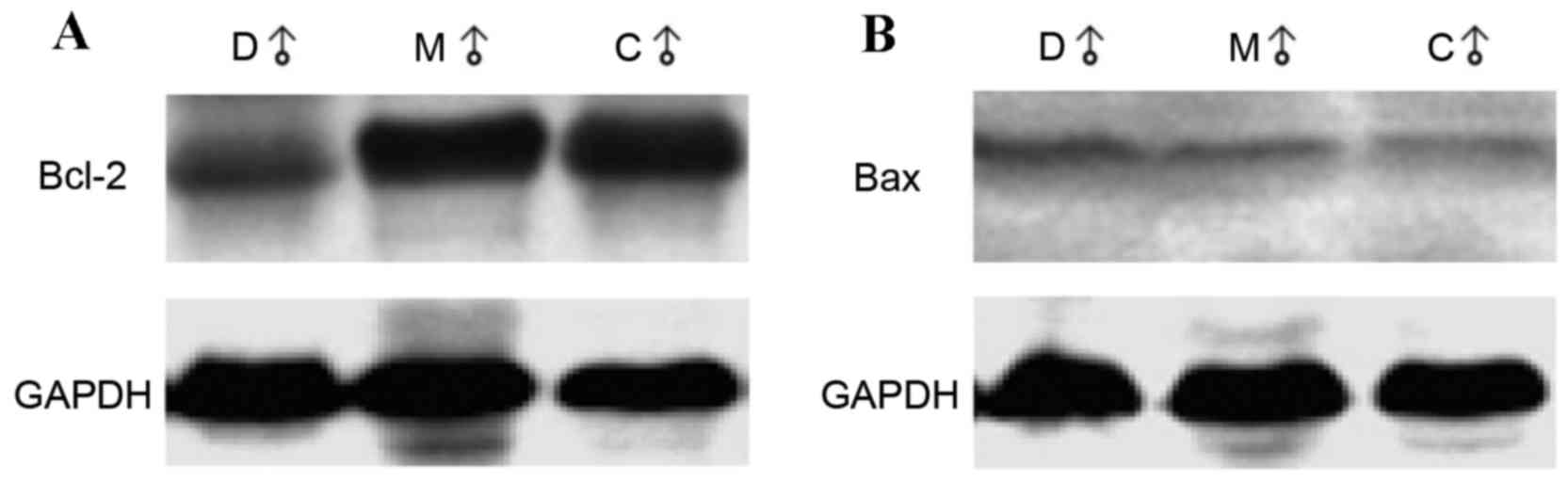
Rats in the melatonin-treated and accelerated-aging
groups demonstrated a significant reduction in Bcl-2 expression
levels when compared with the control group (0.959 and 0.780 vs.
1.217; P=0.006 and P<0.001, respectively; Table IC). The representative western blot
images are shown in Fig. 3A. Bcl-2
levels in the accelerated-aging group were significantly lower than
in the melatonin-treated group (0.780 vs. 0.959; P=0.0497; Table IC). No significant differences in
Bax protein levels were observed among groups. The representative
western blot images are shown in Fig.
3B. The ratio between Bcl-2 and Bax in the control group and
melatonin-treated group were significantly higher than that in the
accelerated-aging group (1.540 and 1.435 vs. 1.162; P=0.004 and
P=0.032, respectively; Table
IC).
Discussion
The results of the present study suggest that
melatonin exerts a beneficial anti-aging effect on myocardial
tissues in rats via several possible mechanisms. ATP levels were
significantly higher in aged rats receiving melatonin when compared
with untreated aged rats and normal controls. TAN levels were
higher in the melatonin-treated group when compared with the other
groups, and were significantly higher than the controls. The
protein expression levels of cyt-c in the cytoplasm was
significantly higher in the accelerated aging group when compared
with the other two groups, however its expression in the
mitochondria was not significantly increased. Whilst Bcl-2 protein
levels were significantly lower in the melatonin-treated and
accelerated-aging groups compared with the controls, Bax protein
expression was not significantly different among the groups.
However, Bcl-2 and Bax ratios were significantly higher in the
control and melatonin-treated groups compared with the
accelerated-aging group.
The results of the present study are consistent with
those of previous studies that have demonstrated efficacy of short-
and long-term melatonin treatment in preventing age-associated
mitochondrial oxidative stress (6). Age-dependent oxidative alterations in
rat heart mitochondria were associated with reduced activity of the
electron transport chain complex and reduced ATP levels; while ATP
levels increased and the bioenergetic status was restored following
chronic melatonin treatment (2).
It has been demonstrated that JAK2/STAT3 activation by melatonin
attenuates mitochondrial oxidative damage induced by myocardial
ischemia/reperfusion (34).
Similarly, melatonin treatment counteracted increased lipid
peroxidation and reduced glutathione in the mitochondria of SAM
mice (6). In addition, the
function of brain mitochondria was maintained by melatonin therapy
by preventing impairment of the mitochondria and increasing ATP
production (35). Melatonin
treatment increased ATP production in rat brain and liver
mitochondria in controls and counteracted cyanide-induced
inhibition of ATP synthesis, suggesting that mitochondrial
homeostasis may be integral to the anti-aging and neuroprotective
properties of melatonin (36).
In the present study, the energy status of myocytes
(by EC measurement) was increased in the melatonin-treated group;
however, this did not reach statistical significance. ATP, ADP and
AMP constitute a cellular energy-containing state in which an
abundance of cellular ATP indicates higher energy levels, while an
abundance of cellular AMP suggests lower energy levels (37). Mitochondrial production of ATP is a
multi-enzyme process requiring oxygen; however, when ROS inhibits
the electron transport chain, mitochondria have insufficient oxygen
and ATP decreases (5). This may
provide an explanation for the aging mechanism and the development
of age-associated degenerative disease. In the present study, ATP
and TAN values were significantly higher in melatonin-treated rats
compared with controls and those subjected to accelerated ageing.
EC values were also higher in melatonin-treated rats when compared
with the other groups, however this did not reach statistical
significance. This may be explained by a decrease in mitochondrial
function during ageing, possibly accompanied by reduced numbers of
mitochondria, precluding the maintenance of energy levels. In a
previous study, melatonin was observed to enhance myocardial
mitochondria generation in aged myocardium, suggesting that its
anti-aging effects may protect the integrity of the mitochondrial
structure and maintain the number and function of the mitochondria
(38). The antioxidant activity of
melatonin may be of particular interest in late-onset
neurodegenerative diseases, such as Alzheimer's disease and
Parkinson's disease, due to the vulnerability of the nervous system
to oxidative damage and neoplastic disease (26). In addition, a previous study
suggested that melatonin exerts oncostatic actions against tumor
development and growth (39). The
results of the present study suggest that the effects of melatonin
are most likely to be a consequence of free-radical scavenging
activity at the level of the mitochondria. Melatonin metabolites
are also free radical scavengers. Galano et al (40) reported continuous protection
exerted by melatonin metabolites through a free radical scavenging
cascade. Melatonin efficiently protects against oxidative stress
via immune-enhancing, anti-inflammatory and homeostatic activities
in the mitochondria, as well as inhibiting cancer progression
(41).
Cyt-c transfers mitochondrial electrons from the
cyt-c reductase complex to the cyt-c oxidase complex, suggesting
that intracellular cyt-c reduction leads to electron transfer
dysfunction, which subsequently leads to ROS generation and reduced
ATP production (42). Release of
cyt-c from the mitochondria to cytoplasm initiates the apoptosis
process (43). In the present
study, no significant differences in the protein expression levels
of cyt-c in the mitochondria and cytoplasm among groups were
observed; however the ratio of cyt-c in cytoplasm vs. the
mitochondria in untreated aged rats was significantly higher than
the ratio in control and melatonin-treated groups. This suggests
that melatonin may maintain the electron transport chain by
elevating cyt-c thereby preserving normal oxidative phosphorylation
in the mitochondria and decreasing electron leakage. This
mechanism, along with suppressed transfer of cyt-c from the
mitochondria to the cytoplasm, may reduce apoptosis, protect
mitochondrial function and slow myocardial aging. Recently, Yang
et al (34) demonstrated
that melatonin conferred a cardio-protective effect by improving
post-ischemic cardiac function, decreasing infarct size, reducing
the apoptotic index, diminishing release of lactate dehydrogenase,
upregulating Bcl-2 and downregulating Bax.
In the present study, Bcl-2 expression was higher in
the control group compared with the other groups, and in the
melatonin-treated compared with untreated accelerated-aging groups.
In addition, Bax protein levels were not significantly different
among groups. However, the Bcl-2/Bax ratio in control and
melatonin-treated groups was significantly higher than that in the
untreated accelerated aging group. Release of cyt-c from the
mitochondria to the cytoplasm is modulated by the Bcl-2 family,
where Bcl-2 suppresses release of cyt-c, and Bax facilitates cyt-c
release (44). The increase of
Bcl-2/Bax in the melatonin-treated group implies reduced release of
cyt-c in the mitochondria. This suggests that melatonin may reduce
the release of cyt c from mitochondria to the cytoplasm via
modulation of the Bcl-2 family. Melatonin modulates the expression
of the Bcl-2 family, which may also prevent translocation of cyt-c
from the mitochondria to the cytoplasm.
The results of the current study are limited by the
small sample size and the short duration of study. In addition,
chronic melatonin use was not evaluated. Rats were sacrificed
early, which did not allow measurement of the effects of melatonin
on rat survival and age-related changes in the heart over time. In
addition, the molecular characteristics of melatonin such as its
interaction with lipids and the stabilization of mitochondrial
membranes were not examined. This may have enabled the elucidation
of the mechanism of melatonin in protecting the mitochondria. In
order to evaluate of the effects of long-term melatonin treatment,
future studies will include a larger sample size with rats aged
over 10 months that will be observed over a longer study period.
The specific actions of melatonin on the electron transport chain
and its complex activity will also be investigated, and its role in
maintaining intramitochondrial homeostasis will be compared with
other antioxidants (e.g. N-acetylcysteine).
In conclusion, the results of the present study
suggest that melatonin exhibits a protective effect on
mitochondrial function in a rat model of accelerated aging.
Melatonin may maintain the electron transfer chain by increasing
mitochondrial cyt-c levels, thus ensuring normal oxidative
phosphorylation, maintaining ATP production and decreasing electron
leakage and free radical production. The results of the current and
future studies may increase our understanding of the anti-aging
effects of melatonin on the mitochondria, thus providing more
information about the aging process as well as strategies for
anti-aging or treatments for age-associated diseases.
Acknowledgements
All authors appreciate the great contribution of
Mrs. Shi-Wen Wang (Institute of Geriatric Cardiology, Chinese PLA
General Hospital) to the manuscript before she passed away.
Glossary
Abbreviations
Abbreviations:
|
AGEs
|
advanced glycation end products
|
|
cyt-c
|
cytochrome c
|
|
EC
|
energy charge
|
|
SD
|
Sprague-Dawley
|
|
TAN
|
total adenylic acid number
|
References
|
1
|
Harman D: The free radical theory of
aging. Antioxid Redox Signal. 5:557–561. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rodríguez MI, Carretero M, Escames G,
López LC, Maldonado MD, Tan DX, Reiter RJ and Acuña-Castroviejo D:
Chronic melatonin treatment prevents age-dependent cardiac
mitochondrial dysfunction in senescence-accelerated mice. Free
Radic Res. 41:15–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takeda T: Senescence-accelerated mouse
(SAM): A biogerontological resource in aging research. Neurobiol
Aging. 20:105–110. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rizvi SI and Jha R: Strategies for the
discovery of anti-aging compounds. Expert Opin Drug Discov.
6:89–102. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Acuña-Castroviejo D, Martín M, Macías M,
Escames G, León J, Khaldy H and Reiter RJ: Melatonin, mitochondria,
and cellular bioenergetics. J Pineal Res. 30:65–74. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rodriguez MI, Escames G, López LC, García
JA, Ortiz F, López A and Acuña-Castroviejo D: Melatonin
administration prevents cardiac and diaphragmatic mitochondrial
oxidative damage in senescence-accelerated mice. J Endocrinol.
194:637–643. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Escames G, Lopez A, García JA, García L,
Acuña-Castroviejo D, García JJ and López LC: The role of
mitochondria in brain aging and the effects of melatonin. Curr
Neuropharmacol. 8:182–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Petrosillo G, Moro N, Paradies V, Ruggiero
FM and Paradies G: Increased susceptibility to Ca(2+)-induced
permeability transition and to cytochrome c release in rat heart
mitochondria with aging: Effect of melatonin. J Pineal Res.
48:340–346. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Judge S and Leeuwenburgh C: Cardiac
mitochondrial bioenergetics, oxidative stress, and aging. Am J
Physiol Cell Physiol. 292:C1983–C1992. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lesnefsky EJ, Moghaddas S, Tandler B,
Kerner J and Hoppel CL: Mitochondrial dysfunction in cardiac
disease: Ischemia-reperfusion, aging, and heart failure. J Mol Cell
Cardiol. 33:1065–1089. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee SJ, Jin Y, Yoon HY, Choi BO, Kim HC,
Oh YK, Kim HS and Kim WK: Ciclopirox protects mitochondria from
hydrogen peroxide toxicity. Br J Pharmacol. 145:469–476.
2005.PubMed/NCBI
|
|
12
|
Poeggeler B, Durand G, Polidori A,
Pappolla MA, Vega-Naredo I, Coto-Montes A, Böker J, Hardeland R and
Pucci B: Mitochondrial medicine: Neuroprotection and life extension
by the new amphiphilic nitrone LPBNAH acting as a highly potent
antioxidant agent. J Neurochem. 95:962–973. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schapira AH: Mitochondrial involvement in
Parkinson's disease, Huntington's disease, hereditary spastic
paraplegia and Friedreich's ataxia. Biochim Biophys Acta.
1410:159–170. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Barlow-Walden LR, Reiter RJ, Abe M, Pablos
M, Menendez-Pelaez A, Chen LD and Poeggeler B: Melatonin stimulates
brain glutathione peroxidase activity. Neurochem Int. 26:497–502.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tan DX, Chen LD, Poeggeler B, Manchester
LC and Reiter RJ: Melatonin: A potent endogenous hydroxyl radical
scavenger. Endocrine J. 1:57–60. 1993.
|
|
16
|
Hardeland R: Melatonin and the theories of
aging: A critical appraisal of melatonin's role in antiaging
mechanisms. J Pineal Res. 55:325–356. 2013.PubMed/NCBI
|
|
17
|
Acuna-Castroviejo D, Escames G, Rodriguez
MI and Lopez LC: Melatonin role in the mitochondrial function.
Front Biosci. 12:947–963. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Reiter RJ: Oxidative processes and
antioxidative defense mechanisms in the aging brain. FASEB J.
9:526–533. 1995.PubMed/NCBI
|
|
19
|
Reiter RJ, Tan DX, Mayo JC, Sainz RM, Leon
J and Czarnocki Z: Melatonin as an antioxidant: Biochemical
mechanisms and pathophysiological implications in humans. Acta
Biochim Pol. 50:1129–1146. 2003.PubMed/NCBI
|
|
20
|
Okatani Y, Wakatsuki A, Shinohara K,
Taniguchi K and Fukaya T: Melatonin protects against oxidative
mitochondrial damage induced in rat placenta by ischemia and
reperfusion. J Pineal Res. 31:173–178. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Okatani Y, Wakatsuki A, Reiter RJ and
Miyahara Y: Melatonin reduces oxidative damage of neural lipids and
proteins in senescence-accelerated mouse. Neurobiol Aging.
23:639–644. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lardone PJ, Alvarez-García O,
Carrillo-Vico A, Vega-Naredo I, Caballero B, Guerrero JM and
Coto-Montes A: Inverse correlation between endogenous melatonin
levels and oxidative damage in some tissues of SAM P8 mice. J
Pineal Res. 40:153–157. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bubenik GA and Konturek SJ: Melatonin and
aging: Prospects for human treatment. J Physiol Pharmacol.
62:13–19. 2011.PubMed/NCBI
|
|
24
|
Reiter RJ, Richardson BA, Johnson LY,
Ferguson BN and Dinh DT: Pineal melatonin rhythm: Reduction in
aging Syrian hamsters. Science. 210:1372–1373. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Reiter RJ, Craft CM, Johnson JE Jr, King
TS, Richardson BA, Vaughan GM and Vaughan MK: Age-associated
reduction in nocturnal pineal melatonin levels in female rats.
Endocrinology. 109:1295–1297. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Karasek M: Melatonin, human aging, and
age-related diseases. Exp Gerontol. 39:1723–1729. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wei H, Li L, Song Q, Ai H, Chu J and Li W:
Behavioural study of the D-galactose induced aging model in
C57BL/6J mice. Behav Brain Res. 157:245–251. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Song X, Bao M, Li D and Li YM: Advanced
glycation in D-galactose induced mouse aging model. Mech Ageing
Dev. 108:239–251. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Deng HB, Cheng CL, Cui DP, Li DD, Cui L
and Cai NS: Structural and functional changes of immune system in
aging mouse induced by D-galactose. Biomed Environ Sci. 19:432–438.
2006.PubMed/NCBI
|
|
30
|
Hammond JB and Kruger NJ: The bradford
method for protein quantitation. Methods Mol Biol. 3:25–32.
1988.PubMed/NCBI
|
|
31
|
Khlyntseva SV, Bazel YR, Vishnikin AB and
Andruch V: Methods for the determination of adenosine triphosphate
and other adenine nucleotides. J Analytic Chem. 64:657–673. 2009.
View Article : Google Scholar
|
|
32
|
Lee SD, Kuo WW, Ho YJ, Lin AC, Tsai CH,
Wang HF, Kuo CH, Yang AL, Huang CY and Hwang JM: Cardiac
Fas-dependent and mitochondria-dependent apoptosis in
ovariectomized rats. Maturitas. 61:268–277. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Karyn M Usher, Steven W Hansen, Jennifer S
Amoo, Allison P Bernstein and Mary Ellen P. McNally: Precision of
internal standard and external standard methods in high performance
liquid chromatography. LCGC Special Issues. 33:40–46. 2015.
|
|
34
|
Yang Y, Duan W, Jin Z, Yi W, Yan J, Zhang
S, Wang N, Liang Z, Li Y, Chen W, et al: JAK2/STAT3 activation by
melatonin attenuates the mitochondrial oxidative damage induced by
myocardial ischemia/reperfusion injury. J Pineal Res. 55:275–286.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Carretero M, Escames G, López LC, Venegas
C, Dayoub JC, García L and Acuña-Castroviejo D: Long-term melatonin
administration protects brain mitochondria from aging. J Pineal
Res. 47:192–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Martín M, Macías M, León J, Escames G,
Khaldy H and Acuña-Castroviejo D: Melatonin increases the activity
of the oxidative phosphorylation enzymes and the production of ATP
in rat brain and liver mitochondria. Int J Biochem Cell Biol.
34:348–357. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Atkinson DE and Fall L: Adenosine
triphosphate conservation in biosynthetic regulation. Escherichia
coli phosphoribosylpyrophosphate synthase. J Biol Chem.
242:3241–3242. 1967.PubMed/NCBI
|
|
38
|
Lopez LC, Escames G, Ortiz F, Ros E and
Acuña-Castroviejo D: Melatonin restores the mitochondrial
production of ATP in septic mice. Neuro Endocrinol Lett.
27:623–630. 2006.PubMed/NCBI
|
|
39
|
Karasek M, Reiter RJ, Cardinali DP and
Pawlikowski M: Future of melatonin as a therapeutic agent. Neuro
Endocrinol Lett. 23:(Suppl 1). S118–S121. 2002.
|
|
40
|
Galano A, Tan DX and Reiter RJ: On the
free radical scavenging activities of melatonin's metabolites, AFMK
and AMK. J Pineal Res. 54:245–257. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Galano A, Tan DX and Reiter RJ: Melatonin
as a natural ally against oxidative stress: A physicochemical
examination. J Pineal Res. 51:1–16. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fang X, Chen M and Chen R: Cytochrome C
and Apoptosis. J Foreign Medical Sciences (Clinical Biochemistry
and Laboratory Medicine). 26:43–46. 2005.
|
|
44
|
Shimizu S, Narita M and Tsujimoto Y: Bcl-2
family proteins regulate the release of apoptogenic cytochrome c by
the mitochondrial channel VDAC. Nature. 399:483–487. 1999.
View Article : Google Scholar : PubMed/NCBI
|