Introduction
Circadian rhythms are endogenous, cyclical 24-h
variations that are associated with numerous physiological
processes. These rhythms are generated by circadian clocks, which
are located in most cell types, including cells of the
cardiovascular system (1,2). The mammalian circadian clock consists
of a network of transcriptional and translational feedback loops
(3). Two basic
helix-loop-helix-PAS domain transcription factors, circadian
locomotor output cycle protein kaput (CLOCK) and brain and muscle
Arnt-like protein 1, are at the core of the major circadian loop.
Clock-controlled genes are able to encode various proteins, and
they markedly influence cellular functions.
Aberrations in the circadian clock system have
pathological consequences. Disruptions to the circadian rhythm and
defects in circadian clock genes are known to be associated with
certain human malignancies (4,5).
Previous studies have indicated that biological oscillations driven
by the circadian clock are able to influence endothelial
dysfunction, pathological vascular remodeling and thrombosis, which
may ultimately result in vascular disease (6,7). Our
previous study revealed that the upregulation of CLOCK in the
vessel walls of veins may be involved in the pathogenesis and
progression of venous disease (8).
However, the exact molecular mechanism underlying this circadian
control and the induction of inflammation remain largely
unknown.
Nuclear factor-κB (NF-κB) belongs to a family of
constitutive and inducible transcription factors that act through
diverse target genes. The NF-κB pathway serves an important role in
controlling the response to cellular stress, and it is also
involved in cell division, transformation, survival, apoptosis,
inflammation and immunity (9–11).
Transcription factor p65 is a member of the NF-κB family, which has
an important function in inflammation and the immune response
(12). Research over the past
decade has identified novel molecular links between the circadian
clock and the NF-κB pathway (13,14).
Our previous study demonstrated that CLOCK may activate the NF-κB
pathway (14); however, in spite
of the substantial evidence supporting the existence of crosstalk
between the circadian clock and the NF-κB pathway, further
investigation is required to identify the associations and
mechanisms. The present study aimed to evaluate the crucial role of
the NF-κB pathway in the CLOCK-induced inflammatory response at the
molecular level.
Materials and methods
Cell culture and treatment
Human umbilical vein endothelial cells (HUVECs) were
purchased from the American Type Culture Collection (Manassas, VA,
USA) and maintained using an Endothelial Cell Growth Medium-2
BulletKit (Lonza Group AG, Basel, Switzerland) containing 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C in an
atmosphere containing 5% CO2 and 95% room air. The Xvivo
Closed Incubation system (Xvivo system 300C; BioSpherix, Parish,
NY, USA) was used to accurately maintain different oxygen tensions
in different chambers. Pyrrolidine dithiocarbamate (PDTC), an
inhibitor of NF-κB, was purchased from Sigma-Aldrich (Merck
Millipore, Darmstadt, Germany) and a stock solution (200 mmol) was
prepared using distilled water. After 24 h of culture, the cells
were divided into separate chambers with 5% O2 and
further incubated for 12 or 24 h; subsequently, they were harvested
for measurement.
Preparation of the retroviral
vector
Gain- and loss-of-function of human CLOCK (hCLOCK)
was established as previously described (15). The hCLOCK-coding sequence was
amplified and subcloned into the pGV186 retroviral vector (GeneChem
Co., Ltd., Shanghai, China). The short hairpin RNA (shRNA)
targeting hCLOCK was designed and constructed by Shanghai GeneChem
Co., Ltd. (Shanghai, China). Vector and shRNA sequences, as well as
successful insertions were confirmed by DNA sequencing (Biosune
Biotechnology Co., Ltd., Shanghai, China). Scramble control (SCR)
served as a control for hCLOCK knockdown. For the production of
viral particles, 293T cells were co-transfected with the lentiviral
vectors, psPAX2 and pMD2.G (Addgene, Inc., Cambridge, MA, USA)
using X-tremeGENE Transfection Reagent (Roche Diagnostics, Basel,
Switzerland), according to the manufacturer's protocol. The viral
supernatant was collected 48 h post-transfection and passed through
a 0.45-µm filter (Sartorius AG, Göttingen, German). HUVECs were
infected with lentiviral particles containing 8 µg/ml Polybrene
Transfection Reagent (Merck Millipore, Darmstadt, Germany). Stable
overexpression or knockdown of hCLOCK was confirmed by western blot
analysis. After 24 h of cultivation in conventional cell culture,
the cells were divided into separate chambers containing 5%
O2 and incubated for 12 or 24 h prior to harvesting.
Enzyme-linked immunosorbent assay
(ELISA)
The expression levels of proinflammatory cytokines,
including interleukin (IL)-1β (catalog no. DLB50), IL-6 (catalog
no. D6050) and tumor necrosis factor-α (TNF-α; catalog no. DTA00C),
in the supernatants were measured using commercial ELISA kits
(R&D Systems Inc., Minneapolis, MN USA) according to the
manufacturer's protocol.
Western blot analysis
The relative expression levels of hCLOCK, IL-1β,
IL-6, intercellular adhesion molecule 1 (ICAM-1), cyclooxygenase 2
(COX-2), TNF-α, phosphorylated-p65 (p-p65) and β-actin were
measured by western blot analysis using standard methods, as
previously described (15).
Briefly, total cell proteins were extracted using
Radioimmunoprecipitation Assay Reagent (Cell Signaling Technology,
Inc., Danvers, MA, USA), and concentrations were measured using the
Bicinchoninic Acid Protein Assay kit (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Equal amounts of protein (50 µg) were separated
by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
and transferred to polyvinylidene difluoride membranes (Bio-Rad
Laboratories, Inc.). Membranes were blocked with 5% skimmed milk in
TBS containing 0.1% Tween-20 at room temperature for 1 h. A mouse
monoclonal anti-hCLOCK (1:500; catalog no. ab98948) antibody was
purchased from Abcam (Cambridge, MA, USA). Rabbit polyclonal
anti-IL-6 antibody (1:1,000; catalog no. 21865-1-AP) was purchased
from Proteintech Group, Inc. (Chicago, IL, USA). Rabbit polyclonal
anti-COX2 (1:1,000; catalog no. 4842), rabbit polyclonal anti-IL-1β
(1:1,000; catalog no. 12703), rabbit polyclonal anti-ICAM-1
(1:1,000; catalog no. 4915), rabbit monoclonal anti-TNF-α (1:1,000;
catalog no. 6945) and rabbit monoclonal anti-p-NF-κB p65 (Ser536;
1:1,000; clone, 93H1, catalog no. 3033) antibodies were purchased
from Cell Signaling Technology, Inc. Mouse polyclonal anti-b-actin
antibody (1:1,000; catalog no. sc-47778) was purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA). Primary antibodies were
incubated with the membranes at 4°C overnight. The membranes were
subsequently incubated with horseradish peroxidase (HRP)-conjugated
goat anti-rabbit IgG (1:2,000; catalog no. CW0103S; CWBiotech,
Beijing, China) or goat anti-mouse IgG (1:2,000; catalog no.
CW0102S, CWBiotech) secondary antibodies at 4°C for 2 h. Bands were
visualized with Enhanced Chemiluminescence reagents (CWBiotech) and
the signal was detected using an ImageQuant™ LAS 4000 Mini
Biomolecular Imager (GE Healthcare Life Sciences, Chalfont, UK).
Band intensities were analyzed using ImageJ software version 1.47
(National Institutes of Health, Bethesda, MD, USA) and were
normalized to the band intensity of b-actin, a cell structure
protein.
Statistical analysis
All results are presented as the mean ± standard
deviation of at least three experiments that were performed in
triplicate. Statistical comparisons among groups were made using a
one-way analysis of variance and two-tailed student's t-test.
Statistical analysis was performed using SPSS 20.0 (IBM SPSS,
Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Effects of hypoxia on the expression
of inflammatory cytokines and the NF-κB signaling pathways
The present study measured the effects of a hypoxic
state on the expression levels of proinflammatory cytokines (IL-1β,
IL-6, ICAM-1, COX-2 and TNF-α) in HUVECs by western blot analysis
over a 24-h period. The relative expression levels of these
cytokines increased significantly at the 24-h time point in hypoxic
environments (Fig. 1A and B). In
addition, the expression of p-p65 increased with longer exposure to
hypoxia (Fig. 1A and B). These
results were confirmed by ELISA on the HUVEC supernatants at 24 h,
which indicated an increased concentration of IL-6 (Fig. 1C), IL-1β (Fig. 1D) and TNF-α (Fig. 1E). These findings demonstrated that
inflammatory cytokines and the NF-κB signaling pathway may be
involved in the hypoxic response.
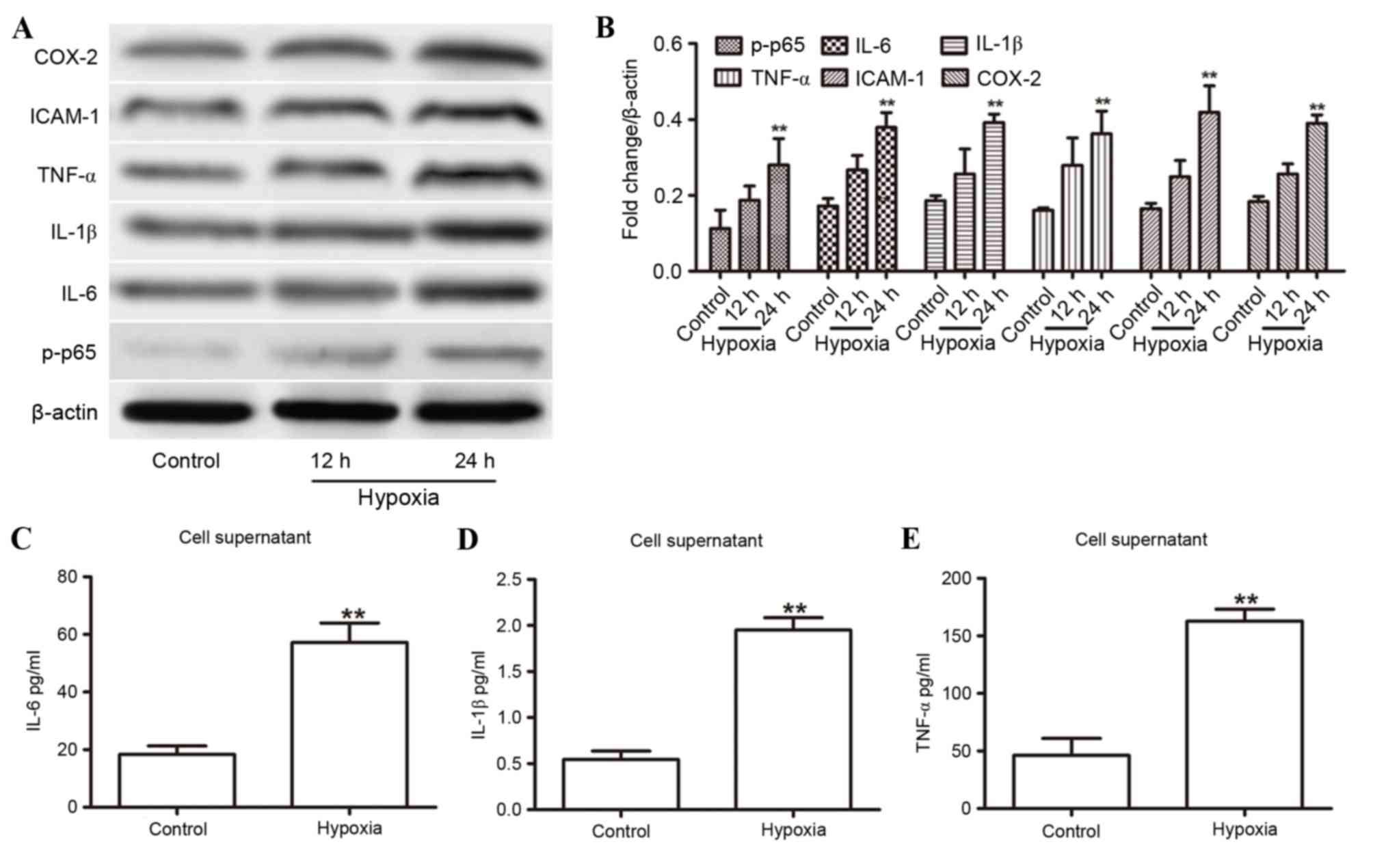 | Figure 1.Hypoxia-induced expression of
proinflammatory cytokines in HUVECs. (A) Western blot analysis of
IL-1β, IL-6, ICAM-1, COX-2, TNF-α and p-p65 expression in the
HUVECs for the indicated duration. (B) Expression levels were
normalized to β-actin. (C-E) ELISA analysis revealed increased
supernatant concentrations of (C) IL-6, (D) IL-1β and (E) TNF-α in
HUVECs with or without 24 h of hypoxia. **P<0.01 compared with
control. COX-2, cyclooxygenase 2; HUVECs, human umbilical vein
endothelial cells; ICAM-1, intercellular adhesion molecule 1; IL,
interleukin; p-p65, phosphorylated-p65; TNF-α, tumor necrosis
factor-α. |
Silencing CLOCK inhibits
hypoxia-induced inflammatory response augmentation
To confirm that hCLOCK is involved in
hypoxia-induced inflammatory responses, hCLOCK was inhibited and
the expression of inflammatory factors was evaluated. HUVECs that
had been transduced with either SCR or shRNA sequences targeting
hCLOCK (shCLOCK) were cultured under hypoxic conditions for 24 h.
IL-1β, IL-6, ICAM-1, COX-2, p-p65 and TNF-α were significantly
downregulated in the hypoxic environment when hCLOCK was knocked
down compared with the SCR group (P<0.05; Fig. 2A and B), albeit the levels were
still higher than those of the control group. There was a
statistically significant increase in the relative IL-6, TNF-α and
IL-1β levels in hypoxic HUVEC supernatants compared with the
normoxic control cells; however, levels were reduced in the shCLOCK
group (P<0.05; Fig. 2C-E).
These findings indicated that hCLOCK is directly involved in
activation of the NF-κB signaling pathway.
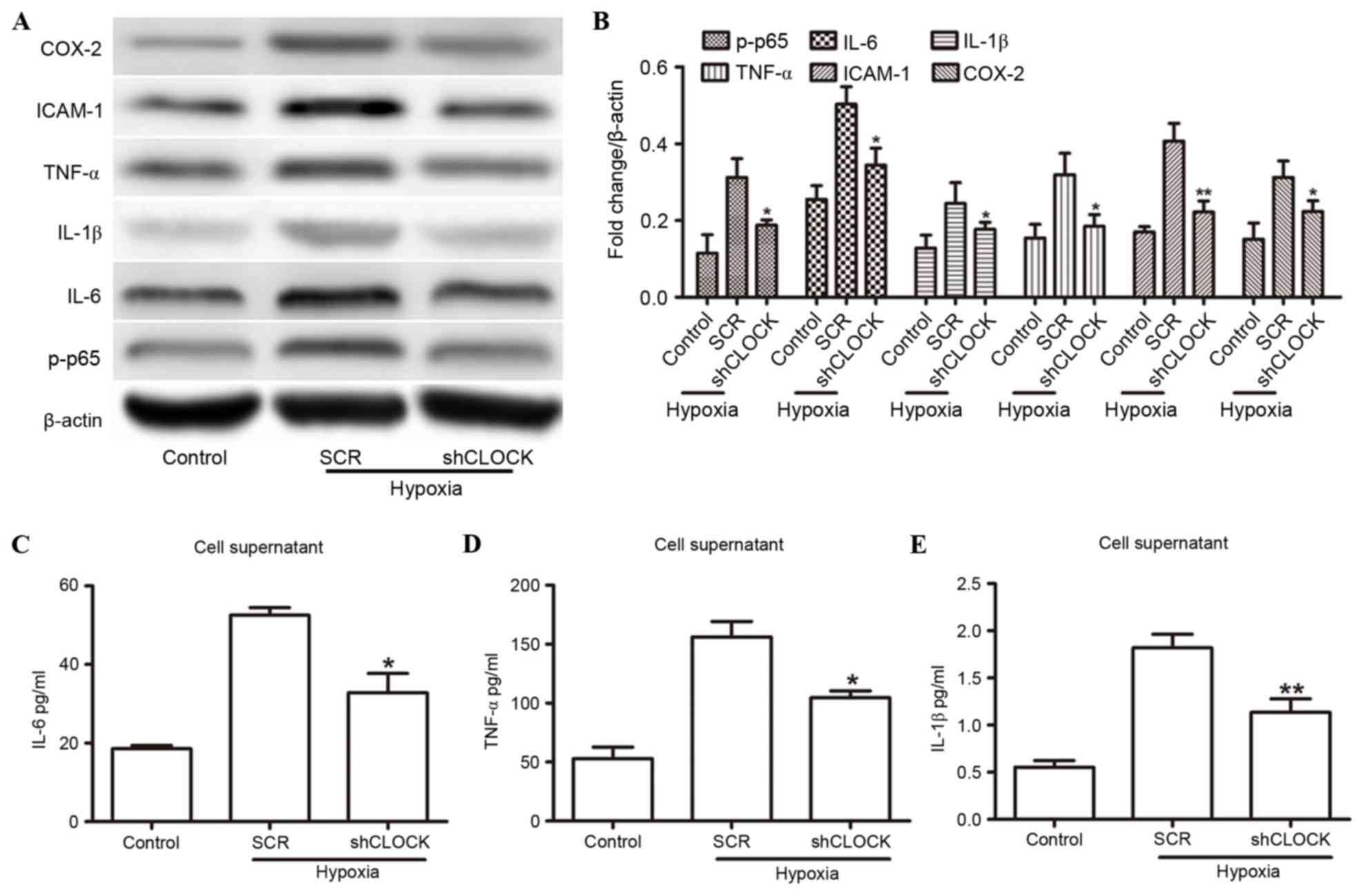 | Figure 2.hCLOCK mediated the hypoxia-induced
activation of NF-κB. (A) Western blot analysis of NF-κB pathway
effectors IL-1β, IL-6, ICAM-1, COX-2, TNF-α and p-p65, in HUVECs
that were transduced with SCR or shCLOCK vector under hypoxic
conditions. (B) Expression levels are normalized to β-actin. (C-E)
ELISA analysis revealed the expression levels of (C) IL-6, (D)
TNF-α and (E) IL-1β in the supernatants from HUVECs that were
transduced with SCR or shCLOCK vector. *P<0.05 and **P<0.01
compared to SCR. COX-2, cyclooxygenase 2; hCLOCK, human circadian
locomotor output cycle protein kaput; HUVECs, human umbilical vein
endothelial cells; ICAM-1, intercellular adhesion molecule 1; IL,
interleukin; NF-κB, nuclear factor-κB; p-p65, phosphorylated-p65;
SCR, scrambled control vector; shCLOCK, short hairpin RNA targeting
hCLOCK; TNF-α, tumor necrosis factor-α. |
Effects of PDTC on hypoxia-induced
inflammatory cytokine expression
The present study focused on the NF-κB signaling
pathway to determine whether NF-κB was a key factor in the
hypoxia-mediated inflammatory response. HUVECs were subjected to
hypoxia with or without exposure to the NF-κB inhibitor PDTC. As
expected, 20 µmol PDTC treatment for 24 h significantly inhibited
the expression levels of IL-1β, IL-6, ICAM-1, COX-2, TNF-α and
p-p65 compared with those treated with control (PBS) solution
(Fig. 3A and B). Similar results
were observed in ELISA analysis on the cell supernatants for IL-6
(Fig. 3C), TNF-α (Fig. 3D) and IL-1β (Fig. 3E). These findings indicated that
inhibition of NF-κB attenuates the expression of hypoxia-induced
inflammatory cytokines.
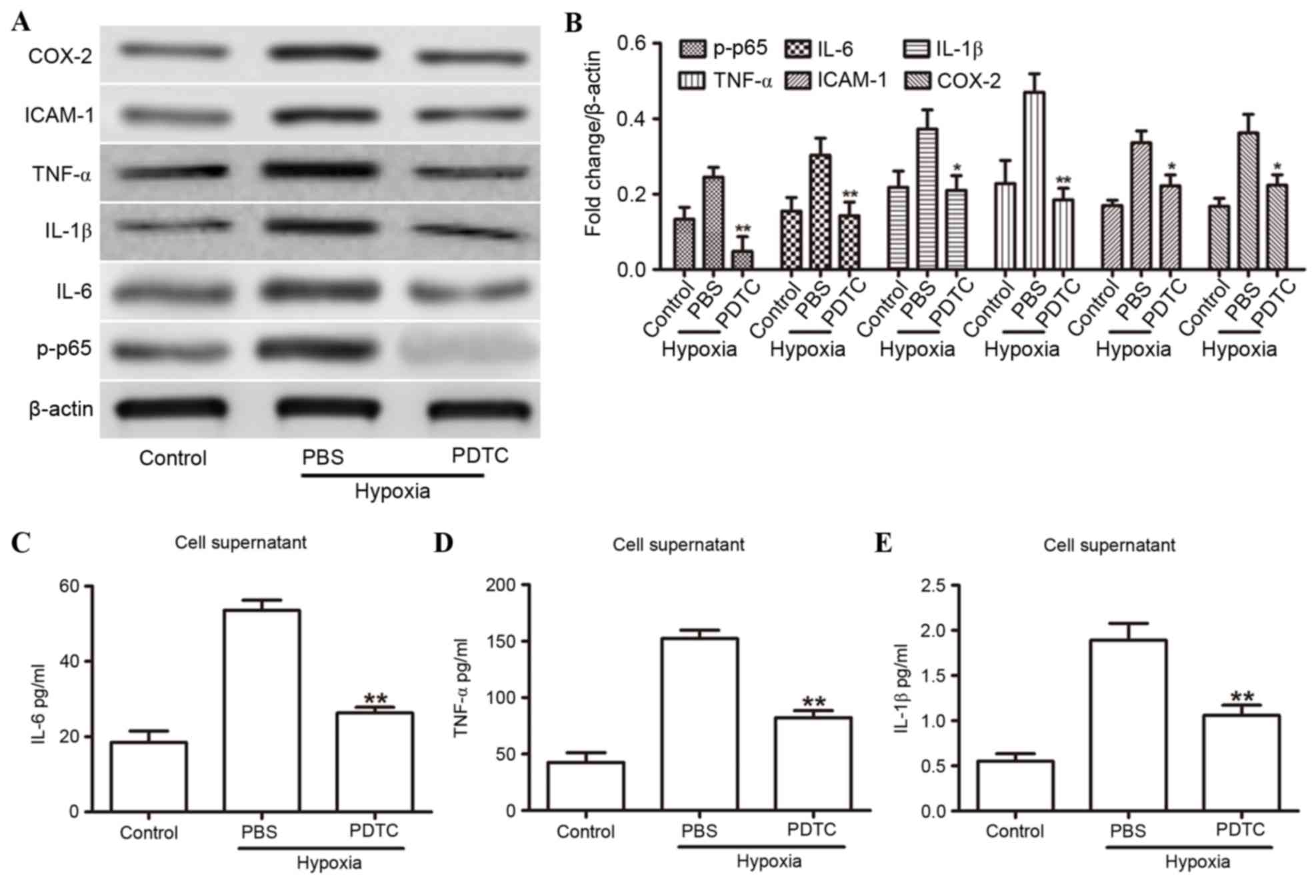 | Figure 3.NF-κB was involved in the
hypoxia-induced inflammatory response. (A) Western blot analysis of
IL-1β, IL-6, ICAM-1, COX-2, TNF-α and p-p65 in HUVECs that were
treated with either PBS or PDTC under hypoxic conditions. (B)
Expression levels are normalized to β-actin. (C-E) ELISA analysis
revealed the expression levels of (C) IL-6, (D) TNF-α and (E) IL-1β
in the supernatants from HUVECs that were treated with either 20
µmol PDTC or PBS under hypoxic conditions. *P<0.05 and
**P<0.01 compared to PBS. COX-2, cyclooxygenase 2; HUVECs, human
umbilical vein endothelial cells; ICAM-1, intercellular adhesion
molecule 1; IL, interleukin; NF-κB, nuclear factor-κB; p-p65,
phosphorylated-p65; PDTC, pyrrolidine dithiocarbamate; TNF-α, tumor
necrosis factor-α. |
NF-κB signaling is necessary for
CLOCK-induced inflammatory cytokine expression
To further investigate whether NF-κB was crucial to
the hCLOCK-mediated inflammatory response, the following culture
experiments were performed: i) HUVECs were transduced with the
control vector and treated with control solution (PBS); ii) HUVECs
were transduced with an hCLOCK-overexpressing vector and treated
with control solution; and iii) HUVECs were transduced with an
hCLOCK-overexpressing vector and treated with 20 µmol PDTC. The
hCLOCK-overexpressing group that was treated with PBS demonstrated
increased levels of IL-1β, IL-6, ICAM-1, COX-2, p-p65 and TNF-α
expression under hypoxic conditions (Fig. 4A-E). However, the expression levels
of IL-1β, IL-6, ICAM-1, COX-2, p-p65 and TNF-α expression levels
were markedly decreased when hCLOCK-overexpressing HUVECs were
treated with PDTC. These findings suggested that NF-κB signaling is
required for CLOCK-induced inflammatory cytokine expression.
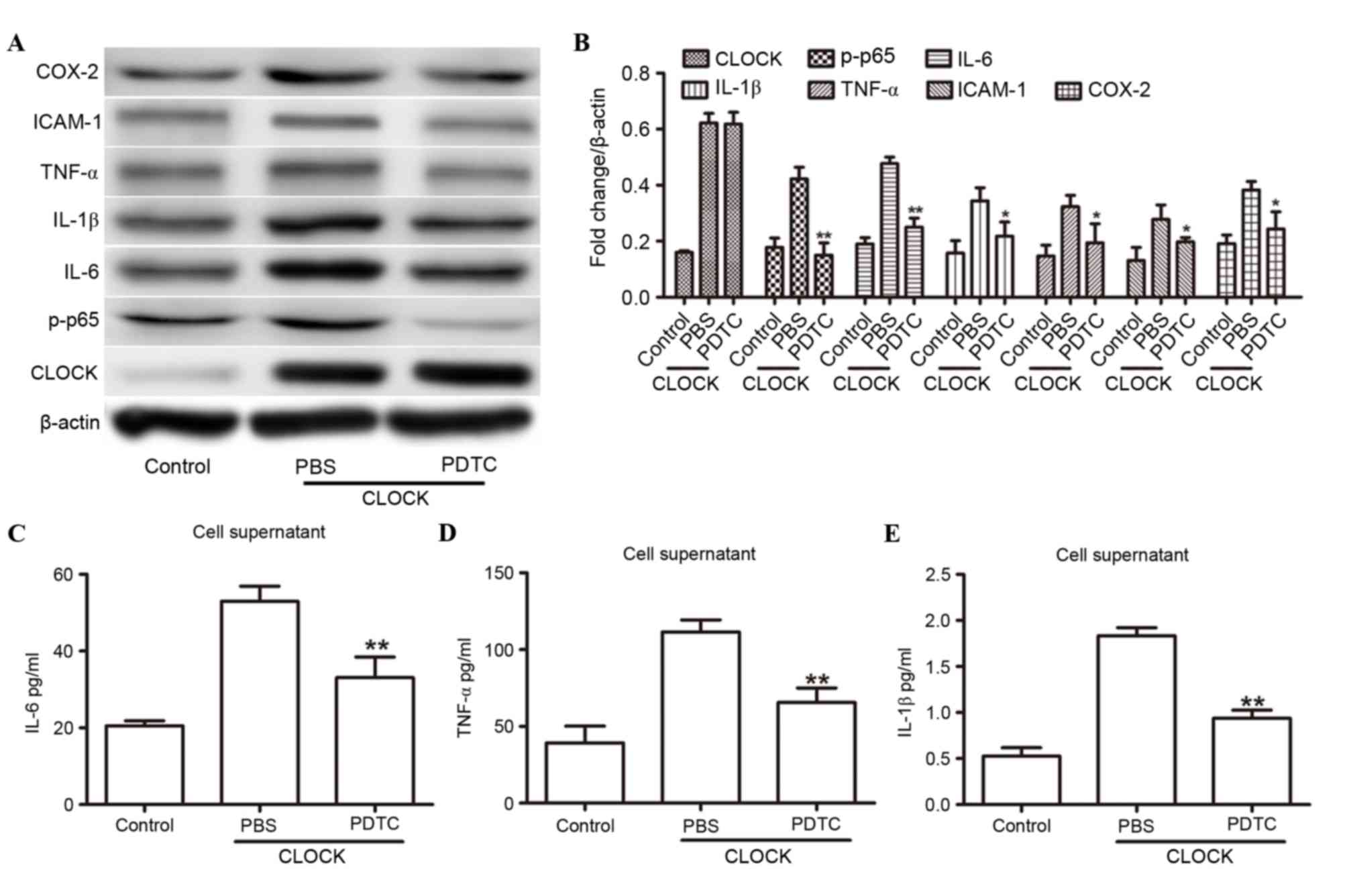 | Figure 4.NF-κB was involved in the
hCLOCK-induced inflammatory response. (A) Western blot analysis of
CLOCK, IL-1β, IL-6, ICAM-1, COX-2, TNF-α and p-p65 in HUVECs that
were transduced with the control or hCLOCK vector, and were then
treated with either PBS or 20 µmol PDTC. (B) Expression levels were
normalized to β-actin. (C-E) ELISA analysis revealed the expression
levels of (C) IL-6, (D) TNF-α and (E) IL-1β in HUVECs transduced
with control vector or hCLOCK and then treated with either PBS or
PDTC. *P<0.05 and **P<0.01 compared to PBS. COX-2,
cyclooxygenase 2; hCLOCK, human circadian locomotor output cycle
protein kaput; HUVECs, human umbilical vein endothelial cells;
ICAM-1, intercellular adhesion molecule 1; IL, interleukin; NF-κB,
nuclear factor-κB; p-p65, phosphorylated-p65; PDTC, pyrrolidine
dithiocarbamate; TNF-α, tumor necrosis factor-α. |
Discussion
The present study was undertaken, in part, to
elucidate the molecular basis for the hCLOCK-induced inflammatory
response pathway. Specifically, the results demonstrated that NF-κB
and proinflammatory cytokine levels were increased in response to
hypoxia; however, silencing hCLOCK reversed the effects and
inhibited the hypoxia-induced inflammatory. In addition,
suppressing NF-κB with PDTC inhibited the proinflammatory cytokine
levels in HUVECs under hypoxic conditions, and that NF-κB was
required for the hCLOCK-mediated inflammatory response.
The mammalian circadian clock controls the rhythmic
expression of numerous downstream genes, which are known as
clock-controlled genes. Clock-controlled genes encode various
proteins and profoundly influence cellular functions, including the
daily rhythm for the synthesis and release of cytokines, chemokines
and cytolytic factors (15,16).
In vivo and in vitro studies have clearly
demonstrated that the circadian clock genes modulate inflammatory
responses (17,18); however, to date, the effects of the
core circadian protein, CLOCK, on inflammation are rarely
mentioned. In the present study, the silencing of hCLOCK inhibited
hypoxia-induced inflammatory response augmentation and
significantly downregulated all of the key effector proteins
examined in a hypoxic environment. These findings indicated that
hCLOCK is directly involved in the inflammatory response induced by
hypoxia. The present study investigated this pathway based on
several areas of prior research that have linked hCLOCK with the
hypoxic and inflammatory responses in the cardiovascular system.
The NF-κB pathway has been implicated in the inflammatory response
(12) and, therefore, the present
study sought to identify the association between the circadian
clock and the NF-κB pathway.
NF-κB belongs to a family of constitutive and
inducible transcription factors. In the majority of cell types,
NF-κB is mainly represented by the p65/p50 heterodimeric complex.
The activated NF-κB complex enters the nucleus, where it binds to
consensus sites in the promoters of specific genes, such as
cytokines and various regulators of cellular survival and
proliferation, and activates their expression (19). Research over the past decade has
uncovered novel molecular links between the circadian clock and the
NF-κB pathway (20). A detailed
mathematical model of NF-κB indicated that the frequencies and
amplitudes of the NF-κB oscillation depend on the strength and
modes of coupling to the circadian clock (21,22).
Mutations in the cryptochrome gene, which encodes one of the core
clock proteins, enhance extrinsic apoptosis by interfering with
NF-κB-mediated transcriptional activation of genes that are
required for anti-apoptosis in response to cytokine stimulation
(13).
The present study confirmed an initial link between
hCLOCK expression and the hypoxia-induced response pathway, and
that the inhibition of NF-κB attenuates the expression of
hypoxia-induced inflammatory cytokines. There are multiple hypoxic
response pathways; however, the role of NF-κB signaling in
hCLOCK-induced inflammation has not, to the best of our knowledge,
been characterized definitively. The NF-κB inhibitor PDTC is a
dithiocarbamate of the pyrrole derivatives (23). This molecule is able to hinder the
dissociation of the inhibitory protein IκB from the NF-κB complex
through antioxidation, thus inhibiting NF-κB activation (24). In addition, PDTC impedes the
translocation of p65 to the nucleus and significantly reduces the
expression of p65 in the nucleus (25). PDTC may also directly reduce the
binding ability between NF-κB and DNA, obstructing the
NF-κB-activated signaling pathway (26). When HUVECs cultured in hypoxic
conditions were exposed to PDTC, the previously overexpressed
hypoxia-induced inflammatory cytokines were downregulated.
Furthermore, when the hCLOCK-overexpressing HUVECs were treated
with PDTC, they exhibited decreased protein levels of IL-1β, IL-6
and TNF-α, which are downstream effectors. The findings of the
present study further indicated that NF-κB signaling was necessary
for CLOCK-induced inflammatory cytokine expression.
The NF-κB pathway profoundly influences human
biology. Excessive or dysregulated activation of the NF-κB pathway
may lead to the development of pathological inflammation, which may
in turn cause acute and chronic diseases. Often, chronic
inflammation is linked to pathologies, such as arthritis, asthma,
septic shock, lung fibrosis, glomerulonephritis, atherosclerosis
and premature aging (27). To
date, research has been performed on multiple links in a complex
chain of biochemical pathways and has ultimately connected
regulators, such as hCLOCK, with inflammation and tumorigenesis.
Since NF-κB is considered to be a plausible target for therapeutic
activation and suppression, the present novel characterization of
the relative roles of the NF-κB signaling pathway in regulating
hCLOCK-induced inflammation may allow for the development of novel
therapeutic tools and strategies for treating inflammatory
disorders.
In conclusion, the results of the present study
linked two major signaling pathways: The circadian clock, which
introduces a temporal variable into numerous physiological
functions, and the NF-κB pathway, which is a key nodal focus in the
inflammatory response. These data suggested that the mechanisms of
inflammation induced by CLOCK primarily involve activation of the
NF-κB signaling pathways. These findings may aid further
elucidation of certain aspects of the complex biology of
inflammation and tumorigenesis.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81570433),
the Project of Shanghai Municipal Commission of Health and Family
Planning (grant no. 20154Y0104) and the Zhongshan Hospital Youth
Talent Training Program (grant no. 201514).
References
|
1
|
Xu C, Lu C, Hua L, Jin H, Yin L, Chen S
and Qian R: Rhythm changes of clock genes, apoptosis-related genes
and atherosclerosis-related genes in apolipoprotein E knockout
mice. Can J Cardiol. 25:473–479. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Takeda N and Maemura K: The role of clock
genes and circadian rhythm in the development of cardiovascular
diseases. Cell Mol Life Sci. 72:3225–3234. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reppert SM and Weaver DR: Coordination of
circadian timing in mammals. Nature. 418:935–941. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ramin C, Devore EE, Pierre-Paul J, Duffy
JF, Hankinson SE and Schernhammer ES: Chronotype and breast cancer
risk in a cohort of US nurses. Chronobiol Int. 30:1181–1186. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Savvidis C and Koutsilieris M: Circadian
rhythm disruption in cancer biology. Mol Med. 18:1249–1260. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Anea CB, Zhang M, Stepp DW, Simkins GB,
Reed G, Fulton DJ and Rudic RD: Vascular disease in mice with a
dysfunctional circadian clock. Circulation. 119:1510–1517. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Paschos GK and FitzGerald GA: Circadian
clocks and vascular function. Circ Res. 106:833–841. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tang X, Guo D, Lin C, Shi Z, Qian R, Fu W,
Liu J, Li X and Fan L: Upregulation of the gene expression of CLOCK
is correlated with hypoxia-inducible factor 1α in advanced varicose
lesions. Mol Med Rep. 12:6164–6170. 2015.PubMed/NCBI
|
|
9
|
Ghosh S, May MJ and Kopp EB: NF-kappa B
and Rel proteins: Evolutionarily conserved mediators of immune
responses. Annu Rev Immunol. 16:225–260. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pahl HL: Activators and target genes of
Rel/NF-kappaB transcription factors. Oncogene. 18:6853–6866. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pasparakis M, Luedde T and
Schmidt-Supprian M: Dissection of the NF-kappaB signalling cascade
in transgenic and knockout mice. Cell Death Differ. 13:861–872.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee JH and Sancar A: Regulation of
apoptosis by the circadian clock through NF-kappaB signaling. Proc
Natl Acad Sci USA. 108:12036–12041. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Monje FJ, Cabatic M, Divisch I, Kim EJ,
Herkner KR, Binder BR and Pollak DD: Constant darkness induces
IL-6-dependent depression-like behavior through the NF-κB signaling
pathway. J Neurosci. 31:9075–9083. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang X, Guo D, Lin C, Shi Z, Qian R, Fu W,
Liu J, Li X and Fan L: hCLOCK causes Rho-Kinase-Mediated
endothelial dysfunction and NF-κB-mediated inflammatory responses.
Oxid Med Cell Longev. 2015:6718392015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Labrecque N and Cermakian N: Circadian
clocks in the immune system. J Biol Rhythms. 30:277–290. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Keller M, Mazuch J, Abraham U, Eom GD,
Herzog ED, Volk HD, Kramer A and Maier B: A circadian clock in
macrophages controls inflammatory immune responses. Proc Natl Acad
Sci USA. 106:21407–21412. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sato S, Sakurai T, Ogasawara J, Takahashi
M, Izawa T, Imaizumi K, Taniguchi N, Ohno H and Kizaki T: A
circadian clock gene, Rev-erbα, modulates the inflammatory function
of macrophages through the negative regulation of Ccl2 expression.
J Immunol. 192:407–417. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lawrence T and Fong C: The resolution of
inflammation: Anti-inflammatory roles for NF-kappaB. Int J Biochem
Cell Biol. 42:519–523. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hayden MS, West AP and Ghosh S: NF-kappaB
and the immune response. Oncogene. 25:6758–6780. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Spengler ML, Kuropatwinski KK, Comas M,
Gasparian AV, Fedtsova N, Gleiberman AS, Gitlin II, Artemicheva NM,
Deluca KA, Gudkov AV and Antoch MP: Core circadian protein CLOCK is
a positive regulator of NF-κB-mediated transcription. Proc Natl
Acad Sci USA. 109:E2457–E2465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
González-Miranda JM: On the effect of
circadian oscillations on biochemical cell signaling by NF-κB. J
Theor Biol. 335:283–294. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang X, Yu W and Zheng L: The dynamics of
NF-κB pathway regulated by circadian clock. Math Biosci. 260:47–53.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cuzzocrea S, Chatterjee PK, Mazzon E, Dugo
L, Serraino I, Britti D, Mazzullo G, Caputi AP and Thiemermann C:
Pyrrolidine dithiocarbamate attenuates the development of acute and
chronic inflammation. Br J Pharmacol. 135:496–510. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu SF and Malik AB: NF-kappa B activation
as a pathological mechanism of septic shock and inflammation. Am J
Physiol Lung Cell Mol Physiol. 290:L622–L645. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Németh ZH, Haskó G and Vizi ES:
Pyrrolidine dithiocarbamate augments IL-10, inhibits TNF-alpha,
MIP-1alpha, IL-12, and nitric oxide production and protects from
the lethal effect of endotoxin. Shock. 10:49–53. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Roy A, Jana A, Yatish K, Freidt MB, Fung
YK, Martinson JA and Pahan K: Reactive oxygen species up-regulate
CD11b in microglia via nitric oxide: Implications for
neurodegenerative diseases. Free Radic Biol Med. 45:686–699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Alonso-Fernández P and De la Fuente M:
Role of the immune system in aging and longevity. Curr Aging Sci.
4:78–100. 2011. View Article : Google Scholar : PubMed/NCBI
|














