Introduction
According to multi-level cooperative study results
of cardiovascular health in random samples of urban and rural
residents (aged 35–74 years) in China, the prevalence rate of
congestive heart failure in females was identified to be 1.0%
(1,2). The north had a higher prevalence rate
compared with the south and as age increased, the prevalence rate
markedly increased; however, there was no clear difference between
urban and rural regions (2). With
the increase of coronary heart disease and high blood
pressure-associated morbidity, accelerating population aging and
the increase of various dangerous factors, the number of patients
presenting with congestive heart failure in China is also
increasing (3). Ischemic heart
disease caused by coronary artery disease has already become the
most common pathogenesis resulting in congestive heart failure and
seriously threatens the health of Chinese patients (4).
Acute myocardial infarction (AMI) causes oxidative
stress reactions and inflammatory responses. These activate
potential matrix metalloproteinases (MMPs) in cardiac muscle
tissues (such as MMP-1, MMP-2, MMP-3 and MMP-9), degrade
extracellular matrix and coronary vessel structures, promote
inflammatory cell homing in the blood to the ischemic myocardium,
as well as participating in enzymolysis and phagocytosis of infarct
cardiac muscle tissues (5,6).
Subsequent to AMI, reactive oxygen species (ROS) and
intracellular components generated by the damaged myocardium
activate Toll-like receptor, NF-κB expression and complement
activation, causing high expression of the vascular endothelial
cell adherence factor in infarct cardiac muscle tissue and the
increase in expression of damage-associated stress factors, and
increasing the number of inflammatory cells in circulation,
including neutrophil granulocytes and macrophages, which return to
the infarct area, participating in enzymolysis and phagocytosis of
the infract cardiac muscle tissue (7,8).
Currently, the inflammatory response peaks one or two weeks after
AMI, and 3 to 4 weeks after AMI, the inflammatory cells become
apoptotic and diminish independently (9).
A previous study demonstrated that myocardial
ischemia may be the initial factor of ventricular remodeling after
AMI (10). Relevant
damage-associated stress factors following AMI contribute to
regulating myocardial cell death and progression of ventricular
remodeling via other approaches (11). For example, AMI promotes Bcl-2
interacting protein 3 to express relevant genes, which results in
apoptosis of myocardial cells via a caspase-dependent pathway
(12). A previous study indicates
that myocardial cells may be prevented from presenting ischemia and
anaerobic conditions, so as to improve ventricular remodeling
(11). Currently, researchers hope
to regulate programmed cell death after AMI and improve the
prognosis of ventricular remodeling.
Ginkgo biloba (also termed ginkgo) is a tall
deciduous tree. The plant dates back to the Carboniferous period,
345 million years ago (13).
Following Quaternary glaciation, it is the sole living
representative of its genus and one of the oldest relic plants in
the world. Ginkgo biloba was initially native to China and was
subsequently introduced to Europe in 1710 (14). Ginkgo biloba extract may directly
lead to anti-oxygenation, elimination of oxygen free radicals,
regulation of superoxide dismutase activity and catalases, as well
as eliminating nitric oxide (NO), thus contributing to protecting
against ischemia damage and damage of vascular endothelial cells,
potentially preventing atherosis (15). In the current study, whether Ginkgo
biloba extract prevents AMI was investigated, in addition to the
molecular mechanisms associated with its anti-inflammation
effect.
Materials and methods
Animals
The current study was performed in strict accordance
with the recommendations from the Guide for Animal Management Rules
from Guangxi Medical University (Nanning, China). C57BL/6 mice
(n=24, 8 mice per group) were purchased from the Department of
Laboratory Animal Science (Guangxi Medical University) and housed
together under specific-pathogen-free conditions (23–24°C;
humidity, 55–60%) in an animal room under a 12-h light/dark cycle
with free access to water and food. All mice were randomly
distributed into three groups as follows: Control, AMI and AMI +
Ginkgo biloba extract (GBE, Guizhou Provincial Biochemical
Engineering Center, Guiyang, China).
Induction of the AMI model
Anesthesia was performed by inhalation of 1.0–2.0%
isoflurane gas and mechanically ventilated on a positive pressure
ventilator. Left thoracotomy was performed and the pericardium was
immediately stripped away to expose the heart. The coronary artery
was identified and occluded with an 8–0 silk ligature; successful
ligation was confirmed when the left ventricle turned pale. The
chest cavity was closed and mice were placed in their cages on a
heating pad. The control mice underwent the same surgical
procedures without ligation. AMI mice were administered normal
saline (200 µl) via daily gavage for 8 weeks. The AMI + GBE group
mice received 100 mg/kg GBE via daily gavage for 8 weeks.
Infarct size assessment
Anesthesia was performed by inhalation of 1.0–2.0%
isoflurane gas and the mice were sacrificed using decollation
following the 8 weeks of the experiment. The hearts were
immediately removed and cut into 1.0 mm vertical sections. These
sections were stained with 1% 2,3,5-triphenyl-tetrazolium chloride
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) in PBS for 10 min
at 37°C. Infarct size areas were determined using a microscope
(model BX53M; Olympus Corporation, Tokyo, Japan). with Image-Pro
Plus software version 6.0 (Media Cybernetics, Inc., Rockville, MD,
USA).
ELISA assay
Anesthesia was performed by inhalation of 1.0–2.0%
isoflurane gas and then blood (100 µl) was collected from the eye
sockets of the mice after treatment with Ginkgo biloba extract.
Serum was collected by centrifugation at 2,000 × g for 10 min at
4°C. ELISA kits were used to determine serum histamine, lactate
dehydrogenase, creatine kinase (CK; A032) and CK-MB (H197),
interleukin (IL)-6 (H007) and IL-1β (H002) levels, and caspase-3/9
activity (G015 and G018) were evaluated using ELISA kits (all from
Nanjing Jiancheng Biology Engineering Institute, Nanjing, China)
according to the manufacturer's instructions.
Western blotting
Anesthesia was performed by inhalation of 1.0–2.0%
isoflurane gas and mice were sacrificed using decollation following
treatment with Ginkgo biloba extract. The hearts were immediately
removed and homogenated using RIPA Lysis Buffer (Beyotime Institute
of Biotechnology, Haimen, China) for 30–40 min at 4°C. Lysates were
centrifuged at 10,000 × g for 10 min to analyze the protein
concentration via BCA assay (Beyotime Institute of Biotechnology)
and then 50–80 µg protein was resolved on 8–10% SDS gel. Following
electrophoresis, the proteins were electrotransferred (2.5A, 25 V
for 30 min) onto a nitrocellulose membranes. Membranes were blocked
with 5% non-fat milk and probed with MMP-9 (cat. no. 13667;
dilution, 1:2,000; Cell Signaling Technology, Inc., Danvers, MA,
USA), TGF-β (cat. no. 5544; dilution, 1:2,000; Cell Signaling
Technology, Inc.), p-p38 (cat. no. 4511; dilution, 1:2,000; Cell
Signaling Technology, Inc.), NF-κB (cat. no. 8242; dilution,
1:2,000; Cell Signaling Technology, Inc.) and GAPDH (cat. no.
AF0006; dilution, 1:3,000; Beyotime Institute of Biotechnology)
antibodies overnight at 4°C. The blot was washed with TBST three
times for 5 min, exposed to horseradish peroxidase-conjugated
secondary antibodies (cat. no. A0208; dilution, 1:5,000; Beyotime
Institute of Biotechnology) for 1 h at 37°C, and finally examined
by chemiluminescence (ECL; GE Healthcare Life Sciences, Little
Chalfont, UK).
Statistical analysis
Data are presented as means ± standard error of the
mean. Comparisons between the two groups were assessed by Student's
t-test or two-way analysis of variance followed by Bonferroni's
post-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
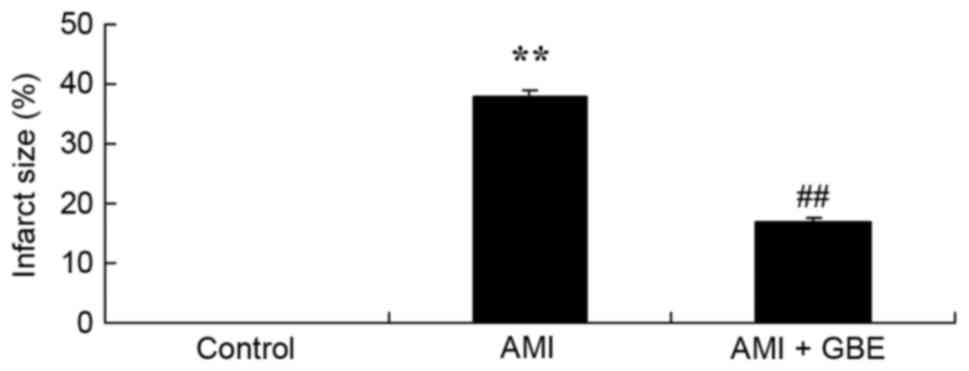
GBE reduces the size of infarct
areas
GBE was observed to reduce the size of infarct areas
in the AMI model mice. A significant increase in the size of the
infarct areas was observed in the AMI model group, when compared
with the control group (Fig. 1).
Subsequently, GBE treatment significantly inhibited the increase of
infarct area size in the AMI mice, when compared with the AMI model
group (Fig. 1).
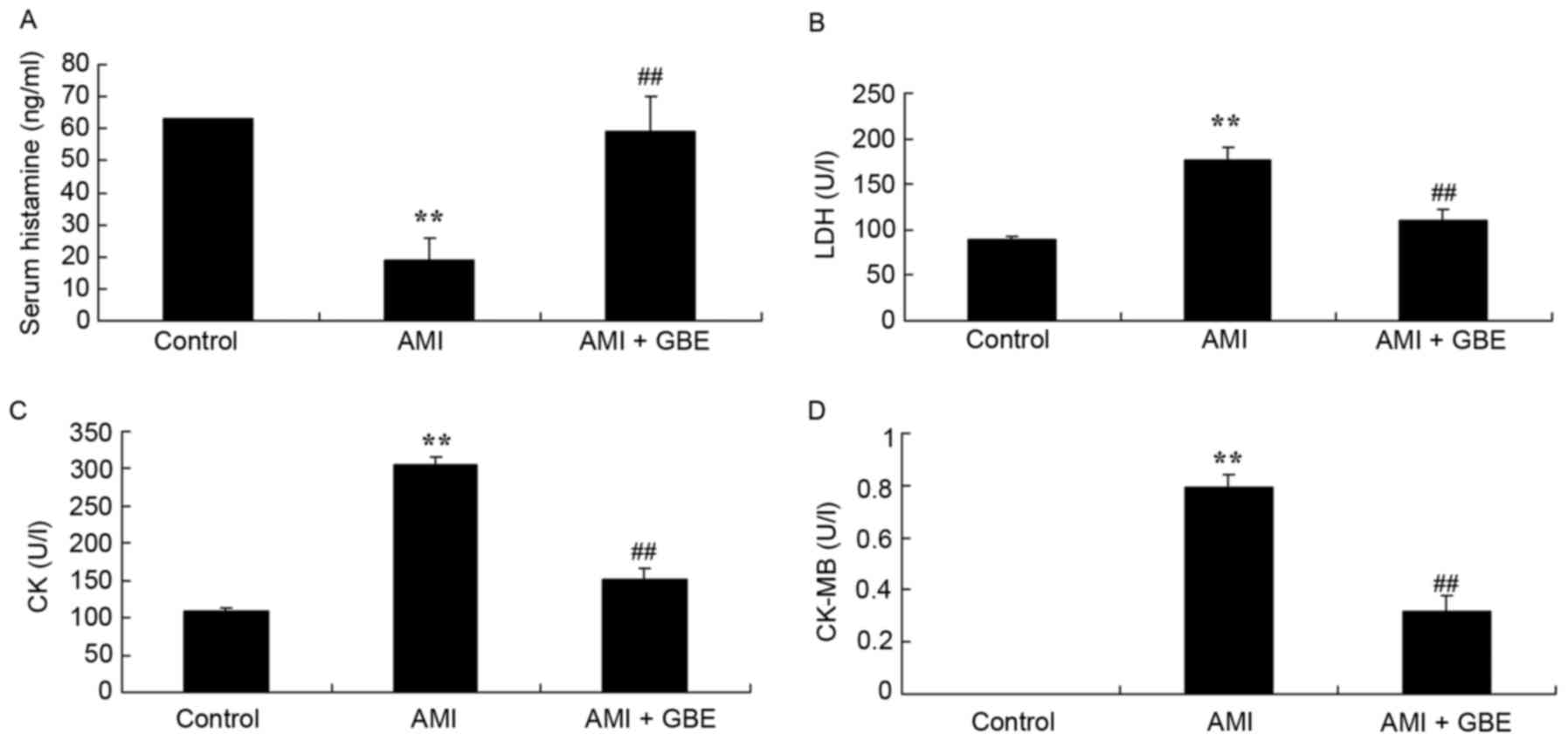
GBE prevents AMI
The effects of GBE on AMI were subsequently
evaluated. As compared with the control group, serum histamine was
significantly decreased, and LDH, CK and CK-MB levels in the AMI
mouse models were significantly increased (Fig. 2). However, GBE treatment
significantly increased the serum histamine level, and decreased
the LDH, CK and CK-MB levels in the AMI mice, when compared with
AMI mouse model group (Fig.
2).
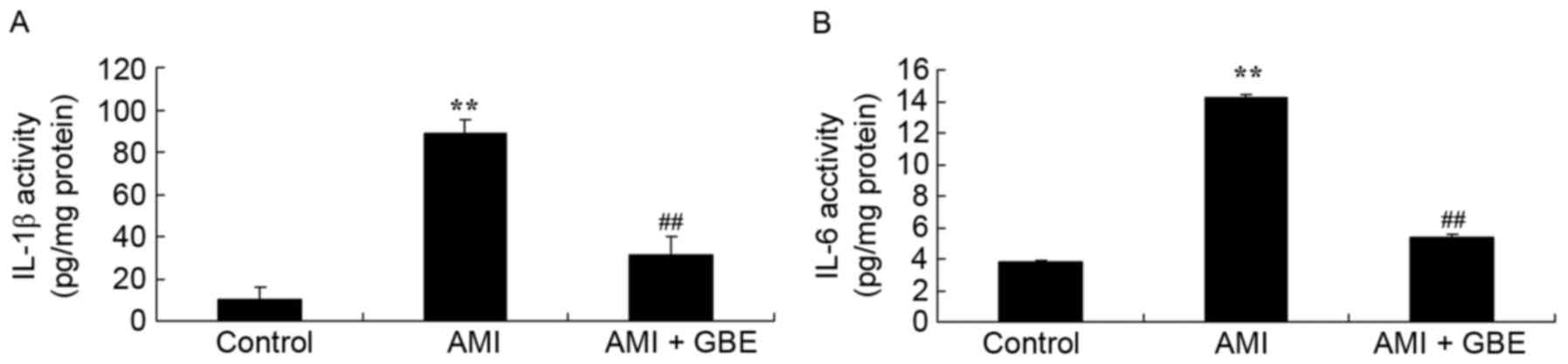
GBE reduces inflammatory
reactions
To investigate the effect of GBE on inflammatory
reactions, the expression levels of IL-6 and IL-1β in tissue
samples were analyzed by ELISA. Significantly increased IL-6 and
IL-1β activity levels were observed in the AMI mice, compared with
the control group (Fig. 3).
Treatment with GBE significantly inhibited the increased IL-6 and
IL-1β activities in the AMI mice (Fig.
3).
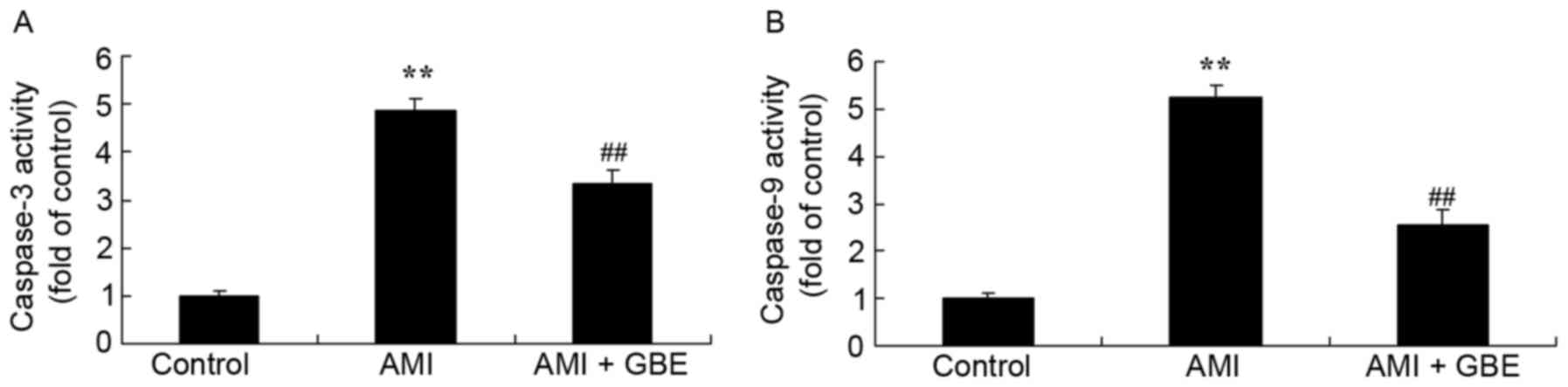
GBE reduces caspase-3/9 activity
In order to investigate the effect of GBE on
apoptosis, caspase-3/9 activities were analyzed by ELISA. Fig. 4 demonstrates the significantly
increased caspase-3/9 activities in the AMI mice as compared with
the control group. GBE treatment significantly reduced the
caspase-3/9 activities in the AMI mice, when compared with AMI
model group (Fig. 4).
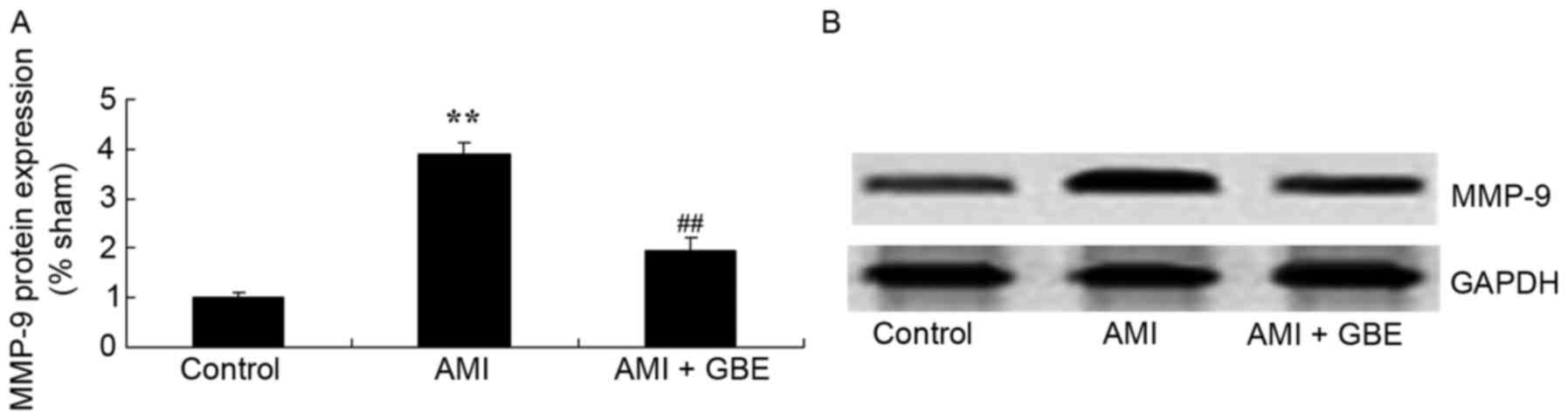
GBE reduces MMP-9 protein expression
levels
To evaluated the underlying mechanism of GBE against
AMI, MMP-9 protein expression levels were analyzed using western
blotting. The results indicated that MMP-9 protein expression was
significantly induced in the AMI mouse model when compared with the
control group. As compared with AMI model group, the group treated
with GBE demonstrated significantly suppressed MMP-9 protein
expression levels (Fig. 5).
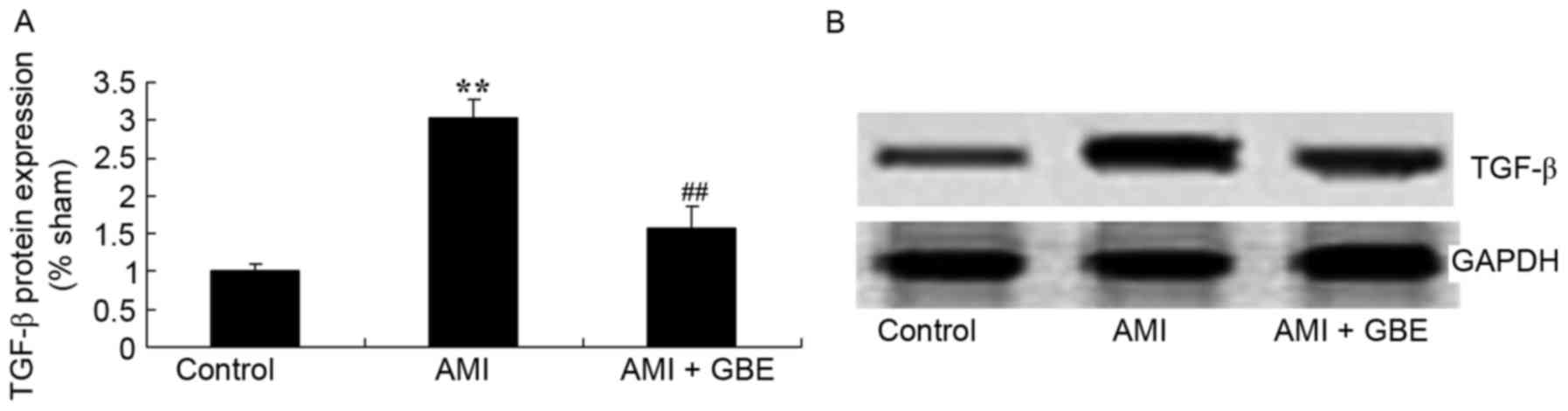
GBE reduces TGF-β protein expression
levels
TGF-β expression was examined to evaluate the
underlying mechanism of GBE against AMI. The level of TGF-β protein
expression observed in the AMI model group was significantly higher
than that of control group. Treatment with GBE significantly
suppressed TGF-β protein expression levels in the AMI mice, when
compared with the AMI model mice (Fig.
6).
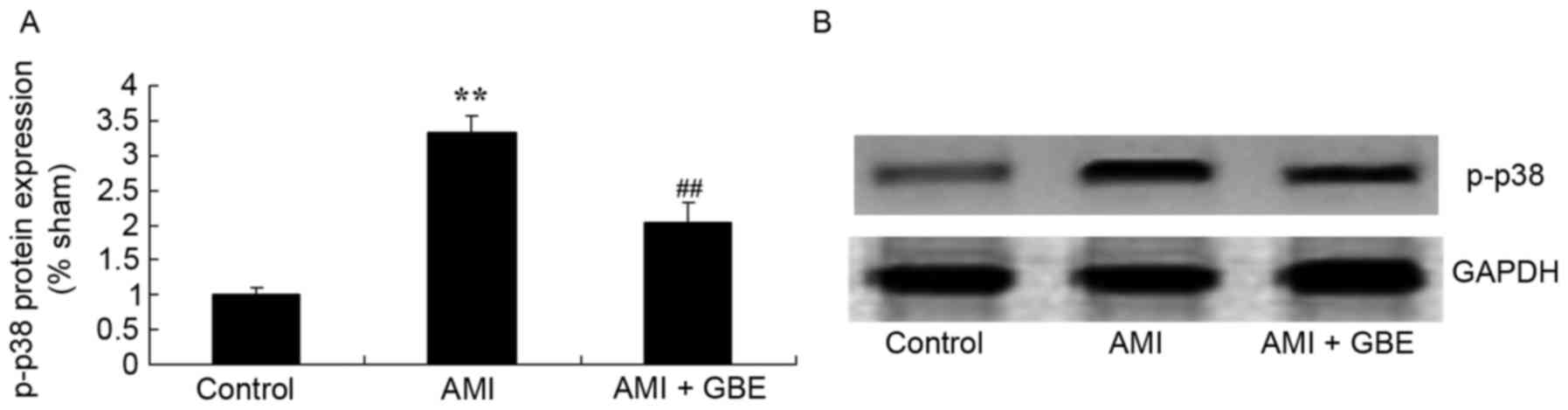
GBE reduces levels of p-p38 protein
expression
Subsequently, the underlying mechanism of the effect
of GBE against AMI was investigated by evaluating p-p38 protein
expression levels using western blotting. The levels of p-p38
protein expression in the AMI model group were greater than that of
the control group. In the AMI mice treated with GBE, p-p38 protein
expression levels were significantly suppressed when compared with
the AMI model mice (Fig. 7).
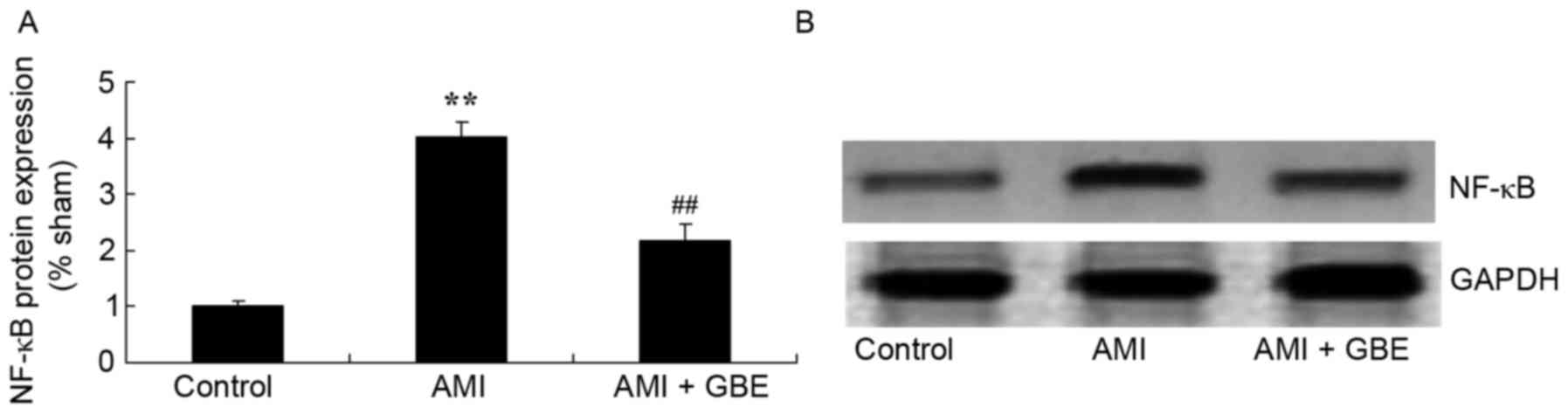
GBE reduces NF-κB protein expression
levels
The underlying mechanism of GBE against AMI was
evaluated by western blotting to detect NF-κB protein expression
levels. A significant increase of NF-κB protein expression was
observed in the AMI model mice when compared with the control group
(Fig. 8). When the AMI mice were
treated with GBE, NF-κB protein expression was significantly
suppressed (Fig. 8).
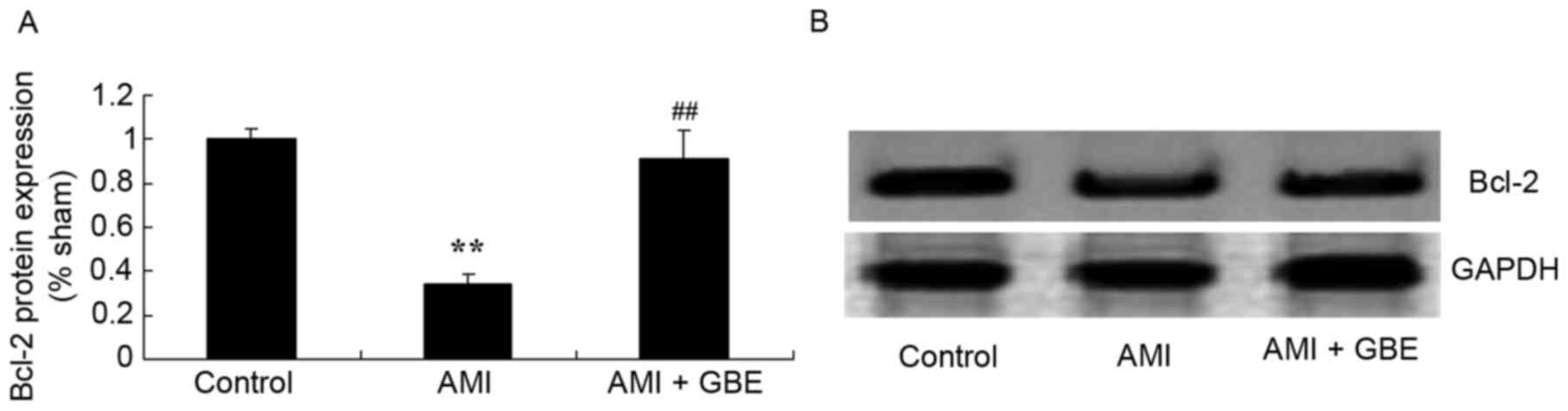
GBE reduces Bcl-2 protein expression
levels
The Bcl-2 protein expression levels were analyzed to
investigate the underlying mechanism of GBE against AMI. Bcl-2
protein expression in the AMI model mice was significantly
inhibited compared with the control group (Fig. 9). Following GBE treatment Bcl-2
protein expression in the AMI mice was significantly increased
compared with the AMI model mice (Fig.
9).
Discussion
AMI leads to ischemic myocardium issues,
particularly in the reperfusion area, where large quantities of ROS
are generated, which cause direct damage to cytomembrane
structures, such as inducing overloading in cells to increase
mitochondrial membrane permeability and causing cell death
(16). Furthermore, ROS promote
the release of inflammatory factors, such as TNF-a, IL-1β and IL-6
in the ischemic region and surrounding area. Apoptosis is induced
via the TNF-a/caspase signaling pathway to promote myocardial
contraction (17). In addition,
ROS and relevant inflammatory factors activate MMPs, degrade the
extracellular matrix (ECM), resulting in sliding cardiac muscle
fibers and finally causing expansion (18,19).
The results of the current study demonstrated that GBE treatment
significantly inhibited the increase of infarct area size,
increased serum histamine levels, decreased LDH, CK and CK-MB
levels, inhibited the increase of IL-6 and IL-1β activities and
reduced caspase-3/9 activities in AMI mice. Li et al
(20) reported that GBE inhibits
experimental rat myocardial remodeling via TGF-β1, MMP-2 and MMP-9
(20).
Dynamic changes in the ECM occur following AMI and
has an important role in ventricular remodeling. During the period
of AMI, transforming growth factor in cardiac muscle tissue of
TGF-β promoting fibrosis factor is activated (21). The fibrosis cell generates into
type I and type III collagenous fibers, which gradually develop
into scar tissue. Meanwhile, cardiac muscle tissue in non-infarct
areas exhibits interstitial and peripheral fibrosis (22). Thus, these results demonstrate that
GBE significantly suppresses TGF-β protein expression in AMI mice.
Li et al (20) reported
that GBE treatment inhibits myocardial remodeling via TGF-β1, MMP-2
and MMP-9 in experimental rats (20).
p38 MAPK is activated by phosphorylation, which
increases the expression of inflammatory factors in rats. This
causes thickening of the heart, interstitial fibrosis, serious
cardiac insufficiency, myocardial apoptosis or mortality (23). p38 MAPK has previously been
demonstrated to increase the expression levels of inflammatory
factors following activation of myocardial ischemia reperfusion, by
reactive activation of p38 MAPK (24). p38 MAPK is associated with
myocardial remodeling and inflammatory factor expression in the
myocardium following AMI (24).
p38 MAPK influences the heart, by stimulating synthesis of
inflammatory factors, promoting cell transformation into
fibroblasts to integrate into the ECM, and by inhibiting MMP
degradation of the ECM. TGF-β1 also promotes hypertrophy of
myocardial cells, differentiation and the increase of the number of
lymphocytes (25). In addition,
TGF-β1 is involved in stimulating tissue fibrosis, causing
increases in fibrocytes, MMPs, collagen deposition and fiber
binding proteins, and results in ventricular remodeling (23). TGF-β1 expression activity in
cardiac muscle tissues of myocardial infarction rats was
demonstrated to be markedly enhanced (26). Corresponding MAP3K7, p38 MAPK, and
p-p38 MAPK protein activity were also clearly enhanced (26). The possible underlying mechanism
involves TGF-β1 activating MAP3K7, thus causing p38 MAPK to be
phosphorylated into p-p38 MAPK and enhancing inflammatory factor
expression levels in rats, finally resulting in cardiac
hypertrophy, interstitial fibrosis, serious cardiac insufficiency,
myocardial apoptosis or mortality (25). The present study demonstrates that
GBE significantly suppressed MMP-9 and p-p38 protein expression in
AMI mice. In addition, Tsai et al (15) demonstrated that GBE reduces
high-glucose-induced endothelial ROS generation via Akt/endothelial
NO synthase and p38 MAPK signaling pathways (15).
NF-κB, a nuclear transcription factor, was initially
identified in mature B cells in 1986, specifically binding with the
enhancer subsequence of the immune globulin κ light-chain gene
(27,28). NF-κB regulates the relevant
processes of AMI, including generation of NO, synthesis of
prostaglandin, calcium and sodium treatment, growth factors,
apoptosis, ECM, stress and reconstruction (29). In the smooth muscle cells of human
atherosclerotic plaque, macrophages and endothelial cells, it has
been demonstrated that activated NF-κB participates in lipid
modification, chemotaxis and attachment (30). Inflammatory factors and tissue
damage result in lesions and unstable plaque. NF-κB participates in
and mediates the process by regulating NO (29). In addition, NF-κB participates in
the immune response and cell apoptosis, and is the key
transcription factor causing inflammatory reactions. An increasing
number of studies demonstrates that NF-κB is important in AMI
(28). The current study
demonstrates that GBE significantly reduced NF-κB protein
expression levels and induced Bcl-2 protein expression in AMI mice.
Furthermore, Wang et al (13) indicated that GBE mitigates liver
fibrosis via NF-κB, p38 MAPK and Bcl-2/Bcl-2-associated X protein
signaling (13).
In conclusion, the present results demonstrates that
treatment with GBE prevents AMI, by increasing serum histamine
levels, decreasing LDH, CK and CK-MB levels, and suppressing
inflammation- and apoptosis-regulating p38 MAPK, NF-κB and Bcl-2
signaling. Therefore, as GBE abrogates the activity of p38 MAPK and
NF-κB signaling pathways in AMI, it may serve as an effective
therapeutic strategy against various types of heart disease.
References
|
1
|
O'Donoghue ML, Glaser R, Cavender MA,
Aylward PE, Bonaca MP, Budaj A, Davies RY, Dellborg M, Fox KA,
Gutierrez JA, et al: Effect of losmapimod on cardiovascular
outcomes in patients hospitalized with acute myocardial infarction:
A randomized clinical trial. JAMA. 315:1591–1599. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Prati F, Romagnoli E, Limbruno U,
Pawlowski T, Fedele S, Gatto L, Di Vito L, Pappalardo A, Ramazzotti
V, Picchi A, et al: Randomized evaluation of intralesion versus
intracoronary abciximab and aspiration thrombectomy in patients
with ST-elevation myocardial infarction: The COCTAIL II trial. Am
Heart J. 170:1116–1123. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Clemmensen P, Grieco N, Ince H, Danchi N,
Goedicke J, Ramos Y, Schmitt J, Goldstein P, et al: MULTIPRAC study
investigators: MULTInational non-interventional study of patients
with ST-segment elevation myocardial infarction treated with
PRimary Angioplasty and Concomitant use of upstream antiplatelet
therapy with prasugrel or clopidogrel-the European MULTIPRAC
registry. Eur Heart J Acute Cardiovasc Care. 4:220–229. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Koltowski L, Koltowska-Haggstrom M,
Filipiak KJ, Kochman J, Golicki D, Pietrasik A, Huczek Z, Balsam P,
Scibisz A and Opolski G: Quality of life in patients with
ST-segment elevation myocardial infarction undergoing percutaneous
coronary intervention-radial versus femoral access (from the OCEAN
RACE Trial). Am J Cardiol. 114:516–521. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xian Y, Wang TY, McCoy LA, Effron MB,
Henry TD, Bach RG, Zettler ME, Baker BA, Fonarow GC and Peterson
ED: Association of discharge aspirin dose with outcomes after acute
myocardial infarction: Insights from the treatment with ADP
receptor inhibitors: Longitudinal assessment of treatment patterns
and events after acute coronary syndrome (TRANSLATE-ACS) study.
Circulation. 132:174–181. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Aarsetøy H, Brügger-Andersen T, Hetland Ø,
Grundt H and Nilsen DW: Long term influence of regular intake of
high dose n-3 fatty acids on CD40-ligand, pregnancy-associated
plasma protein A and matrix metalloproteinase-9 following acute
myocardial infarction. Thromb Haemost. 95:329–336. 2006.PubMed/NCBI
|
|
7
|
Fan Q, Chen, Fang X, Lau WB, Xue L, Zhao
L, Zhang H, Liang YH, Bai X, Niu HY, et al: Aging might augment
reactive oxygen species (ROS) formation and affect reactive
nitrogen species (RNS) level after myocardial ischemia/reperfusion
in both humans and rats. Age (Dordr). 35:1017–1026. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lima-Neto LG, Hirata RD, Luchessi AD,
Silbiger VN, Cavichioli D, Dos Santos ES, Sousa AG, Sprovieri SR,
De Sousa EB Jr, Dos Santos FC, et al: Chlamydophila pneumonia and
increased TLR4 gene expression in leukocytes are associated with
acute myocardial infarction. J Biol Regul Homeost Agents.
28:449–460. 2014.PubMed/NCBI
|
|
9
|
Moreira DM, da Silva RL, Vieira JL, Fattah
T, Lueneberg ME and Gottschall CA: Role of vascular inflammation in
coronary artery disease: Potential of anti-inflammatory drugs in
the prevention of atherothrombosis. Inflammation and
anti-inflammatory drugs in coronary artery disease. Am J Cardiovasc
Drugs. 15:1–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shi ZY, Liu Y, Dong L, Zhang B, Zhao M,
Liu WX, Zhang X and Yin XH: Cortistatin improves cardiac function
after acute myocardial infarction in rats by suppressing myocardial
apoptosis and endoplasmic reticulum stress. J Cardiovasc Pharmacol
Ther. Apr 18–2016.(Epub ahead of print).
|
|
11
|
Sheu JJ, Chua S, Sun CK, Chang LT, Yen CH,
Wu CJ, Fu M and Yip HK: Intra-coronary administration of
cyclosporine limits infarct size, attenuates remodeling and
preserves left ventricular function in porcine acute anterior
infarction. Int J Cardiol. 147:79–87. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lazou A, Iliodromitis EK, Cieslak D,
Voskarides K, Mousikos S, Bofilis E and Kremastinos DT: Ischemic
but not mechanical preconditioning attenuates ischemia/reperfusion
induced myocardial apoptosis in anaesthetized rabbits: The role of
Bcl-2 family proteins and ERK1/2. Apoptosis. 11:2195–2204. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Wang R, Wang Y, Peng R, Wu Y and
Yuan Y: Ginkgo biloba extract mitigates liver fibrosis and
apoptosis by regulating p38 MAPK, NF-κB/IκBα, and Bcl-2/Bax
signaling. Drug Des Devel Ther. 9:6303–6317. 2015.PubMed/NCBI
|
|
14
|
Zhu X, Li Z, Li C, Zhang J, Zou Z and Wang
J: Ginkgo biloba extract and aspirin synergistically attenuate
activated platelet-induced ROS production and LOX-1 expression in
human coronary artery endothelial cells. Phytomedicine. 20:114–119.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tsai HY, Huang PH, Lin FY, Chen JS, Lin SJ
and Chen JW: Ginkgo biloba extract reduces high-glucose-induced
endothelial reactive oxygen species generation and cell adhesion
molecule expression by enhancing HO-1 expression via Akt/eNOS and
p38 MAP kinase pathways. Eur J Pharm Sci. 48:803–811. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bashar T and Akhter N: Study on oxidative
stress and antioxidant level in patients of acute myocardial
infarction before and after regular treatment. Bangladesh Med Res
Counc Bull. 40:79–84. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang CH, Sheu JJ, Tsai TH, Chua S, Chang
LT, Chang HW, Lee FY, Chen YL, Chung SY, Sun CK, et al: Effect of
tacrolimus on myocardial infarction is associated with
inflammation, ROS, MAP kinase and Akt pathways in mini-pigs. J
Atheroscler Thromb. 20:9–22. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen W, Spitzl A, Mathes D, Nikolaev VO,
Werner F, Weirather J, Špiranec K, Röck K, Fischer JW, Kämmerer U,
et al: Endothelial actions of ANP enhance myocardial inflammatory
infiltration in the early phase after acute infarction. Circ Res.
119:237–248. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hedström E, Aström-Olsson K, Ohlin AK,
Ohlin H and Arheden H: Initial results of inflammatory response,
matrix remodeling, and reactive oxygen species following PCI in
acute ischemic myocardial injury in man. J Invasive Cardiol.
23:371–376. 2011.PubMed/NCBI
|
|
20
|
Li W, Luo Z, Liu X, Fu L, Xu Y, Wu L and
Shen X: Effect of Ginkgo biloba extract on experimental cardiac
remodeling. BMC Complement Altern Med. 15:2772015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ayça B, Sahin I, Kucuk SH, Akin F, Kafadar
D, Avşar M, Avci II, Gungor B, Okuyan E and Dinckal MH: Increased
transforming growth factor-β levels associated with cardiac adverse
events in hypertrophic cardiomyopathy. Clin Cardiol. 38:371–377.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Talasaz AH, Khalili H, Jenab Y, Salarifar
M, Broumand MA and Darabi F: N-Acetylcysteine effects on
transforming growth factor-β and tumor necrosis factor-α serum
levels as pro-fibrotic and inflammatory biomarkers in patients
following ST-segment elevation myocardial infarction. Drugs R D.
13:199–205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang X, Lv H, Gu Y, Wang X, Cao H, Tang Y,
Chen H and Huang C: Protective effect of lycopene on cardiac
function and myocardial fibrosis after acute myocardial infarction
in rats via the modulation of p38 and MMP-9. J Mol Histol.
45:113–120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Arabacilar P and Marber M: The case for
inhibiting p38 mitogen-activated protein kinase in heart failure.
Front Pharmacol. 6:1022015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matsumoto-Ida M, Takimoto Y, Aoyama T,
Akao M, Takeda T and Kita T: Activation of TGF-beta1-TAK1-p38 MAPK
pathway in spared cardiomyocytes is involved in left ventricular
remodeling after myocardial infarction in rats. Am J Physiol Heart
Circ Physiol. 290:H709–H715. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Q, Feng J, Wang J, Zhang X, Zhang D,
Zhu T, Wang W, Wang X, Jin J, Cao J, et al: Disruption of TAB1/p38α
interaction using a cell-permeable peptide limits myocardial
ischemia/reperfusion injury. Mol Ther. 21:1668–1677. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bliksøen M, Mariero LH, Torp MK, Baysa A,
Ytrehus K, Haugen F, Seljeflot I, Vaage J, Valen G and Stensløkken
KO: Extracellular mtDNA activates NF-κB via toll-like receptor 9
and induces cell death in cardiomyocytes. Basic Res Cardiol.
111:422016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Haar L, Ren X, Liu Y, Koch SE, Goines J,
Tranter M, Engevik MA, Nieman M, Rubinstein J and Jones WK: Acute
consumption of a high-fat diet prior to ischemia-reperfusion
results in cardioprotection through NF-κB-dependent regulation of
autophagic pathways. Am J Physiol Heart Circ Physiol.
307:H1705–H1713. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ding HS, Yang J, Chen P, Yang J, Bo SQ,
Ding JW and Yu QQ: The HMGB1-TLR4 axis contributes to myocardial
ischemia/reperfusion injury via regulation of cardiomyocyte
apoptosis. Gene. 527:389–393. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zeng M, Wei X, Wu Z, Li W, Li B, Zhen Y,
Chen J, Wang P and Fei Y: NF-κB-mediated induction of autophagy in
cardiac ischemia/reperfusion injury. Biochem Biophys Res Commun.
436:180–185. 2013. View Article : Google Scholar : PubMed/NCBI
|