Introduction
Accumulating evidence suggests that oxidative stress
may contribute to diabetes mellitus (DM)-associated cardiovascular
complications. Hyperglycemia and elevated levels of free fatty
acids and lipids promote oxidative stress in DM, which results in
cardiovascular damage (1).
Hyperglycemia, in particular, has been demonstrated to induce
inflammation and apoptosis in retinal, cardiovascular and kidney
cells; all of which have been implicated in DM-associated
complications (2–4). DM-associated oxidative stress is the
result of high levels of reactive oxygen species (ROS) (5–7).
Increased glucose levels promote ROS production via the following
two major mechanisms: i) The action of nicotinamide adenine
dinucleotide phosphate (NADPH) oxidase located on the plasma
membrane, and ii) dysfunction of the mitochondrial electron
transport chain (8–10). ROS activates multiple biochemical
pathways, including the formation of advanced glycation end
products (11,12), protein kinase C activation
(13,14) and the hexosamine pathway (15), which propagate further ROS
production and increases oxidative stress. Therefore, prevention of
ROS production through inhibiting mitochondrial dysfunction has
been proposed as an alternative approach to conventional
antioxidant therapies to treat diabetes and vascular complications
(16).
Hyperglycemia-induced apoptosis and inflammatory
responses have been recognized as cardiovascular complications of
DM (4), and multiple signaling
pathways have been implicated in hyperglycemia-induced apoptosis of
cardiovascular cells (3,17,18).
By contrast, high glucose levels activate cellular defenses against
oxidative stress. Nuclear factor (erythroid-derived 2)-like-2 is a
redox sensitive transcription factor that has been implicated in
the anti-oxidative stress response to hyperglycemia (19). Neuregulin protects human umbilical
vein endothelial cells against hyperglycemia-induced apoptosis via
activation of the cluster of differentiation 98 heavy chain through
the mitogen-activated protein kinase pathway (20). In addition, upregulated angiotensin
(1–7) preserves endothelial function by
reducing oxidative stress in DM (21), and glucagon-like peptide-1
suppresses hyperglycemia-induced oxidative stress in patients with
DM (22). Therefore, investigating
the molecular pathways underlying cellular defenses against
hyperglycemia-induced oxidative stress is vital.
Silent information regulator T1 (SIRT1), also known
as the nicotinamide adenine dinucleotide-dependent deacetylase
sirtuin-1, is a member of a family of highly conserved proteins
that regulate a variety of metabolic functions, particularly
glucose homeostasis in mammals (23). SIRT1 is involved in alleviating
mitochondrial dysfunction and oxidative stress (24). In the present study, the promotion
of mitochondrial dysfunction and apoptosis by hyperglycemia was
examined in human umbilical vein endothelial (HUV-EC-C) cells.
SIRT1 activity was manipulated using the SIRT1 activator
resveratrol (RSV) and the SIRT1 inhibitor, sirtinol. The effect of
increased and decreased SIRT1 activity on hyperglycemia-induced
mitochondrial dysfunction and apoptosis in HUV-EC-C cells was then
examined. The data revealed the protective effects of SIRT1 against
hyperglycemia-induced apoptosis, which functions through the
alleviation of mitochondrial dysfunction and oxidative stress.
Materials and methods
Cell culture and treatment
The HUV-EC-C human umbilical vein endothelial cell
line was purchased from the American Type Culture Collection
(Manassas, VA, USA) and propagated in Kaighn's modification of
Ham's F-12 medium (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and 1X penicillin/streptomycin
(100 units/ml penicillin and 100 µg/ml streptomycin; Sigma-Aldrich;
Merck KGaA) at 37°C and 5% CO2. For the D-glucose
(Sigma-Aldrich; Merck KGaA) treatment, HUV-EC-C cells (at >85%
confluence) were treated with 5 (as a control), 15, 50 or 150 mg/ml
D-glucose for 0, 1, 2, 4, 6, 8, 16, 24, 32 or 48 h at 37°C. Control
cells were treated with 5 mg/ml D-glucose as this was required for
HUV-EC-C cell culture. The SIRT1 activator (RSV; 5 µM) and the
SIRT1 inhibitor (sirtinol; 10 µM), were obtained from Sigma-Aldrich
(Merck KGaA), and were used to regulate SIRT1 activity in
D-glucose-treated HUV-EC-C cells (>85% confluence), and were
incubated with cells for 8, 16, 24 or 48 h at 37°C and 5%
CO2.
Apoptosis assay via annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI)
staining
Following D-glucose treatment, apoptosis was
examined in HUV-EC-C cells using an annexin V-FITC/PI staining kit
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. Briefly, treated HUV-EC-C cells were harvested,
re-suspended in binding buffer (106 cells/ml) and
incubated with a mixture of 5 µl annexin V-FITC and 10 µl PI for 15
min in the dark at room temperature. Stained cells were visualized
with a live cell imaging system (Olympus Corporation, Tokyo, Japan)
and photographed using a velocity demo imaging system (PerkinElmer,
Waltham, MA, USA). The number of apoptotic cells was expressed as
the percentage of annexin V(+)/PI(−) cells out of the total number
cells.
Nuclear/cytosol protein fractionation,
isolation and western blotting assay
A Nuclear/Cytosol Fractionation kit (BioVision,
Inc., Milpitas, CA, USA) was used to isolate the nuclear and
cytosol protein fractions in HUV-EC-C cells. In brief,
post-treatment, HUV-EC-C cells (~5×106) were detached
via centrifugation for 5 min at 600 × g at 4°C and were washed
three times with ice-cold 1X phosphate buffered saline. The
HUV-EC-C cells (~5×106) were carefully resuspended in
500 µl ice-cold, 1X cytosol extraction buffer [containing
dithiothreitol (DTT)/protease inhibitors] by pipetting up and down.
Following centrifugation at 800 × g for 10 min at 4°C, the
supernatant (cytoplasmic fraction) was pipetted and stored at −80°C
for future use. The nuclear pellet was carefully resuspended in 100
µl ice-cold 1X nuclear extraction buffer (containing DTT/protease
inhibitors) by pipetting up and down, before samples were
centrifuged at 14,000 × g for 30 min at 4°C. The supernatant
(nuclear protein extract) was carefully transferred to another
tube, and stored at −80°C for future use.
For the western blotting assay, protein samples (15
µg per lane) were successively subjected to electrophoresis on 12%
SDS-PAGE gels and transferred to nitrocellulose membranes (EMD
Millipore, Billerica, MA, USA). Membranes were blocked with 2% FBS
at 4°C overnight, and incubated with primary rabbit polyclonal
antibodies against cytochrome c (cat. no. ab90529; 1:500; Abcam,
Cambridge, UK), caspase 3 (cat. no. ab44976; 1:600; Abcam), poly
(ADP-ribose) polymerase-1 (PARP; cat. no. ab32071; 1:400; Abcam),
SIRT1 (cat. no. S5447; Sigma-Aldrich; Merck KGaA) or β-actin [cat.
no. ab95437; diluted with 5% bovine serum albumin (Gibco; Thermo
Fisher Scientific, Inc.) to 1:1,000; Abcam] for 2 h at 37°C. The
membranes were then incubated with horseradish
peroxidase-conjugated goat anti-rabbit immunoglobulin G secondary
antibody (cat. no. HAF008; 1:1,000; R&D Systems, Inc.,
Minneapolis, MN, USA) for 2 h at 37°C. ECL™ Western Blotting
Detection Reagents (GE Healthcare Life Sciences, Little Chalfont,
UK) were used to detect the target protein bands.
Measurement of intracellular ROS and
mitochondrial superoxide
Intracellular ROS levels in ~2×106 cells
were monitored using the fluorescent probe 2′,7′-dichlorofluorescin
(DCFH) diacetate (5 µmol/l; Sigma-Aldrich; Merck KGaA), which was
oxidized to the highly fluorescent compound DCF. DCF-positive cells
were visualized under a live cell imaging system (Olympus
Corporation) with excitation measured at 485 nm and emission at 530
nm. In order to confirm that D-glucose treatment generates ROS, the
conversion of dihydroethidium (DHE) to ethidium by oxidation was
measured, and the ratio of DHE/ethidium was determined. DHE
excitation was measured at 355 nm and emission at 430 nm, while
ethidium excitation was measured at 518 nm and emission at 605 nm.
Mitochondrial superoxide was quantified with MitoSOX™ Red
(Invitrogen; Thermo Fisher Scientific, Inc.), which is
live-cell-permeable and indicates mitochondrial localized
superoxide. HUV-EC-C cells (at >85% confluence) were incubated
with 5 µM MitoSOX™ Red at 37°C for 20 min according to the
manufacturer's protocol. The MitoSOX™ Red fluorescence was detected
with an excitation at 510 nm and emission at 580 nm (FACSCanto flow
cytometer). Data were processed by using CellQuest Pro software
(version, 5.1; BD Biosciences, Franklin Lakes, NJ, USA). The data
were obtained from five independent images taken from each group,
and each experiment was repeated in triplicate.
Measurement of mitochondrial size and
mitochondrial membrane potential (MMP)
HUV-EC-C cells (~2×106) were stained with
20 nM MitoTracker Orange CMTMRos (Thermo Fisher Scientific, Inc.)
at 25°C for 2 h, and mitochondrial size was measured in four fields
per group using an Olympus FV500 confocal fluorescence microscope
(Olympus Corporation) at 540 nm excitation. A JC-1 Mitochondrial
Membrane Potential assay kit (Cayman Chemical Company, Ann Arbor,
MI, USA) was used to measure the MMP of treated HUV-EC-C cells,
according to manufacturer's instructions. Briefly, the treated
cells (~2×106) were incubated with JC-1 staining
solution (5 µg/ml) for 20 min at 37°C. The fluorescence intensity
of mitochondrial JC-1 monomers (514 nm excitation, 529 nm emission)
and aggregates (585 nm excitation, 590 nm emission) were detected
using a live cell imaging system (Olympus Corporation).
Measurement of adenosine
5′-diphosphate (ADP):adenosine trisphosphate (ATP) ratios
ATP and ADP levels were measured using an ADP
Colorimetric/Fluorometric assay kit (BioVision, Inc., Milpitas, CA,
USA) and a luminescence plate reader (PerkinElmer, Inc.). Following
D-glucose treatment, HUV-EC-C cells cultured in 12-well plates
(~2×105 cells/well) were lysed using the kit's nucleotide-releasing
reagent and transferred to 96-well white-walled plates according to
the manufacturer's instructions. ATP was measured in the sample,
then ADP was converted to ATP and the measurement was repeated to
permit the calculation of the ADP:ATP ratio.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total cellular RNA from HUV-EC-C cells
(~5×106) was isolated using an mRNA Isolation and
Purification kit (Clontech Laboratories, Inc., Mountain View, CA,
USA) containing RNasin® Plus RNase Inhibitor (Promega
Corporation, Madison, WI, USA). Reverse transcription of total RNA
(2 mol/µl) and qPCR was performed with the One Step SYBR
PrimeScript Plus RT-PCR kit (Perfect Real Time; Takara Bio, Otsu,
Japan). The reaction mix (20 µl) contained the following: 2X One
Step RT-PCR Buffer III (10 µl); TaKaRa Ex Taq HS (5 U/µl; 0.4 µl);
PrimeScript RT Enzyme Mix II (0.4 µl); forward/reverse primer (10
µM; 0.4 µl each); probe (0.8 µl); target RNA (2 µl); and RNase Free
dH2O (5.6 µl). The PCR reaction was performed as
follows: Stage 1 for reverse transcription reaction, 42°C for 5 min
and 95°C for 10 sec for 1 cycle; Stage 2 for PCR reaction, 95°C for
5 sec and 60°C for 20 sec for 40 cycles. The following primers were
obtained from Sangon Biotech Co., Ltd. (Shanghai, China): SIRT1,
5′-GAATACCTGACTTCAGGTCA-3′ (forward) and 5′-GAATACCTGACTTCAGGTCA-3′
(reverse); β-actin, 5′-TGTCCACCTTCCAGCAGATGT-3′ (forward) and
5′-AGCTCAGTAACAGTCCGCCTAGA-3′ (reverse). The experiment was
repeated three times. Relative quantification was determined using
the 2-∆∆Cq method with β-actin as a reference gene (25).
SIRT1 activity assay
SIRT1 activity was measured using a SIRT1 Activity
assay kit (Fluorometric; Abcam), according to the manufacturer's
protocol. Cells (~2×106) were lysed using Pierce
Universal Nuclease (Thermo Fisher Scientific, Inc.). Freshly lyzed
cellular solution was immunoprecipitated using a
Dynabeads® Co-Immunoprecipitation kit (Thermo Fisher
Scientific, Inc.) and an anti-SIRT1 antibody (cat. no. S5447;
Sigma-Aldrich; Merck KGaA). Subsequently, fluorosubstrate peptide
solution and protein A agarose beads were simultaneously added into
the reaction mix, and the NAD-dependent deacetylase activity was
measured based on fluorescence intensity at 2 min intervals at
excitation/emission =350/460 nm (BX-URA2; Olympus Corporation) and
was presented as the percentage of the control group.
Statistical analysis
SPSS statistical software (version, 16.0; SPSS,
Inc., Chicago, IL, USA) was utilized to analyze statistical
differences. Normally distributed quantitative data was expressed
as the mean ± standard error, while data that was not normally
distributed was logarithmically transformed for further analysis.
The Student's t-test or two-way analysis of variance and Dunnett's
post-hoc test were used to determine significant differences
between two groups and among multiple groups, respectively.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Hyperglycemia induces apoptosis in
human umbilical vein endothelial HUV-EC-C cells
To investigate the influence of hyperglycemia on the
viability of human endothelial cells, HUV-EC-C cells were treated
with a range of D-glucose concentrations for different durations,
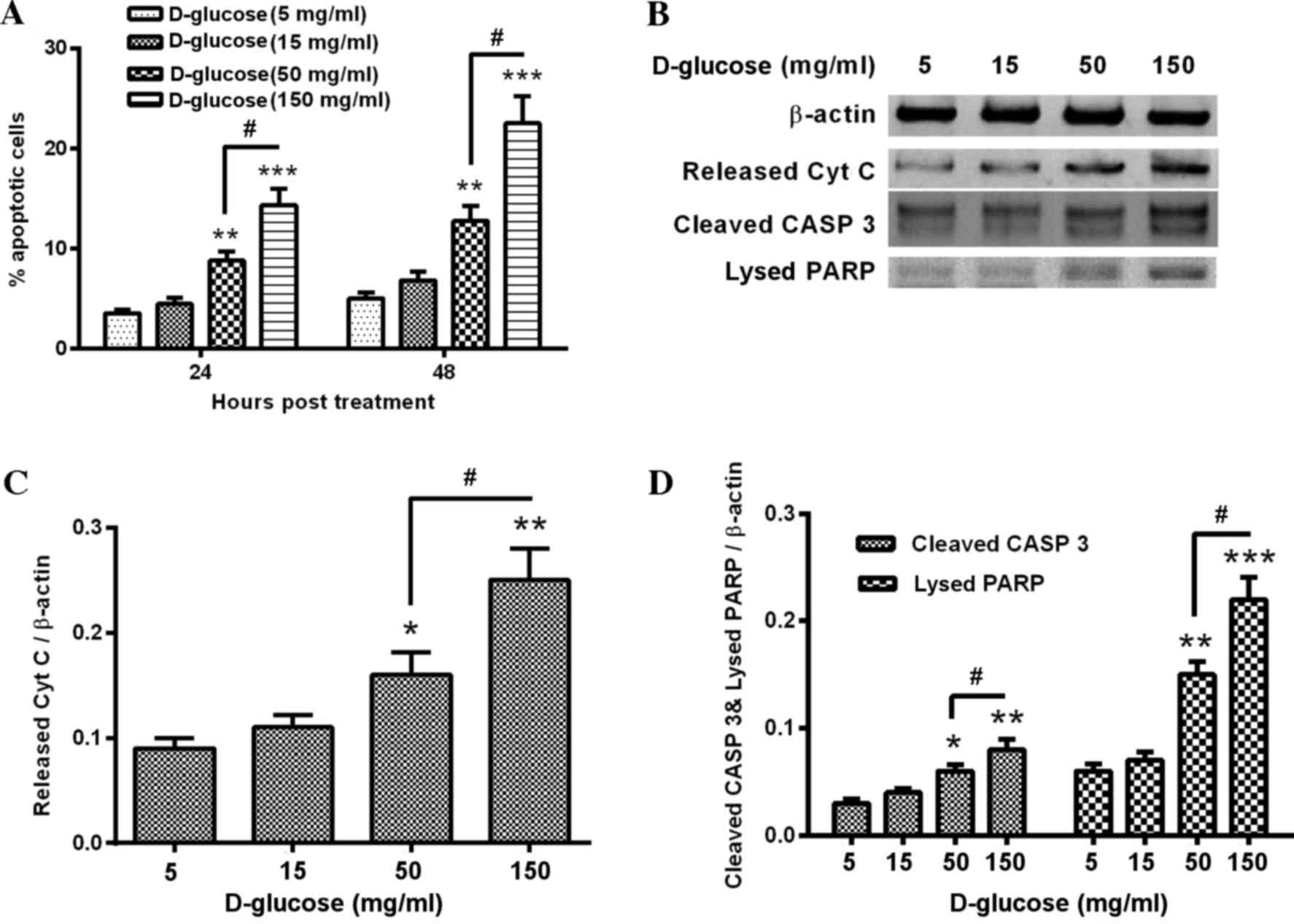
in order to evaluate hyperglycemia-induced apoptosis. Treatment
with 50 and 150 mg/ml D-glucose significantly increased the
percentage of apoptotic HUV-EC-C cells when compared with control
cells (50 mg/ml, P<0.01 and P<0.01at 24 and 48 h,
respectively; 150 mg/ml, P<0.001 and P<0.001 at 24 and 48 h,
respectively; Fig. 1A). The
induction of apoptosis was dose-dependent, with significantly
increased apoptosis levels in 150 mg/ml D-glucose treated cells
when compared with 50 mg/ml D-glucose-treated cells (P<0.05;
Fig. 1A). Western blot analysis
revealed a notable increase in apoptosis markers, including
released cytochrome C, cleaved caspase 3 and lysed PARP in 150
mg/ml D-glucose-treated cells following 24 h treatment when
compared with the controls (Fig.
1B). The release of cytochrome C from mitochondria was
significantly upregulated by 50 or 150 mg/ml D-glucose treatment
when compared with control cells (P<0.05 and P<0.01,
respectively; Fig. 1C). This
effect was also dose-dependent, with increased cytochrome C levels
in 150 mg/ml D-glucose treated cells compared with 50 mg/ml
D-glucose treated cells (P<0.05; Fig. 1C). Similar increases in expression
were demonstrated in D-glucose treated cells compared with controls
for cleaved caspase 3 (50 mg/ml, P<0.05; 150 mg/ml, P<0.01;
Fig. 1D) and lysed PARP (50 mg/ml,
P<0.01; 150 mg/ml, P<0.001; Fig.
1D). The effect of D-glucose on cleaved caspase 3 and lysed
PARP levels were dose-dependent, with significantly greater
increases in 150 mg/ml-treated cells when compared with 50
mg/ml-treated cells (cleaved caspase 3, P<0.05; lysed PARP,
P<0.05; Fig. 1D). Therefore,
hyperglycemia was confirmed to induce apoptosis in HUV-EC-C
cells.
Hyperglycemia promotes ROS
accumulation in HUV-EC-C cells
Previous studies have demonstrated an association
between oxidative stress and hyperglycemia-induced apoptosis in
cardiac myocytes (26) and
endothelial cells (27).
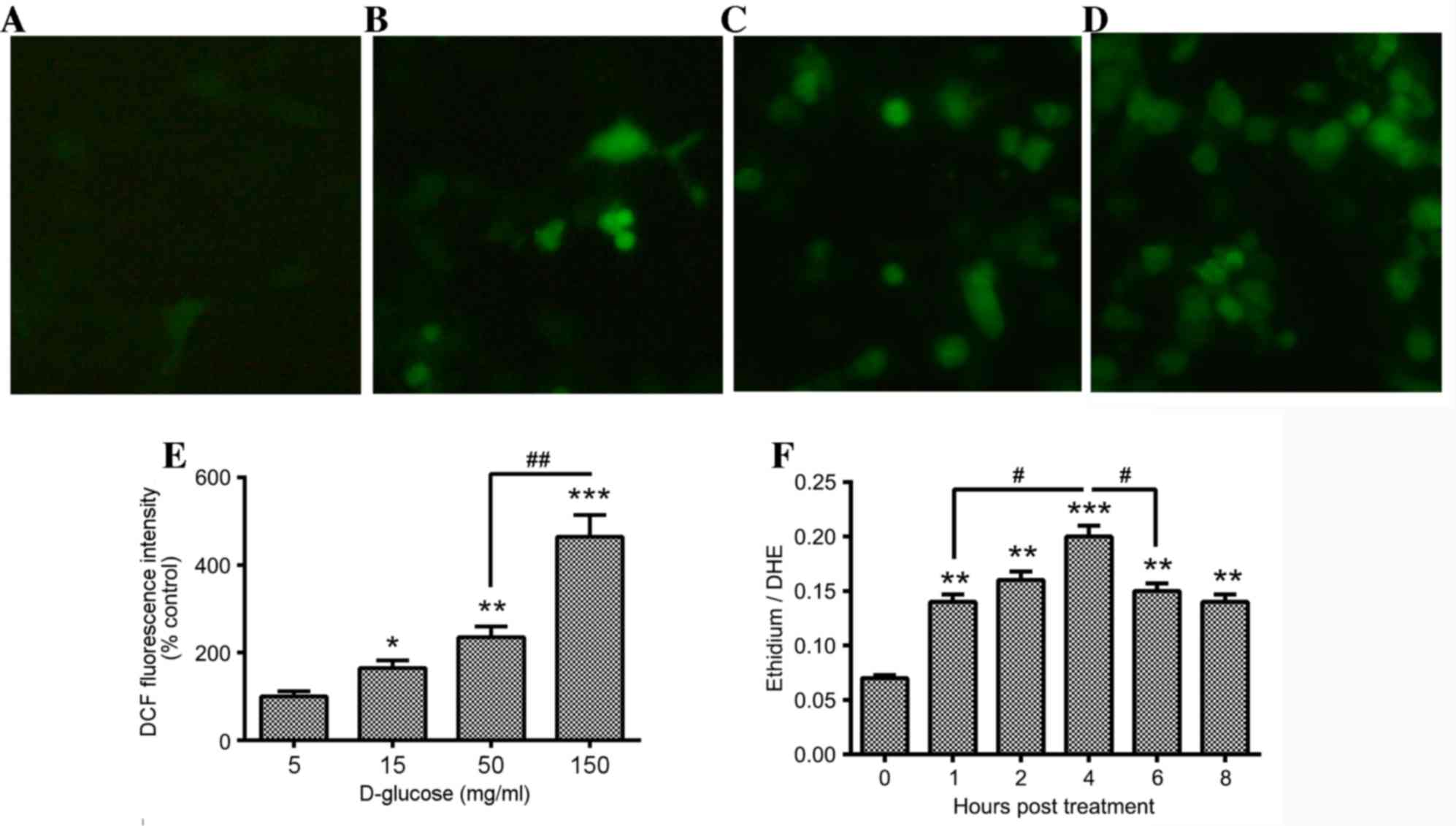
Therefore, the accumulation of ROS in D-glucose-treated HUV-EC-C
cells was examined using DCFH, which oxidizes to form the highly
fluorescent compound DCF. A small number of DCF-positive (green
fluorescence) HUV-EC-C cells were present in the control group
(Fig. 2A). However, there were
significantly increased levels of DCF-positive cells in the 15
(Fig. 2B) 50 (Fig. 2C) and 150 mg/ml (Fig. 2D) D-glucose-treated groups when
compared with the control cells (P<0.05, P<0.01 and
P<0.001, respectively; Fig.
2E). This effect was dose-dependent, with significantly
increased levels of DCF-positive cells in the 150 mg/ml treatment
group compared with the 50 mg/ml treated group (P<0.01; Fig. 2E). To determine the time-course of
ROS production in the D-glucose-treated HUV-EC-C cells, the
conversion of DHE to ethidium by oxidation was measured by assaying
the ratio of DHE to ethidium using a fluorimeter. ROS generation
increased in a dose-dependent manner in 50 mg/ml D-glucose-treated
HUV-EC-C cells from 1–8 h following treatment, with increased
levels of ROS at 1, 2 and 4 h following treatment when compared
with 0 h (P<0.01, P<0.01 and P<0.001, respectively;
Fig 2F). In addition,
significantly increased levels of ROS were observed at 4 h
following treatment compared with 1 h following treatment
(P<0.05; Fig. 2F), with
decreased ROS generation at 6 and 8 h following treatment when
compared with 4 h following treatment (P<0.05 at 6 h vs. 4 h;
Fig. 2F). The ratio of ethidium to
DHE peaked at 4 h following D-glucose treatment (Fig. 2F).
Hyperglycemia promotes mitochondrial
dysfunction in HUV-EC-C cells
To further investigate the effect of oxidative
stress exerted by hyperglycemia in the HUV-EC-C cells,
mitochondrial function was examined in D-glucose-treated HUV-EC-C
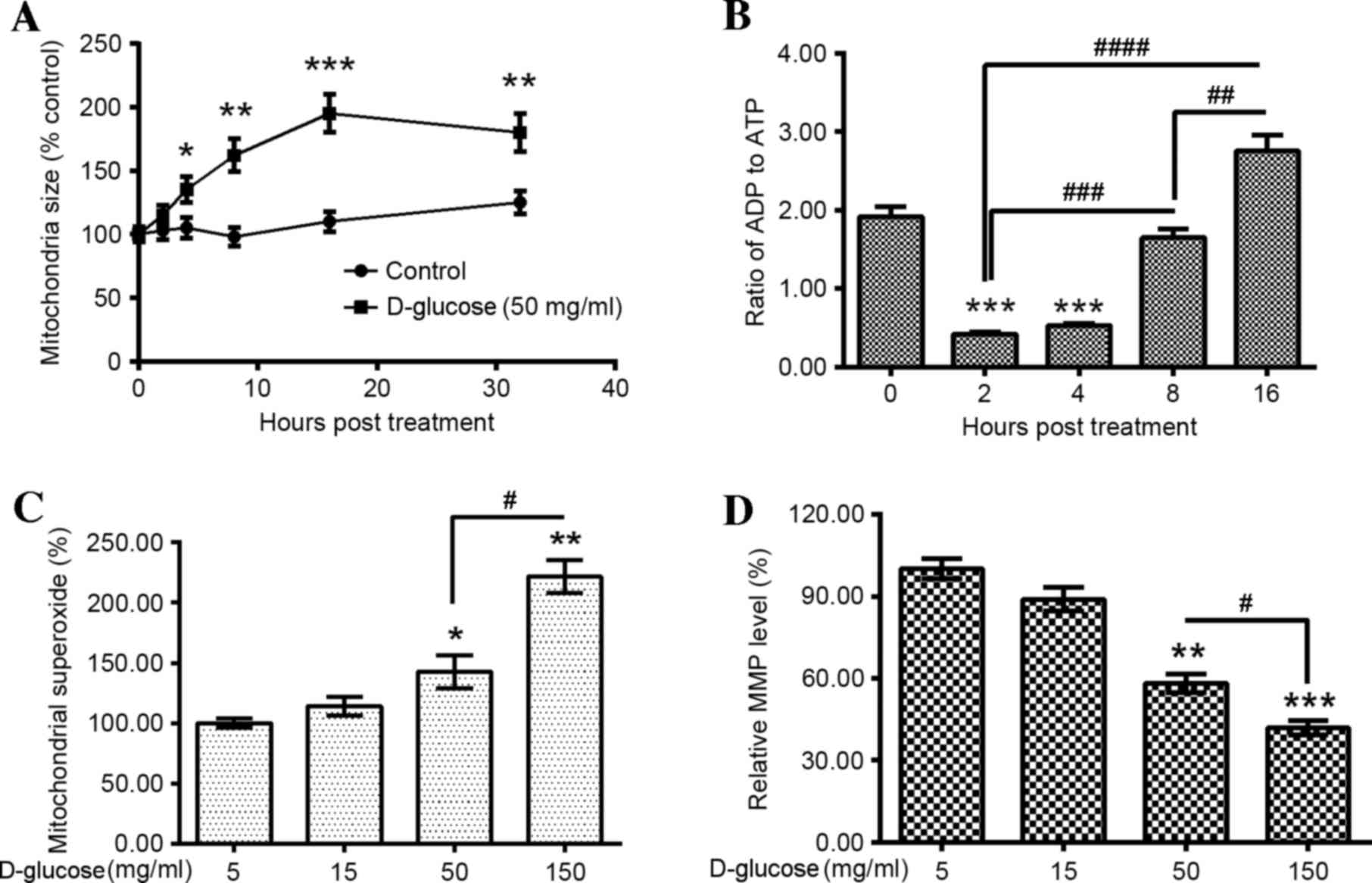
cells. An assay that measured glucose-induced mitochondrial
swelling was initially performed. Mitochondrial size significantly
increased from 4–32 h following 50 mg/ml D-glucose treatment when
compared with the 0 h time-point (4 h, P<0.05; 8 h, P<0.01;
16 h, P<0.001; 32 h, P<0.01; Fig. 3A), with mitochondrial swelling
peaking at 16 h following 50 mg/ml D-glucose treatment. The ratio
of ADP to ATP was then determined in the D-glucose-treated HUV-EC-C
cells. The ADP:ATP ratio decreased significantly at 2 and 4 h
following treatment when compared with the 0 h time-point
(P<0.001 and P<0.001, respectively; Fig. 3B). However, a significant increase
in the ADP: ATP ratio was observed at 8 and 16 h following
treatment when compared with the 2 h time-point (P<0.001 and
P<0.0001, respectively; Fig.
3B). This effect was time-dependent, with a significantly
greater increase at 16 h following treatment when compared to 8 h
(P<0.01; Fig. 3B).
In addition, the mitochondrial localized superoxide
and MMP were examined in D-glucose treated HUV-EC-C cells.
Mitochondrial superoxide was significantly upregulated by 50 and
150 mg/ml D-glucose treatment compared with 5 mg/ml
D-glucose-treated controls (P<0.05 and P<0.01, respectively;
Fig. 3C). This effect was
dose-dependent, with a significant increase in mitochondrial
superoxide observed in HUV-EC-C cells treated with 150 mg/ml
D-glucose when compared with 50 mg/ml D-glucose (P<0.05;
Fig. 3C). In addition, the MMP was
significantly reduced by 50 and 150 mg/ml D-glucose treatment when
compared with the controls (P<0.01 and P<0.001, respectively;
Fig. 3D). This effect was
dose-dependent, with a significantly greater reduction in MMP in
150 mg/ml D-glucose-treated cells when compared with 50 mg/ml
D-glucose-treated cells (P<0.05; Fig. 3D). These results suggest that
hyperglycemia promotes mitochondrial dysfunction in HUV-EC-C
cells.
Activation of SIRT1 inhibits
hyperglycemia-induced mitochondrial dysfunction and apoptosis in
HUV-EC-C cells
In order to investigate the involvement of SIRT1 in
hyperglycemia-induced mitochondrial dysfunction and apoptosis in
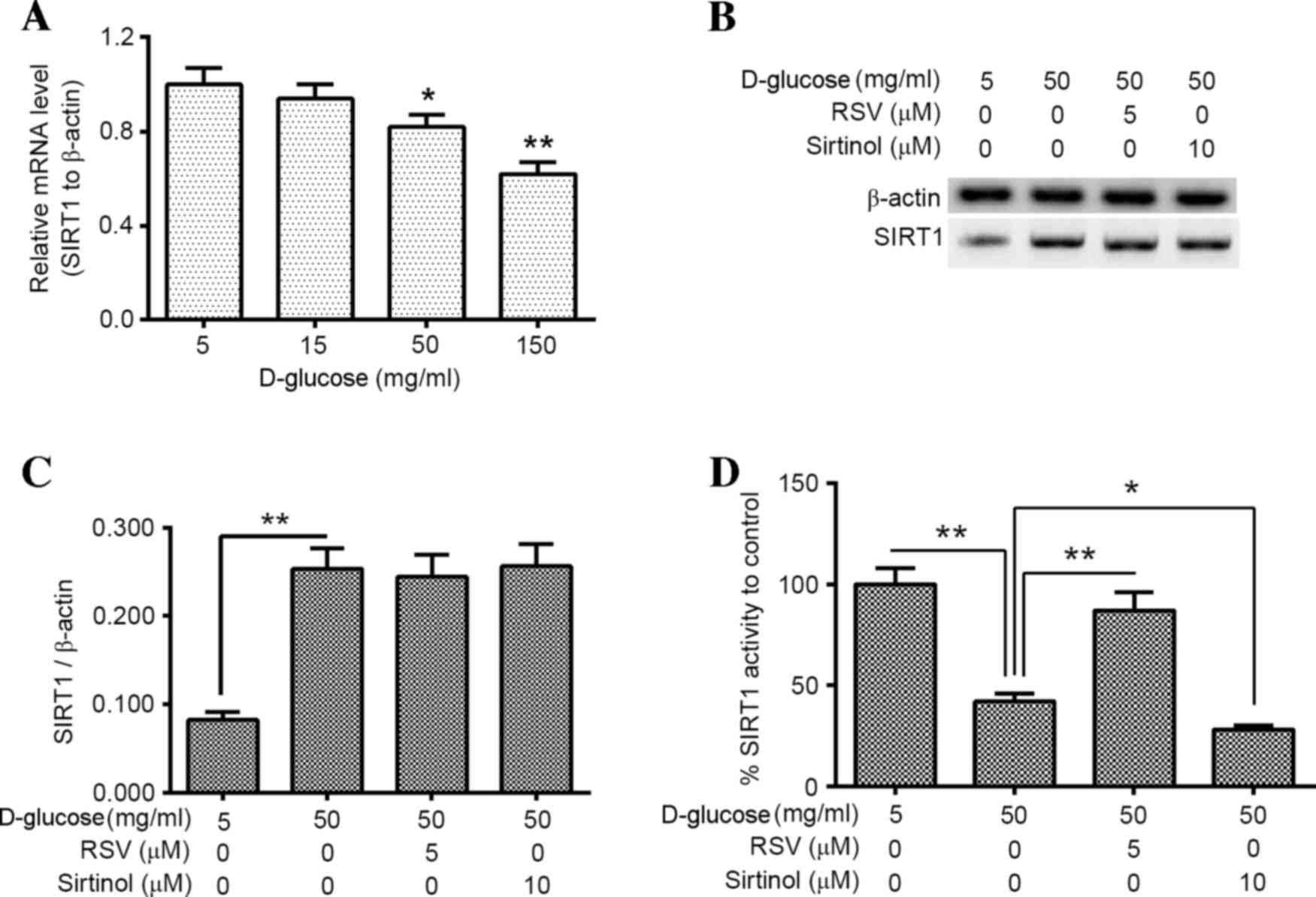
HUV-EC-C cells, the influence of hyperglycemia on SIRT1 expression
and activity in HUV-EC-C cells exposed to high glucose was
examined. SIRT1 activity was then manipulated with the SIRT1
activator (RSV) and inhibitor (sirtinol), and mitochondrial
function and induction of apoptosis were examined in
D-glucose-treated HUV-EC-C cells. SIRT1 mRNA expression levels were
significantly decreased following 50 and 150 mg/ml D-glucose
treatment when compared with the controls (P<0.05 and P<0.01,
respectively; Fig. 4A). However,
the western blotting assay demonstrated a significant increase in
SIRT1 protein expression levels following 50 mg/ml D-glucose
treatment when compared with controls (P<0.01; Fig. 4B and C). In addition, SIRT1
activity was determined in 50 mg/ml D-glucose-treated and untreated
HUV-EC-C cells. The results demonstrated thatSIRT1 activity was
significantly downregulated in 50 mg/ml D-glucose-treated HUV-EC-C
cells when compared with the controls (P<0.01; Fig. 4D). Although 5 µM RSV or 10 µM
sirtinol treatment failed to alter SIRT1 protein expression levels
in50 mg/ml D-glucose-treated HUV-EC-C cells (Fig. 4B and C), SIRT1 activity was
significantly increased following 50 mg/ml D-glucose plus 5 µM RSV
treatment when compared with 50 mg/ml D-glucose-only treated cells
(P<0.01; Fig. 4D), and
significantly reduced in 50 mg/ml D-glucose plus 10 µM sirtinol
treated cells when compared with 50 mg/ml D-glucose-only treated
cells (P<0.05; Fig. 4D).
Regulation of SIRT1 activity by RSV or sirtinol
significantly inhibited and increased hyperglycemia-induced
apoptosis and mitochondrial dysfunction, respectively. The
percentage of apoptotic cells was significantly decreased in 50
mg/ml D-glucose plus 5 µM RSV-treated HUV-EC-C cells when compared
with 50 mg/ml D-glucose-only treated cells at 24 and 48 h following
treatment (P<0.05 and P<0.05, respectively; Fig. 5A). By contrast, the percentage of
apoptotic cells was significantly increased in 50 mg/ml D-glucose
plus 10 µM sirtinol-treated HUV-EC-C cells when compared with 50
mg/ml D-glucose-only treated cells at 24 and 48 h following
treatment (P<0.05 and P<0.05, respectively; Fig. 5A). Western blotting assay results
indicated that the protein levels of apoptosis-associated markers,
including cytochrome c, cleaved caspase 3 and lyzed PARP, were
markedly decreased following 50 mg/ml D-glucose plus 5 µM RSV
treatment and notably increased following 50 mg/ml D-glucose plus
10 µM sirtinol treatment when compared with cells treated with 50
mg/ml D-glucose alone (Fig. 5B).
Hyperglycemia-induced accumulation of ROS was reduced by 50 mg/ml
D-glucose plus 5 µM RSV treatment and increased by 50 mg/ml
D-glucose plus 10 µM sirtinol treatment when compared with cells
treated with 50 mg/ml D-glucose alone (Fig. 5C). In addition, mitochondrial
swelling was significantly inhibited by 50 mg/ml D-glucose plus 5
µM RSV treatment, and significantly increased by 50 mg/ml D-glucose
plus 10 µM sirtinol treatment, when compared with cells treated
with 50 mg/ml D-glucose only (P<0.05 and P<0.05,
respectively; Fig. 5D). The
production of mitochondrial localized superoxide was significantly
decreased by 50 mg/ml D-glucose plus 5 µM RSV treatment, and
significantly increased by 50 mg/ml D-glucose plus 10 µM sirtinol
treatment, when compared with cells treated with 50 mg/ml D-glucose
only (P<0.05 and P<0.05, respectively; Fig. 5E). These results suggest that
activation of SIRT1 prevents hyperglycemia-induced mitochondrial
dysfunction and apoptosis in HUV-EC-C cells.
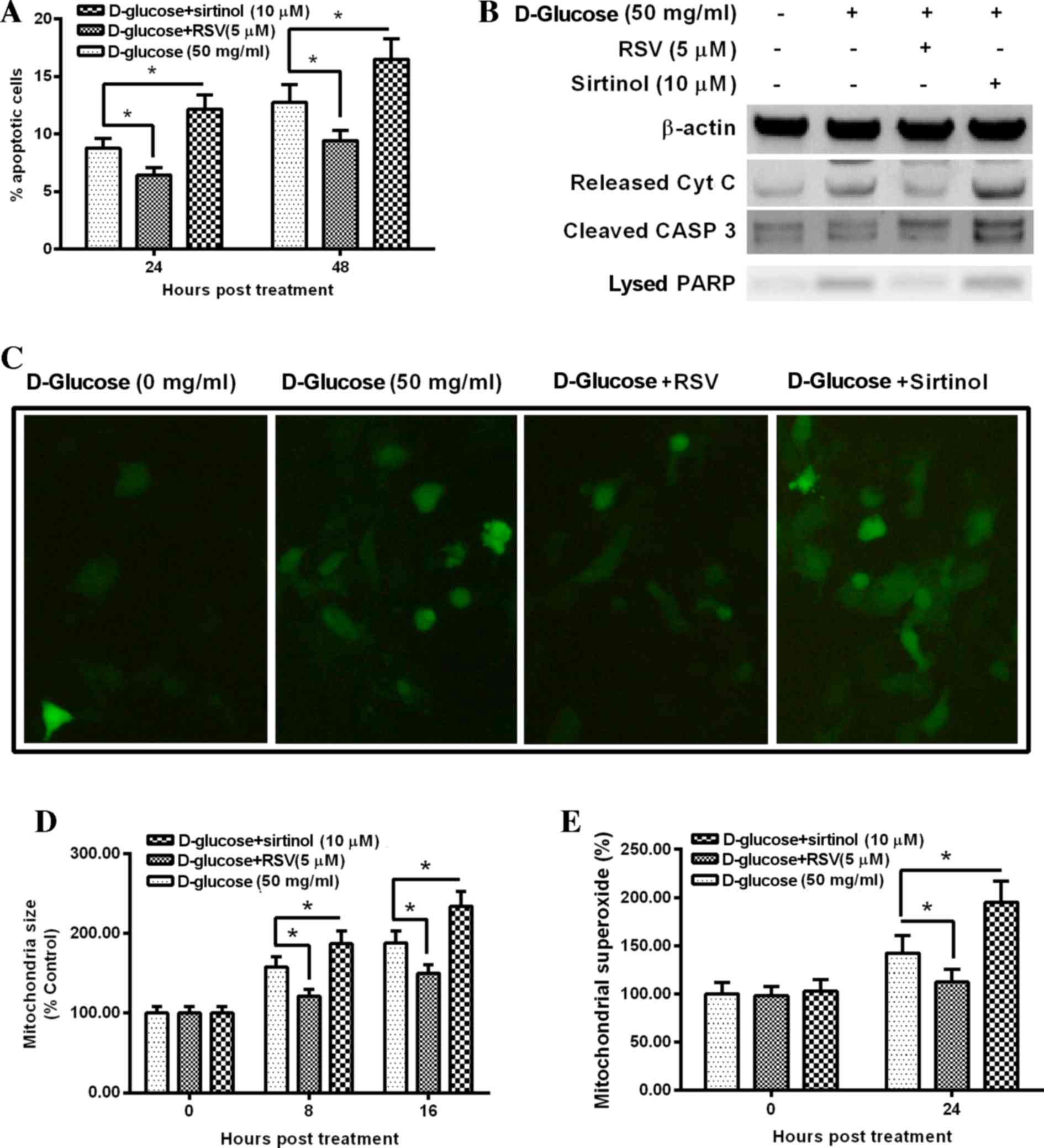 | Figure 5.Manipulation of SIRT1 activity
regulates hyperglycemia-induced apoptosis and mitochondrial
dysfunction in HUV-EC-C cells. (A) Apoptosis of D-glucose-treated
HUV-EC-C cells, which were treated with or without RSV or sirtinol.
(B) Western blot analysis of Cyt C, cleaved CASP 3 and lysed PARP
protein levels in D-glucose-treated HUV-EC-C cells, which were
treated with or without RSV or sirtinol for 24 h. (C)
Representative fluorescence images (magnification, ×40) of DCF
stained HUV-EC-C cells treated with D-glucose and with or without
RSV or sirtuin, with quantification of (D) mitochondrial size and
(E) mitochondrial superoxide levels. Data are presented as the mean
± standard deviation of four independent experiments. *P<0.05
vs. 50 mg/ml D-glucose. RSV, resveratrol; Cyt C, cytochrome C; CASP
3, caspase 3; PARP, poly(ADP-ribose) polymerase-1; DCF,
2′,7′-dichlorofluorescin. |
Discussion
DM-associated mitochondrial dysfunction has been
demonstrated to be markedly upregulated by hyperglycemia in
hepatocytes (28,29), skeletal muscle cells (30), cardiomyocytes (31,32),
retinal cells (33) and in renal
proximal tubular cells (34).
Dysfunctional mitochondria are one of the two major sources of
increased ROS production under hyperglycemic conditions (8–10).
The results of the present study confirmed that hyperglycemia
promotes oxidative stress and mitochondrial dysfunction in human
endothelial HUV-EC-C cells. ROS accumulation, mitochondrial
swelling, the ratio of ADP to ATP and the amount of mitochondrial
localized superoxide were increased by treatment with >50 mg/ml
D-glucose. In addition, the MMP was significantly decreased by
>50 mg/ml D-glucose treatment. Taken together, these results
confirm that hyperglycemia promoted oxidative stress and
mitochondrial dysfunction in HUV-EC-C cells. Dysfunctional
mitochondria have previously been demonstrated to induce apoptotic
damage to hepatocytes (28),
cardiomyocytes (31,32) and retinal cells (33). Consistent with these results, the
present study confirmed that hyperglycemia induces apoptosis in
HUV-EC-C cells, which suggests that hyperglycemia-induced oxidative
stress and mitochondrial dysfunction may lead to apoptotic death of
HUV-EC-C cells.
SIRT1 is the mammalian homolog of the yeast protein
Sir2, and is thought to be involved in the regulation of aging
(35). However, to the best of our
knowledge, only limited data supports the regulatory involvement of
SIRT1 in aging and longevity in mammals, although it has been
confirmed as a key regulator of cellular senescence and metabolic
pathways in response to nutrient status (35). SIRT1 is highly expressed in the
vasculature during blood vessel growth (36). Notably, SIRT1 alleviates oxidative
stress and mitochondrial dysfunction (24). Previous reports have demonstrated
that the SIRT1 activator, RSV, attenuates high-fat diet-induced
insulin resistance by influencing skeletal muscle lipid transport
and ameliorating mitochondrial function (37). Despite the potential of SIRT1
agonists to alleviate oxidative stress and mitochondrial
dysfunction, there remains a need to increase the specificity of
SIRT1 in ameliorating endothelial function in the context of
hyperglycemia. The results of the present study confirmed the
prevention of hyperglycemia-induced apoptosis by RSV and the
increase in hyperglycemia-induced apoptosis by sirtinol, via the
amelioration or enhancement of oxidative stress and mitochondrial
dysfunction in D-glucose-treated endothelial HUV-EC-C cells. The
present study expands upon previous observations of SIRT1
involvement in hyperglycemia-induced oxidative stress and
mitochondrial dysfunction in endothelial cells (27,36),
and the results suggest that manipulation of SIRT1 signaling may
ameliorate oxidative stress-mediated injury to endothelial cells by
hyperglycemia during vascular complications in either type I or
type II diabetes.
In conclusion, the present study confirmed that
oxidative stress, mitochondrial dysfunction and apoptosis are
increased by hyperglycemia in human umbilical vein endothelial
cells (HUV-EC-C). In addition, the results demonstrated that
manipulation of SIRT1 activity using a SIRT1 activator (RSV), or a
SIRT1 inhibitor (sirtinol), ameliorated or enhanced
hyperglycemia-induced mitochondrial dysfunction and apoptosis in
HUV-EC-C cells, respectively. The results confirm the protective
effects of SIRT1 against the hyperglycemia-induced apoptosis
potentially via the alleviation of mitochondrial dysfunction and
oxidative stress.
References
|
1
|
Chen F, Qian LH, Deng B, Liu ZM, Zhao Y
and Le YY: Resveratrol protects vascular endothelial cells from
high glucose-induced apoptosis through inhibition of NADPH oxidase
activation-driven oxidative stress. CNS Neurosci Ther. 19:675–681.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang XM, Yao M, Liu SX, Hao J, Liu QJ and
Gao F: Interplay between the Notch and PI3K/Akt pathways in high
glucose-induced podocyte apoptosis. Am J Physiol Renal Physiol.
306:F205–F213. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Muto T, Tien T, Kim D, Sarthy VP and Roy
S: High glucose alters Cx43 expression and gap junction
intercellular communication in retinal Müller cells: Promotes
Müller cell and pericyte apoptosis. Invest Ophthalmol Vis Sci.
55:4327–4337. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pan Y, Wang Y, Zhao Y, Peng K, Li W, Wang
Y, Zhang J, Zhou S, Liu Q, Li X, et al: Inhibition of JNK
phosphorylation by a novel curcumin analog prevents high
glucose-induced inflammation and apoptosis in cardiomyocytes and
the development of diabetic cardiomyopathy. Diabetes. 63:3497–3511.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cave AC, Brewer AC, Narayanapanicker A,
Ray R, Grieve DJ, Walker S and Shah AM: NADPH oxidases in
cardiovascular health and disease. Antioxid Redox Signal.
8:691–728. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Du XL, Edelstein D, Rossetti L, Fantus IG,
Goldberg H, Ziyadeh F, Wu J and Brownlee M: Hyperglycemia-induced
mitochondrial superoxide overproduction activates the hexosamine
pathway and induces plasminogen activator inhibitor-1 expression by
increasing Sp1 glycosylation. Proc Natl Acad Sci USA.
97:12222–12226. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nishikawa T, Edelstein D, Du XL, Yamagishi
S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP,
et al: Normalizing mitochondrial superoxide production blocks three
pathways of hyperglycaemic damage. Nature. 404:787–790. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang WF, Xu YY, Xu KP, Wu WH, Tan GS, Li
YJ and Hu CP: Inhibitory effect of selaginellin on high
glucose-induced apoptosis in differentiated PC12 cells: Role of
NADPH oxidase and LOX-1. Eur J Pharmacol. 694:60–68. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sachse A and Wolf G: Angiotensin
II-induced reactive oxygen species and the kidney. J Am Soc
Nephrol. 18:2439–2446. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Coppey LJ, Davidson EP, Rinehart TW,
Gellett JS, Oltman CL, Lund DD and Yorek MA: ACE inhibitor or
angiotensin II receptor antagonist attenuates diabetic neuropathy
in streptozotocin-induced diabetic rats. Diabetes. 55:341–348.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hammes HP, Martin S, Federlin K, Geisen K
and Brownlee M: Aminoguanidine treatment inhibits the development
of experimental diabetic retinopathy. Proc Natl Acad Sci USA.
88:11555–11558. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Du Y, Miller CM and Kern TS: Hyperglycemia
increases mitochondrial superoxide in retina and retinal cells.
Free Radic Biol Med. 35:1491–1499. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xia P, Inoguchi T, Kern TS, Engerman RL,
Oates PJ and King GL: Characterization of the mechanism for the
chronic activation of diacylglycerol-protein kinase C pathway in
diabetes and hypergalactosemia. Diabetes. 43:1122–1129. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Koya D and King GL: Protein kinase C
activation and the development of diabetic complications. Diabetes.
47:859–866. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nerlich AG, Sauer U, Kolm-Litty V, Wagner
E, Koch M and Schleicher ED: Expression of
glutamine:fructose-6-phosphate amidotransferase in human tissues:
Evidence for high variability and distinct regulation in diabetes.
Diabetes. 47:170–178. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Perez-Matute P, Zulet MA and Martinez JA:
Reactive species and diabetes: Counteracting oxidative stress to
improve health. Curr Opin Pharmacol. 9:771–779. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qin X, Zhang Z, Xu H and Wu Y: Notch
signaling protects retina from nuclear factor-κB- and
poly-ADP-ribose-polymerase-mediated apoptosis under high-glucose
stimulation. Acta Biochim Biophys Sin (Shanghai). 43:703–711. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Q and Steinle JJ: DNA-PK
phosphorylation of IGFBP-3 is required to prevent apoptosis in
retinal endothelial cells cultured in high glucose. Invest
Ophthalmol Vis Sci. 54:3052–3057. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He M, Nitti M, Piras S, Furfaro AL,
Traverso N, Pronzato MA and Mann GE: Heme oxygenase-1-derived
bilirubin protects endothelial cells against high glucose-induced
damage. Free Radic Biol Med. 89:91–98. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He S, Zhang J, Qi X, Wang D, Wang X and
Zhou S: Neuregulin protects human umbilical vein endothelial cell
via activating CD98hc through MAPK pathway. Int J Clin Exp Med.
8:6702–6712. 2015.PubMed/NCBI
|
|
21
|
Zhang Y, Liu J, Luo JY, Tian XY, Cheang
WS, Xu J, Lau CW, Wang L, Wong WT, Wong CM, et al: Upregulation of
angiotensin (1–7)-mediated signaling preserves endothelial function
through reducing oxidative stress in diabetes. Antioxid Redox
Signal. 23:880–892. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang R, Lu L, Guo Y, Lin F, Chen H, Chen W
and Chen M: Effect of glucagon-like peptide-1 on
high-glucose-induced oxidative stress and cell apoptosis in human
endothelial cells and its underlying mechanism. J Cardiovasc
Pharmacol. 66:135–140. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen YR, Lai YL, Lin SD, Li XT, Fu YC and
Xu WC: SIRT1 interacts with metabolic transcriptional factors in
the pancreas of insulin-resistant and calorie-restricted rats. Mol
Biol Rep. 40:3373–3380. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gerhart-Hines Z, Rodgers JT, Bare O, Lerin
C, Kim SH, Mostoslavsky R, Alt FW, Wu Z and Puigserver P: Metabolic
control of muscle mitochondrial function and fatty acid oxidation
through SIRT1/PGC-1alpha. EMBO J. 26:1913–1923. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Duan J, Wei G, Guo C, Cui J, Yan J, Yin Y,
Guan Y, Weng Y, Zhu Y, Wu X, et al: Aralia taibaiensis protects
cardiac myocytes against high glucose-induced oxidative stress and
apoptosis. Am J Chin Med. 43:1159–1175. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao H, Ma T, Fan B, Yang L, Han C, Luo J
and Kong L: Protective effect of trans-δ-viniferin against high
glucose-induced oxidative stress in human umbilical vein
endothelial cells through the SIRT1 pathway. Free Radic Res.
50:68–83. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiang X, Tang X, Zhang P, Liu G and Guo H:
Cyanidin-3-O-β-glucoside protects primary mouse hepatocytes against
high glucose-induced apoptosis by modulating mitochondrial
dysfunction and the PI3K/Akt pathway. Biochem Pharmacol.
90:135–144. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xu Z, Zhang L, Li X, Jiang Z, Sun L, Zhao
G, Zhou G, Zhang H, Shang J and Wang T: Mitochondrial
fusion/fission process involved in the improvement of catalpol on
high glucose-induced hepatic mitochondrial dysfunction. Acta
Biochim Biophys Sin (Shanghai). 47:730–740. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang HH, Ma XJ, Wu LN, Zhao YY, Zhang PY,
Zhang YH, Shao MW, Liu F, Li F and Qin GJ: SIRT1 attenuates high
glucose-induced insulin resistance via reducing mitochondrial
dysfunction in skeletal muscle cells. Exp Biol Med (Maywood).
240:557–565. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang M, Niu X, Hu J, Yuan Y, Sun S, Wang
J, Yu W, Wang C, Sun D and Wang H: Lin28a protects against
hypoxia/reoxygenation induced cardiomyocytes apoptosis by
alleviating mitochondrial dysfunction under high glucose/high fat
conditions. PLoS One. 9:e1105802014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang H and Wu K, You Q, Huang R, Li S and
Wu K: Naringin inhibits high glucose-induced cardiomyocyte
apoptosis by attenuating mitochondrial dysfunction and modulating
the activation of the p38 signaling pathway. Int J Mol Med.
32:396–402. 2013.PubMed/NCBI
|
|
33
|
Roy S, Trudeau K, Roy S, Tien T and
Barrette KF: Mitochondrial dysfunction and endoplasmic reticulum
stress in diabetic retinopathy: Mechanistic insights into high
glucose-induced retinal cell death. Curr Clin Pharmacol. 8:278–284.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Munusamy S and MacMillan-Crow LA:
Mitochondrial superoxide plays a crucial role in the development of
mitochondrial dysfunction during high glucose exposure in rat renal
proximal tubular cells. Free Radic Biol Med. 46:1149–1157. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bordone L and Guarente L: Calorie
restriction, SIRT1 and metabolism: Understanding longevity. Nat Rev
Mol Cell Biol. 6:298–305. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Potente M, Ghaeni L, Baldessari D,
Mostoslavsky R, Rossig L, Dequiedt F, Haendeler J, Mione M, Dejana
E, Alt FW, et al: SIRT1 controls endothelial angiogenic functions
during vascular growth. Genes Dev. 21:2644–2658. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen LL, Zhang HH, Zheng J, Hu X, Kong W,
Hu D, Wang SX and Zhang P: Resveratrol attenuates high-fat
diet-induced insulin resistance by influencing skeletal muscle
lipid transport and subsarcolemmal mitochondrial β-oxidation.
Metabolism. 60:1598–1609. 2011. View Article : Google Scholar : PubMed/NCBI
|