Introduction
Osteoarthritis (OA) is a prevalent degenerative
joint disease, which is characterized by progressive destruction of
the articular cartilage and subchondral bone. Numerous etiological
factors are implicated in the pathogenesis of OA; however,
cartilage destruction appears to be due to chondrocyte apoptosis
(1). It has previously been
suggested that microRNAs (miRNAs/miR) are involved in the
pathogenesis of OA (2). miRNAs are
single-stranded, non-coding small RNAs that negatively regulate the
expression of target genes in a post-transcriptional manner;
>700 miRNAs have been identified to serve a role in the
regulation of cellular processes, including proliferation,
apoptosis and differentiation (3,4).
Previous studies that used miRNA microarrays to compare the
expression of miRNAs between normal and OA human articular
cartilage revealed that miRNAs were comprehensively involved in OA
generation (5,6). Diaz-Prado et al (6) previously generated cartilage-specific
Dicer-null mice, which exhibited reduced chondrocyte proliferation
and severe growth defects, thus indicating the importance of miRNAs
in regulating cartilage function. However, the expression and
functional role of miRNAs in OA remains to be fully elucidated.
Therefore, the miRNA expression pattern and functions that are
essential for chondrocyte development and OA damage require further
investigation. The inflammatory cytokine interleukin (IL)-1β, which
is a major catabolic inducer, is one of the most prominent
catabolic cytokines, which serves a crucial role in OA pathogenesis
(7,8). IL-1β promotes the production of
proteases, such as matrix metallo proteinases (MMPs), and inhibits
the synthesis of proteoglycans and collagens by chondrocytes
(9,10). Previous studies have suggested that
various miRNAs are involved in IL-1β-induced OA cartilage
destruction, including miR-9, miR-98 (11), miR-140 (12), miR-146a (13) and miR-558 (14).
Previous studies have investigated the role of
miR-98 in the regulation of tumor growth, invasion, angiogenesis
and anti-inflammation in various disease models. Siragam et
al (15) defined a regulatory
role for miR-98 in tumor angiogenesis and invasion via the
suppression of activin A receptor type 1B and MMP11 expression. Du
et al (16) suggested that
miR-93, miR-98 and miR-197 exert a negative regulatory effect on
the expression of tumor suppressor gene FUS1 in lung cancer. Li
et al (17) demonstrated
that the expression levels of miR-98 were reduced in melanoma
tissues at a higher tumor stage and in melanoma with metastasis,
and suggested that miR-98 inhibited melanoma metastasis via a novel
miR-98-IL-6-negative feedback loop. However, the expression pattern
and functional role of miR-98 in chondrocyte apoptosis of OA
cartilage remains to be fully elucidated. The present study aimed
to determine the molecular mechanism underlying the pathogenesis of
OA. Therefore, the expression levels of miR-98 in normal
chondrocytes and IL-1β-treated OA chondrocytes were detected. In
addition, the role of miR-98 in an IL-1β-induced OA disease model
was identified, suggesting its potential involvement in the
regulation of apoptosis. The role of miR-98 in the regulation of
chondrocyte proliferation and apoptosis was investigated using an
in vitro OA model. The findings of the present study
confirmed that miR-98 targeted the 3′-untranslated region (3′-UTR)
of B-cell lymphoma 2 (Bcl-2). These findings suggested that miR-98
serves a crucial role in the coordinated regulation of the
expression of apoptosis-associated proteins in chondrocytes in
response to IL-1β-induction.
Materials and methods
Mouse primary chondrocyte culture
The present study was approved by the Ethics
Committee of the First People's Hospital of Yunnan Province
(Kunming, China). A total of 30 2-week old male C57BL/6 mice (~10
g) were purchased from the Shanghai Animal Center, Chinese Academy
of Sciences and housed in a specific pathogen free facility with a
constant humidity and temperature at 12:12 h light:dark cycle with
free access to food and water for 2 days in the animal research
facility in accordance with an approved protocol. Mice were
anesthetized with diethyl ether (60297; Sinopharm Chemical Reagent
Co., Ltd., Shanghai, China) and sacrificed, then cartilage sections
of mice were aseptically removed and washed in PBS (pH 7.4; Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 1%
penicillin-streptomycin (Gibco; Thermo Fisher Scientific, Inc.).
Subsequently, cartilage slices were minced with sterile ophthalmic
scissors and transferred into digestion buffer containing 0.25%
trypsin (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) for 30 min
at 37°C. The supernatant containing trypsin was discarded following
centrifugation at 4°C for 10 min at 1,000 × g and the trypsinized
cartilage was digested again using 2 mg/l type IV collagenase
(Sigma-Aldrich; Merck KGaA) for 4–6 h at 37°C. Following the
digestion, the cells were washed with Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.) containing 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and
centrifuged at 800 × g for 10 min at 4°C prior to being cultured.
The quantity of chondrocytes obtained was determined using a
hemocytometer, and 0.4% Trypan blue dye (Sigma-Aldrich, Merck KGaA)
was used to assess the viability of the cells. Chondrocytes were
cultured in a 25 cm2 culture flask (Corning, Inc.,
Corning, NY, USA) in DMEM supplemented with 1% glutamine and 10%
FBS at 37°C in 5% CO2. Half of the medium was replaced
once every 3 days with an equal quantity of fresh medium. Cells
between passages 3 and 10 were used in the present study.
Normal chondrocytes were treated with 10 ng/ml IL-1β
for 12, 24 and 48 h for OA chondrocyte preparation. The cell
treatment groups including the control group, IL-1β treatment
group, IL-1β treatment with miR-98 inhibitor transfection group and
corresponding control, miR-98 mimic transfection group and miR-98
mimic control group.
Transfections with miRNA mimics and
inhibitors
In order to manipulate the cellular function of
miR-98 in mouse chondrocytes, the present study used specific
antisense oligonucleotides to miRNAs in order to inhibit miRNA
function, and specific miRNA precursors to increase miRNA
expression. Briefly, following culture of mouse chondrocytes to 90%
confluence, they were transfected with 3–100 nM miR-98 mimic,
miR-98 inhibitor or negative controls (Sangon Biotech Co., Ltd.,
Shanghai, China) using Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
The expression levels of miR-98 were detected 24 h
post-transfection in order to confirm the optimal concentration of
miR-98 mimic, miR-98 inhibitor or negative controls had been used.
The final concentration used in the present study was 50 nM.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol, and the concentration of total RNA was
determined using NanoDrop 2000 (Thermo Fisher Scientific, Inc.).
cDNA synthesis for the detection of target genes and miRNA was
performed with 1 µg total RNA using PrimeScript™ 1st Strand cDNA
Synthesis kit (Takara Bio, Inc., Otsu, Japan) according to the
manufacturer's protocol. The mRNA expression levels of Fas cell
surface death receptor (Fas), caspase-3, caspase-8,
Bcl-2-associated X protein (Bax), Bcl-2 and GAPDH, and the
expression levels of miR-98 and U6 were analyzed using SYBR Green
PCR Master Mix kit (Takara Bio, Inc.) according to the
manufacturer's protocol. Primer sequences are presented in Table I. All oligonucleotide primers were
synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). qPCR
reactions (denaturation: 95°C, 30 sec 1 cycle; PCR reaction: 95°C,
5 sec, 60°C, 40 sec 45 cycles) were performed on an ABI7900
Real-Time system (Applied Biosystems; Thermo Fisher Scientific,
Waltham, MA, USA). The Cq values were analyzed using the
comparative Cq (ΔΔCq) method (18)
and the quantity of target mRNA was obtained by normalizing to the
endogenous references (GAPDH or U6), relative to the control. All
experiments were repeated at least six times.
 | Table I.Primer sequences for quantitative
polymerase chain reaction. |
Table I.
Primer sequences for quantitative
polymerase chain reaction.
| Gene | Sense (5′-3′) | Antisense
(5′-3′) |
|---|
| miR-98 |
ACACTCCAGCTGGGTGAGGTAGTAAGT |
CTCAACTGGTGTCGTGGAGTC |
| U6 |
CTCGCTTCGGCAGCACATTGC |
AACGCTTCACGAATTTGCGT |
| Fas |
AGACTGCGTGCCCTGCCAAGA |
GGCCTGCCTGTTCAGTAACT |
| Caspase-3 |
CTCGCTCTGGTACGGATGTG |
TCCCATAAATGACCCCTTCATCA |
| Caspase-8 |
CGTGCCTAATGGCGTTAACCA |
AGCCTTGGCCAGCCGACCTT |
| Bax |
GACCCGGTGCCTCAGGATGC |
GTCTGTGTCCACGGCGGCAA |
| Bcl-2 |
GCTACCGTCGTGACTTCGC |
CCCCACCGAACTCAAAGAAGG |
| GAPDH |
GACCCCTTCATTGACCTCAACTACA |
GTCCACCACCCTGTTGCTGTAGCCA |
Western blotting
Whole cell lysates were obtained using RIPA Protein
Extraction reagent (Beyotime Institute of Biotechnology, Inc.,
Haimen, China) plus EDTA-free Protease Inhibitor Cocktail tablets
(Roche Diagnostics, Basel, Switzerland). Protein concentrations
were detected using the BCA Protein Assay kit. Equal amounts of
protein (25 µg) were separated by 12.5% SDS-PAGE. Total proteins
were electrotransferred onto polyvinylidene fluoride membranes
(Merck KGaA) and blocked with 3% bovine serum albumin (Beyotime
Institute of Biotechnology, Inc., Haimen, China) in Tris-HCl
buffered saline following electrophoresis. Bcl-2 (1,5071; 28 kD;
1:2,000) and β-actin antibodies (12262; 42 kD; 1:2,000) were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Following the primary antibody incubation at 4°C overnight,
membranes were incubated with horseradish peroxidase-conjugated
species-specific secondary antibody (sc-2031, 1:5,000; Santa Cruz
Biotechnology, Inc.) at 37°C for 1 h and the proteins were
visualized using an enhanced chemiluminescence kit (Pierce; Thermo
Fisher Scientific, Inc.). The results of the western blotting were
analyzed using Gel-Pro Analyzer version 4.0 (Media Cybernetics,
Inc., Rockville, MD, USA). All experiments were repeated at least
three times. Densitometric levels of target protein were
semi-quantified and normalized to β-actin, relative to
untransfected cells.
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) staining
TUNEL staining was performed 24 h post-transfection
according to the manufacturer's protocol (Merck KGaA). The
chondrocytes were fixed in cold 4% paraformaldehyde in PBS for 20
min at room temperature and washed three times in cold PBS.
Subsequently equilibration buffer was added at room temperature for
10 min. The fixed and permeabilized samples were incubated with
DNaseI (300 U/ml in 50 mM Tris-HCl, pH 7.5) for 10 min at room
temperature in order to induce DNA strand breaks prior to labeling
procedures. Reaction mix (10–50 µl/well to immerse all samples) was
added to the cells and incubated at 37°C for 2 h in a humidified
atmosphere in the dark. Subsequently, the chondrocytes were rinsed
with PBS three times for 5 min. Hoechst 33342 solution was applied
for 1 min in order to visualize all nuclei under a fluorescent
microscope (Leica Microsystems GmbH, Wetzlar, Germany).
Cell proliferation assay
The cell proliferation rate was evaluated using an
MTT proliferation assay. Briefly, following exposure to IL-1β
(PHC0813; Thermo Fisher Scientific, Inc.) or transfection with
recombinant plasmids, cells were cultured in 96-well plates and
were washed twice with PBS at 24 and 48 h. Subsequently, 0.5 mg/ml
MTT was added to each well and the cells were incubated for 4 h at
37°C in 5% CO2. The medium was replaced with 200 µl
dimethyl sulfoxide, the plates were agitated for 15 min at room
temperature and the absorbance was determined at 490 nm using a
microplate spectrophotometer.
Luciferase reporter constructs and
luciferase assay
cDNA oligonucleotides (Sangon Biotech Shanghai, Co.,
Ltd., Shanghai, China) containing the putative miR-98 target site
according to miRBase sequence database (http://www.mirbase.org) within the 3′-UTR of the
target gene were synthesized with Hind III and Sac I restriction
enzyme digestion (Takara Biotechnology Co., Ltd., Dalian, China)
sites and cloned into the multiple cloning site of the pMIR-REPORT
Luciferase vector (Ambion; Thermo Fisher Scientific, Inc.). An
additional pMIR-REPORT Luciferase construct containing mutant
3′-UTR was generated as a control. Subsequently, HEK293 cells (Cell
bank of Chinese Academy of Sciences, Shanghai, China) were cultured
in 24-well plates with DMEM and 10% FBS for 24 h and then were
transfected with each reporter construct, alongside miR-98 mimic or
miR-98 inhibitor, using Lipofectamine 2000 reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. Firefly luciferase activity was quantified at 48 h
post-transfection using the dual-luciferase reporter assay system
(Thermo Fisher Scientific, Inc.).
Statistical analysis
Statistical analysis was conducted with SPSS 18.0
(SPSS, Inc., Chicago, IL, USA). All of the experiments were
performed with samples in triplicate or greater. Data are presented
as the mean ± standard deviation of six independent experiments.
One-way analysis of variance with Tukey HSD post hoc t tests were
used to determine levels of statistical significance. Analysis of
variance was used to determine statistical significance of the
differences between the treatment groups. P<0.05 was considered
to indicate a statistically significant difference.
Results
Relative expression of miR-98 and
apoptotic factors in chondrocytes following IL-1β treatment
In order to investigate whether the expression of
miR-98 was induced by IL-1β, mouse primary chondrocytes were
incubated with IL-1β (10 ng/ml) for 12, 24 and 48 h. The expression
levels of miR-98 were upregulated 2.75-fold at 24 h and 3.61-fold
at 48 h. The expression of miR-98 was significantly increased
following incubation with 10 ng/ml IL-1β for 24 and 48 h compared
with in the 0 h treatment group (P<0.05; Fig. 1A). The effects of IL-1β on
apoptotic factors were also determined by examining their
expression levels, including Fas, caspase-3, caspase-8, Bax and
Bcl-2. Exposure of primary chondrocytes to IL-1β (10 ng/ml) for 48
h significantly reduced the expression levels of Bcl-2 at the mRNA
and protein level (P<0.05; Fig. 1A
and B). The mRNA expression levels of Fas, caspase-3, caspase-8
and Bax were significantly greater following IL-1β stimulation for
48 h (P<0.05; Fig. 1A).
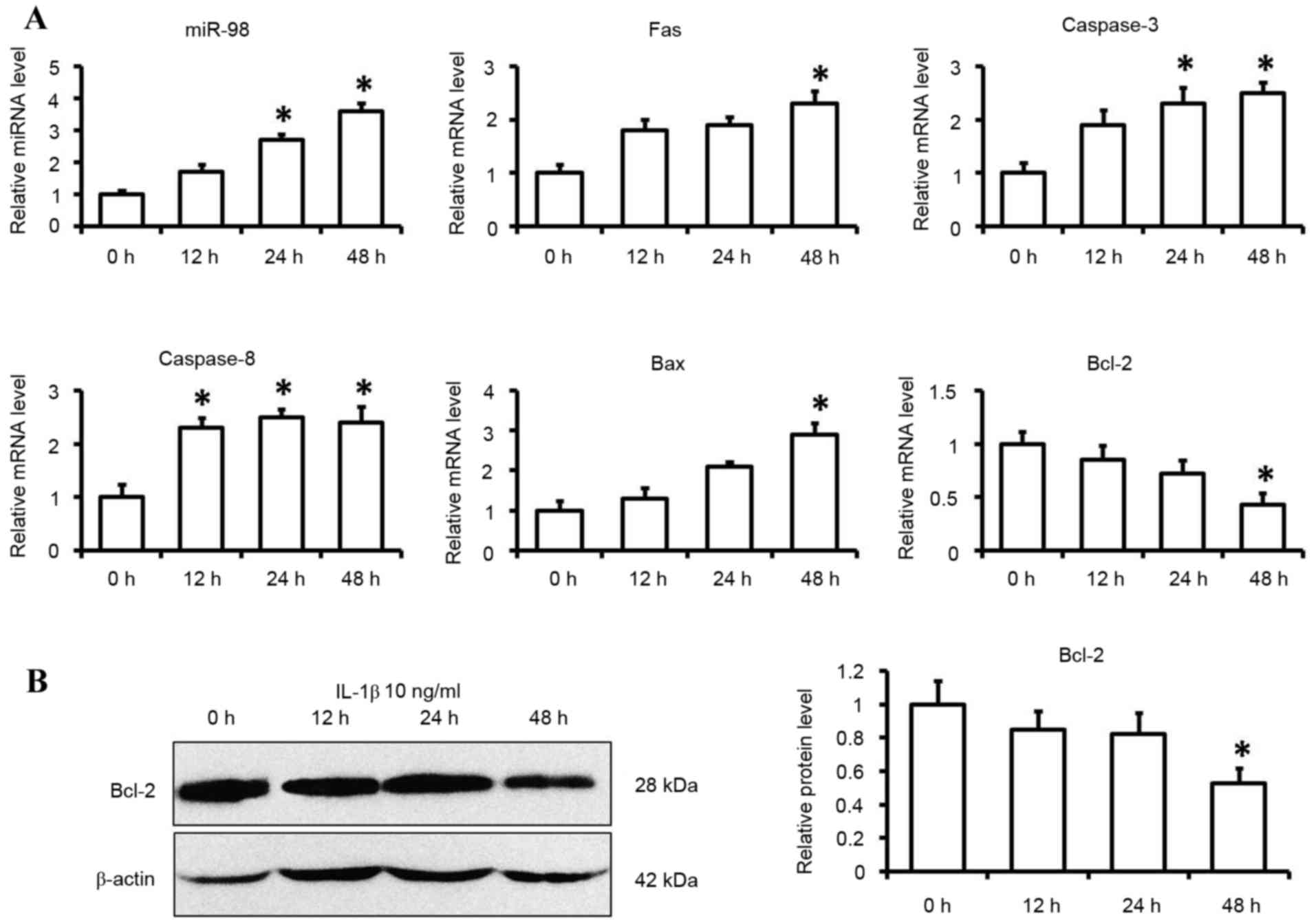 | Figure 1.Expression levels of miR-98 and
apoptotic factors in IL-1β-induced chondrocytes. Cells were
cultured with IL-1β (10 ng/ml) or control medium for 12, 24 and 48
h. (A) miR-98, Fas, caspase-3, caspase-8, Bax and Bcl-2 mRNA
expression levels were determined by reverse
transcription-quantitative polymerase chain reaction using U6 and
GAPDH as internal controls. (B) Bcl-2 protein levels were
determined by western blotting. Each data point was normalized to
the control. Data are presented as the mean ± standard error from
six independent experiments. *P<0.05 vs. control group (0 h).
miR-98, microRNA-98; IL-1β, interleukin 1β; Fas, Fas cell surface
death receptor; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X
protein. |
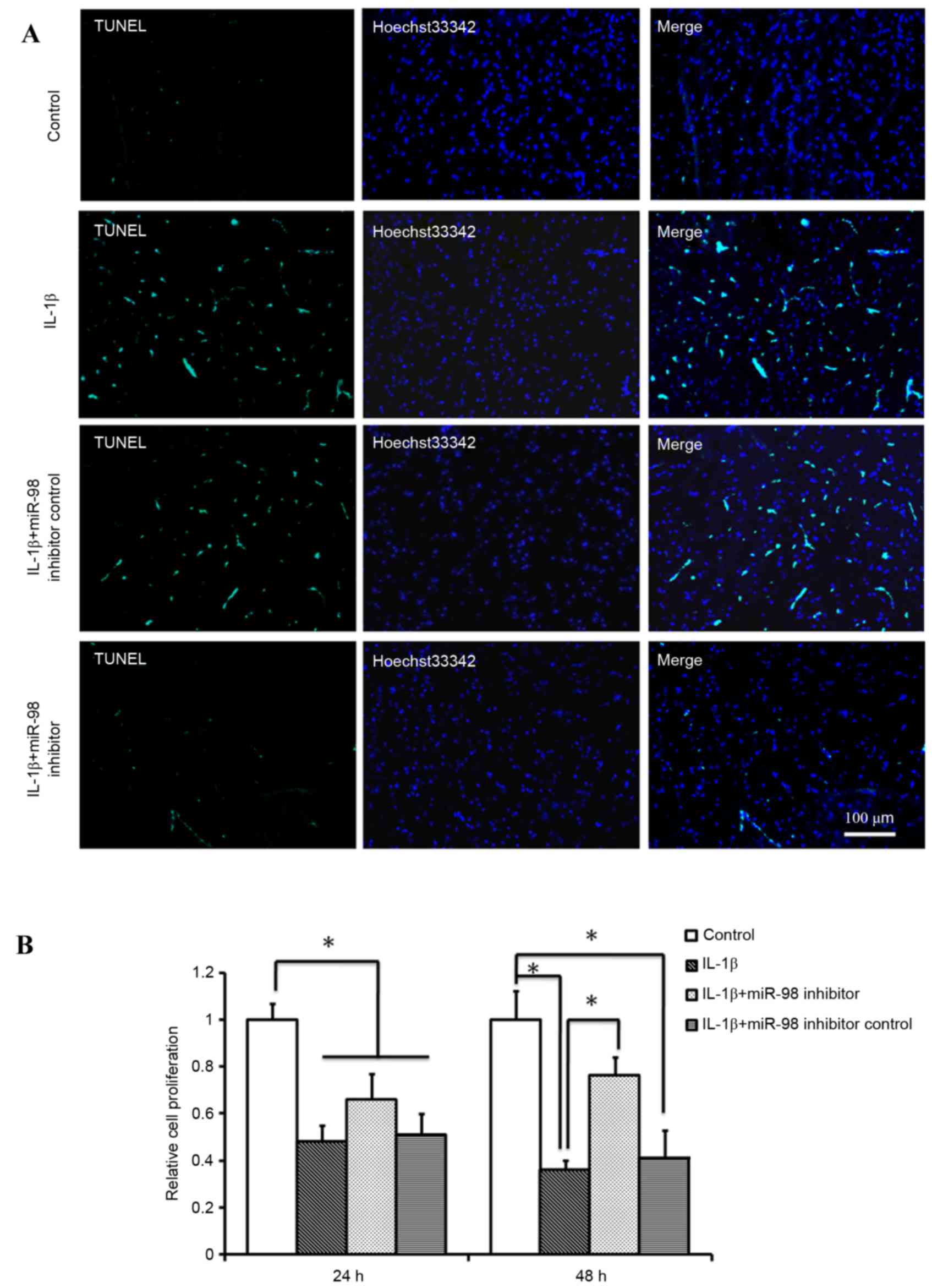
miR-98 affects apoptosis and
proliferation of chondrocytes
In order to determine the effects of silencing
miR-98 on mouse chondrocyte apoptosis and proliferation, miR-98
inhibitor was transfected into primary chondrocytes, and the
chondrocytes were subsequently treated with 10 ng/ml IL-1β for
24and 48 h. A TUNEL assay was performed to detect the number of
apoptotic cells. As presented in Fig.
2A, transfection with the miR-98 inhibitor reduced the number
of TUNEL-positive cells compared with transfection with miR-98
inhibitor control or treatment withIL-1β only. Therefore, it is
possible that downregulation of miR-98 expression in OA may be
effective in the prevention of IL-1β-induced chondrocyte apoptosis.
An MTT assay was used to examine the proliferation rate of
chondrocytes under various conditions and it was determined that
the rate of cell proliferation was significantly reduced following
treatment withIL-1β at 24 and 48 h and this decrease was more
pronounced after 48 h compared with at 24 h (P<0.05; Fig. 2B). However, transfection with the
miR-98 inhibitor significantly alleviated the IL-1β-induced
decrease in cell proliferation at 48 h (P<0.05; Fig. 2B).
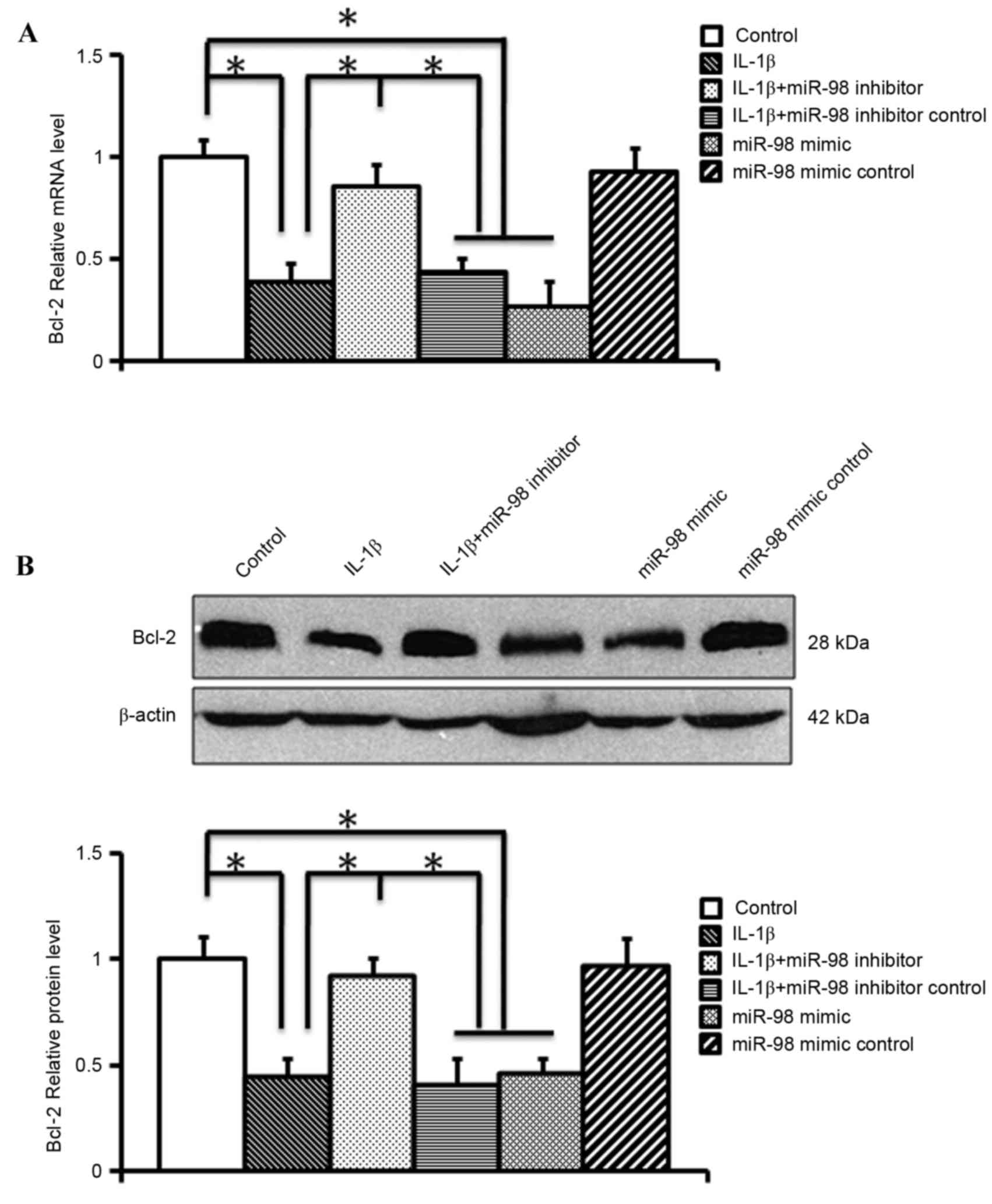
miR-98 negatively regulatesBcl-2
expression in mouse chondrocytes
The mRNA expression levels of miR-98 and apoptotic
factors were compared following exposure to IL-1β in cultured
chondrocytes using RT-qPCR, and an increase inmiR-98 expression was
identified with a corresponding decrease in Bcl-2 expression
following exposure to IL-1β. In order to examine the effects of
silencing miR-98 on Bcl-2 regulation, RT-qPCR and western blotting
were performed following transfection of mouse chondrocytes with
miR-98 inhibitor or mimic for 24 h. RT-qPCR (Fig. 3A) and western blotting (Fig. 3B) revealed that the miR-98 mimic
significantly reduced the mRNA and protein expression levels of
Bcl-2 (P<0.05). Conversely, the miR-98 inhibitor significantly
alleviated the IL-1β-induced downregulation of Bcl-2 mRNA and
protein expression (P<0.05; Fig.
3). These findings indicated that Bcl-2 may be a target gene of
miR-98.
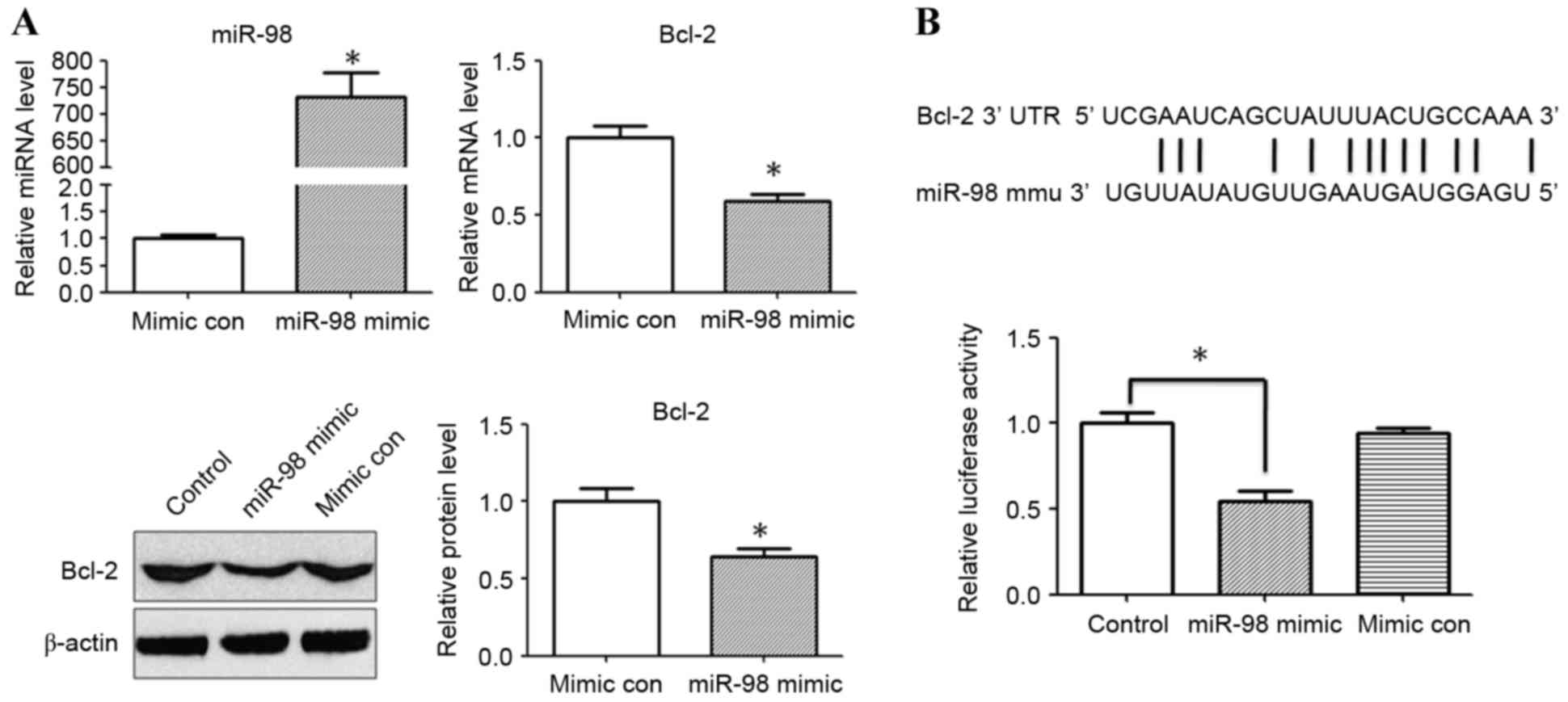
miR-98 targets the 3′-UTR of Bcl-2 and
suppresses translation
The present study hypothesized that miR-98 may
potentially target Bcl-2, and the results revealed that
transfection with a miR-98 inhibitor significantly increased the
expression levels of Bcl-2 following treatment with IL-1β. To
validate the hypothesis that miR-98 regulates Bcl-2 expression
following IL-1β stimulation, miR-98 mimic/mimic control were
transfected into HEK293 cells. To confirm the efficiency of
transfection, the expression levels of miR-98 were quantified using
RT-qPCR. The mRNA and protein expression levels of Bcl-2 were also
evaluated 24 h after miR-98 mimic/mimic control transfection. The
relative expression levels of miR-98 were significantly increased
following transfection of HEK293 cells with miR-98 mimic
(P<0.05; Fig. 4A).RT-qPCR and
western blot analysis revealed that the relative expression of
Bcl-2 was significantly reduced in the miR-98 mimic transfection
group compared with the mimic control group (P<0.05; Fig. 4A). A dual luciferase reporter assay
was performed to investigate whether miR-98 directly binds to the
3′-UTR of Bcl-2. It was determined that co-transfection with miR-98
mimic and pMIR-Bcl-2-wild type reporter plasmids significantly
reduced the luciferase activity in HEK293 cells compared with in
the control group (P<0.05; Fig.
4B). The results for the control luciferase assay was identical
with the mimic control (data not shown).
Discussion
OA is the most prevalent arthropathy and frequently
leads to chondrocyte apoptosis. Age, obesity, history of joint
trauma, repeated injury, overuse and joint dysplasia are possibly
involved in the etiology of OA, which may lead to serious joint
pain, functional impairment or irreversible functional disability,
along with reduced quality of life (19). The pathogenesis of OA is primarily
associated with age and has been confirmed to increase with the
median age of the population (20). Previous studies have indicated that
numerous human diseases are associated with alterations in the
expression of miRNAs, and several reports on miRNA profiling of
human cartilage lesions have already been published (5,21).
Various algorithms may be used to predict potential mRNA targets;
however, only a few miRNAs have been validated and assigned to
specific mRNAs. To the best of our knowledge, the present study is
the first to suggest that miR-98 serves an important role in
chondrocyte apoptosis by regulating Bcl-2 expression in an IL-1β
conditional cultured system in vitro. Previous studies
identified various miRNAs that may be involved in chondrogenesis
and OA (14,22). The role of miR-98 in tumor
apoptosis remains to be elucidated. Previous studies have reported
that miR-98 expression is associated with tumor cell growth
(15,23). Clinical studies have suggested that
miR-98 expression is associated with head and neck cancer
development (24) and may be
downregulated in nasopharyngeal carcinoma (25). In addition, microarrays and qPCR
have confirmed that miR-98 is upregulated in primary breast cancer
specimens (26). However,
contradictory findings have been reported in two other recent
studies. Chen et al (27)
demonstrated that miR-25 was upregulated in OA chondrocytes,
whereas Miyaki et al (28)
stated that this miRNA was downregulated in OA cartilage. Using an
in vitro model of mouse OA chondrocytes, the present study
detected the expression levels of miR-98 using RT-qPCR; the results
demonstrated that the expression levels of miR-98 were increased in
OA chondrocytes following in vitro treatment with IL-1β, as
previously reported (11). The
expression levels of apoptotic factors following IL-1β stimulation
were also detected in order to determine the effects of miR-98 on
the apoptosis of mouse chondrocytes.
Removal of undesirable cells is a biological process
that functions to maintain homeostasis, and is termed apoptosis or
programmed cell death (29).
Apoptosis is controlled by two primary molecular signaling
pathways: The extrinsic and intrinsic pathways (30,31).
The extrinsic apoptotic pathway refers to Fas ligand (FasL) binding
to Fas receptor (32,33). This forms the death-inducing
signaling complex, which contains the Fas-associated death domain,
caspase-8 and caspase-10. Subsequently, cleavage and activation of
executive caspase-3 is induced by cleaved caspase-8; activated
capase-3 consequently cleaves DNA molecules, leading to apoptosis
(34,35). It has previously been demonstrated
that the Fas/FasL pathway exerts a principle role in the induction
of apoptosis (36), and in many
tumor and inflammation related diseases an alteration of the
Fas/FasL pathway has been observed. The Bcl-2 protein family, which
includes anti-apoptotic proteins, such as Bcl-2, Bcl-extra large
and myeloid cell leukemia 1, and pro-apoptotic members, including
Bcl-2 antagonist/killer 1 (Bak), Bax and Bcl-2-like protein 11, are
important mediators of the intrinsic pathway. Overexpression of any
of the Bcl-2-like pro-survival members, or the loss of the
multi-Bcl-2-homology domain proteins Bak and Bax, may inhibit
apoptosis through the intrinsic pathway (37). The present study used RT-qPCR to
identify the mRNA expression levels of apoptotic factors. Fas,
caspase-3, caspase-8 and Bax expression levels were all increased
following IL-1β stimulation; however, a decrease in Bcl-2 and a
corresponding increase in miR-98 expression was detected following
IL-1β exposure. Western blot analysis confirmed that Bcl-2 protein
expression levels were significantly reduced. In order to determine
whether reduced Bcl-2 expression was regulated by the upregulation
of miR-98 RT-qPCR and western blotting were performed, and revealed
that IL-1β treatment reduced Bcl-2 expression via
post-transcriptional gene regulation involving the activation of
miR-98. A dual luciferase reporter assay confirmed that miR-98 may
target the 3′-UTR of Bcl-2, which leads to translational
suppression.
Chondrocytes are an important component of
arthrosis, which serve a role in maintaining normal cartilage
homeostasis and structural integrity, and account for only 5% of
the total cartilage volume; production of the extracellular matrix
and its enzymatic degradation are maintained by chondrocytes
(38). The tilting of this balance
in favor of catabolic events may lead to the loss of articular
cartilage, as observed in OA. Therefore, maintaining the integrity
of the articular cartilage is the primary function of chondrocytes.
In addition, chondrocyte damage may lead to matrix degeneration,
and may be associated with the onset and progression of OA
(39,40). Previous studies have indicated that
chondrocyte apoptosis may be associated with OA pathogenesis, and
studies performed in situ have identified a higher number of
apoptotic chondrocytes in OA compared with in normal samples
(41,42). Therefore, the effective prevention
of chondrocyte apoptosis may be a potential therapeutic strategy
for the treatment of OA. In the present study, miR-98 inhibited the
upregulation of Bcl-2 expression, which may be one of the primary
causes of articular chondrocyte apoptosis. However, the function of
miR-98 in chondrocyte apoptosis via the inhibition of its target
genes remains to be fully elucidated, as computational analyses
frequently state that one miRNA may have hundreds of target genes
(43). Therefore, chondrocyte
apoptosis may be involved in numerous molecular networks associated
with apoptosis and growth.
In conclusion, the present study demonstrated that
the expression of miR-98 was induced by IL-1β treatment in mouse
primary chondrocytes and that a miR-98 inhibitor may prevent the
downregulation of Bcl-2 induced by IL-1β in chondrocytes. These
results suggested that silencing miRNA-98 may prevent chondrocyte
apoptosis.
Acknowledgements
The authors would like to thank Professor Jiesheng
Gao (The Second Xiangya Hospital of Central South University,
Changsha, China) for his guidance. The present study was supported
by the National Natural Science Foundation of China (grant no.
81260286).
References
|
1
|
Aigner T, Söder S, Gebhard PM, McAlinden A
and Haag J: Mechanisms of disease: Role of chondrocytes in the
pathogenesis of osteoarthritis-structure, chaos and senescence. Nat
Clin Pract Rheumatol. 3:391–399. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li YH, Tavallaee G, Tokar T, Nakamura A,
Sundararajan K, Weston A, Sharma A, Mahomed NN, Gandhi R, Jurisica
I and Kapoor M: Identification of synovial fluid microRNA signature
in knee osteoarthritis: Differentiating early- and late-stage knee
osteoarthritis. Osteoarthritis Cartilage. 24:1577–1586. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen CZ, Li L, Lodish HF and Bartel DP:
MicroRNAs modulate hematopoietic lineage differentiation. Science.
303:83–86. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen CZ and Lodish HF: MicroRNAs as
regulators of mammalian hematopoiesis. Semin Immunol. 17:155–165.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Iliopoulos D, Malizos KN, Oikonomou P and
Tsezou A: Integrative microRNA and proteomic approaches identify
novel osteoarthritis genes and their collaborative metabolic and
inflammatory networks. PLoS One. 3:e37402008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Diaz-Prado S, Cicione C, Muiños-Lopez E,
Hermida-Gómez T, Oreiro N, Fernández-López C and Blanco FJ:
Characterization of microRNA expression profiles in normal and
osteoarthritic human chondrocytes. BMC Musculoskelet Disord.
13:1442012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goldring MB and Marcu KB: Cartilage
homeostasis in health and rheumatic diseases. Arthritis Res Ther.
11:2242009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mayan MD, Gago-Fuentes R,
Carpintero-Fernandez P, Fernandez-Puente P, Filgueira-Fernandez P,
Goyanes N, Valiunas V, Brink PR, Goldberg GS and Blanco FJ:
Articular chondrocyte network mediated by gap junctions: Role in
metabolic cartilage homeostasis. Ann Rheum Dis. 74:275–284. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Daheshia M and Yao JQ: The interleukin
1beta pathway in the pathogenesis of osteoarthritis. J Rheumatol.
35:2306–2312. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kapoor M, Martel-Pelletier J, Lajeunesse
D, Pelletier JP and Fahmi H: Role of proinflammatory cytokines in
the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 7:33–42.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jones SW, Watkins G, Le Good N, Roberts S,
Murphy CL, Brockbank SM, Needham MR, Read SJ and Newham P: The
identification of differentially expressed microRNA in
osteoarthritic tissue that modulate the production of TNF-alpha and
MMP13. Osteoarthritis Cartilage. 17:464–472. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miyaki S, Nakasa T, Otsuki S, Grogan SP,
Higashiyama R, Inoue A, Kato Y, Sato T, Lotz MK and Asahara H:
MicroRNA-140 is expressed in differentiated human articular
chondrocytes and modulates interleukin-1 responses. Arthritis
Rheum. 60:2723–2730. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamasaki K, Nakasa T, Miyaki S, Ishikawa
M, Deie M, Adachi N, Yasunaga Y, Asahara H and Ochi M: Expression
of MicroRNA-146a in osteoarthritis cartilage. Arthritis Rheum.
60:1035–1041. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Park SJ, Cheon EJ and Kim HA: MicroRNA-558
regulates the expression of cyclooxygenase-2 and IL-1β-induced
catabolic effects in human articular chondrocytes. Osteoarthritis
Cartilage. 21:981–989. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Siragam V, Rutnam ZJ, Yang W, Fang L, Luo
L, Yang X, Li M, Deng Z, Qian J, Peng C and Yang BB: MicroRNA
miR-98 inhibits tumor angiogenesis and invasion by targeting
activin receptor-like kinase-4 and matrix metalloproteinase-11.
Oncotarget. 3:1370–1385. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Du L, Schageman JJ, Subauste MC, Saber B,
Hammond SM, Prudkin L, Wistuba II, Ji L, Roth JA, Minna JD and
Pertsemlidis A: miR-93, miR-98, and miR-197 regulate expression of
tumor suppressor gene FUS1. Mol Cancer Res. 7:1234–1243. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li F, Li XJ, Qiao L, Shi F and Liu W, Li
Y, Dang YP, Gu WJ, Wang XG and Liu W: miR-98 suppresses melanoma
metastasis through a negative feedback loop with its target gene
IL-6. Exp Mol Med. 46:e1162014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Scharstuhl A, Schewe B, Benz K, Gaissmaier
C, Bühring HJ and Stoop R: Chondrogenic potential of human adult
mesenchymal stem cells is independent of age or osteoarthritis
etiology. Stem Cells. 25:3244–3251. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hung VW, Zhu TY, Cheung WH, Fong TN, Yu
FW, Hung LK, Leung KS, Cheng JC, Lam TP and Qin L: Age-related
differences in volumetric bone mineral density, microarchitecture,
and bone strength of distal radius and tibia in Chinese women: A
high-resolution pQCT reference database study. Osteoporos Int.
26:1691–1703. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Betel D, Wilson M, Gabow A, Marks DS and
Sander C: The microRNA.org resource: Targets and expression.
Nucleic Acids Res. 36:(Database issue). D149–D153. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin EA, Kong L, Bai XH, Luan Y and Liu CJ:
miR-199a, a bone morphogenic protein 2-responsive MicroRNA,
regulates chondrogenesis via direct targeting to Smad1. J Biol
Chem. 284:11326–11335. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wendler A, Keller D, Albrecht C, Peluso JJ
and Wehling M: Involvement of let-7/miR-98 microRNAs in the
regulation of progesterone receptor membrane component 1 expression
in ovarian cancer cells. Oncol Rep. 25:273–279. 2011.PubMed/NCBI
|
|
24
|
Hebert C, Norris K, Scheper MA, Nikitakis
N and Sauk JJ: High mobility group A2 is a target for miRNA-98 in
head and neck squamous cell carcinoma. Mol Cancer. 6:52007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Alajez NM, Shi W, Hui AB, Bruce J,
Lenarduzzi M, Ito E, Yue S, O'Sullivan B and Liu FF: Enhancer of
Zeste homolog 2 (EZH2) is overexpressed in recurrent nasopharyngeal
carcinoma and is regulated by miR-26a, miR-101, and miR-98. Cell
Death Dis. 1:e852010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yan LX, Huang XF, Shao Q, Huang MY, Deng
L, Wu QL, Zeng YX and Shao JY: MicroRNA miR-21 overexpression in
human breast cancer is associated with advanced clinical stage,
lymph node metastasis and patient poor prognosis. RNA.
14:2348–2360. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen CG, Thuillier D, Chin EN and Alliston
T: Chondrocyte-intrinsic Smad3 represses Runx2-inducible matrix
metalloproteinase 13 expression to maintain articular cartilage and
prevent osteoarthritis. Arthritis Rheum. 64:3278–3289. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Miyaki S and Asahara H: Macro view of
microRNA function in osteoarthritis. Nat Rev Rheumatol. 8:543–552.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jin Z and El-Deiry WS: Overview of cell
death signaling pathways. Cancer Biol Ther. 4:139–163. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Carrington PE, Sandu C, Wei Y, Hill JM,
Morisawa G, Huang T, Gavathiotis E, Wei Y and Werner MH: The
structure of FADD and its mode of interaction with procaspase-8.
Mol Cell. 22:599–610. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tibbetts MD, Zheng L and Lenardo MJ: The
death effector domain protein family: Regulators of cellular
homeostasis. Nat Immunol. 4:404–409. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Barnhart BC, Lee JC, Alappat EC and Peter
ME: The death effector domain protein family. Oncogene.
22:8634–8644. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Harper N, Hughes M, MacFarlane M and Cohen
GM: Fas-associated death domain protein and caspase-8 are not
recruited to the tumor necrosis factor receptor 1 signaling complex
during tumor necrosis factor-induced apoptosis. J Biol Chem.
278:25534–25541. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Houston A and O'Connell J: The Fas
signalling pathway and its role in the pathogenesis of cancer. Curr
Opin Pharmacol. 4:321–326. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thomas LR, Johnson RL, Reed JC and
Thorburn A: The C-terminal tails of tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL) and Fas receptors have opposing
functions in Fas-associated death domain (FADD) recruitment and can
regulate agonist-specific mechanisms of receptor activation. J Biol
Chem. 279:52479–52486. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kunes P, Krejsek J, Brtko M, Mandak J,
Kolackova M, Kudlova M Trojackova and Andrys C: Neutrophil
apoptosis by Fas/FasL: Harmful or advantageous in cardiac surgery?
Thorac Cardiovasc Surg. 57:1–6. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Borner C: The Bcl-2 protein family:
Sensors and checkpoints for life-or-death decisions. Mol Immunol.
39:615–647. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Findlay DM and Atkins GJ:
Osteoblast-chondrocyte interactions in osteoarthritis. Curr
Osteoporos Rep. 12:127–134. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hayes AJ, MacPherson S, Morrison H,
Dowthwaite G and Archer CW: The development of articular cartilage:
Evidence for an appositional growth mechanism. Anat Embryol (Berl).
203:469–479. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pacifici M, Koyama E, Iwamoto M and
Gentili C: Development of articular cartilage: What do we know
about it and how may it occur? Connect Tissue Res. 41:175–184.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hashimoto S, Ochs RL, Rosen F, Quach J,
McCabe G, Solan J, Seegmiller JE, Terkeltaub R and Lotz M:
Chondrocyte-derived apoptotic bodies and calcification of articular
cartilage. Proc Natl Acad Sci USA. 95:3094–3099. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kim HA and Blanco FJ: Cell death and
apoptosis in osteoarthritic cartilage. Curr Drug Targets.
8:333–345. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Krek A, Grun D, Poy MN, Wolf R, Rosenberg
L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M
and Rajewsky N: Combinatorial microRNA target predictions. Nat
Genet. 37:495–500. 2005. View
Article : Google Scholar : PubMed/NCBI
|