Introduction
Hepatocellular carcinoma (HCC) represents the fifth
most common type of cancer in males and the ninth most common in
females (1). The prognosis of HCC
is poor due to the highly aggressive nature of the disease,
particularly for HCC patients with recurrent and/or metastatic
tumors (2,3).
Increasing knowledge regarding the molecular
alterations that initiate malignancy has led to the development of
novel compounds targeting pathways aberrantly activated in cancer.
Of these, the multiple receptor tyrosine kinase inhibitor
sorafenib, which primarily targets platelet-derived growth factor
receptor and vascular endothelial growth factor receptor, is the
worldwide standard therapy for the treatment of advanced HCC.
However, although sorafenib has significantly improved the
treatment protocol, the overall response rate is disappointingly
low and resistance inevitably develops, further limiting the
efficacy of the drug (4).
Recent studies have provided strong evidence to
suggest that phosphatidylinositol 3-kinase (PI3K)/Akt/mechanistic
target of rapamycin (mTOR) signaling is upregulated in HCC, and
activation of this pathway triggers rapid HCC development and
progression (5,6). In clinical practice, mTOR
hyperactivity has been associated with high-grade tumors and tumors
with poor prognosis (7).
Therefore, mTOR inhibition may be considered a plausible strategy
in mitigating HCC. The present study aimed to investigate the
antitumor effects of two mTOR inhibitors, everolimus and
temsirolimus. Temsirolimus has been approved for first-line
treatment of patients with poor prognosis renal cell carcinoma
(RCC), whereas the oral mTOR inhibitor, everolimus, has been
recommended for patients with advanced progressive RCC or for
patients with failed vascular endothelial growth factor-targeted
therapy (8). In the present study,
experiments were conducted on a panel of HCC cell lines, and aimed
to determine the effects of mTOR inhibitors on tumor growth and
invasion. In addition, integrin α and β subtype expression was
investigated, since these receptors are not only involved in tumor
growth and invasion, but also in cellular differentiation.
Materials and methods
Cell culture
HepG2 and HuH7 cells were purchased from CLS Cell
Lines Service GmbH (Eppelheim, Germany). Hep3B cells were purchased
from DSMZ (Braunschweig, Germany). HepG2 cells were grown and
subcultured in Dulbecco's modified Eagle's medium:Nutrient Mixture
F-12 medium (Thermo Fisher Scientific GmbH, Dreieich, Germany)
supplemented with 10% fetal bovine serum (FBS; Thermo Fisher
Scientific GmbH), 20 mM Hepes-buffer and gentamicin (0.5 ml/l).
HuH7 and Hep3B cells were incubated in RPMI-1640 medium,
supplemented with 10% FBS, 20 mM HEPES-buffer, 1% glutamax, and 1%
penicillin/streptomycin (all Gibco; Thermo Fisher Scientific GmbH)
at 37°C in a humidified incubator containing 5% CO2.
mTOR inhibitors
Everolimus (Novartis Pharma AG, Basel, Switzerland)
and temsirolimus (LC Laboratories, Woburn, MA, USA) were dissolved
in dimethyl sulfoxide (DMSO) as a 10 mM stock solution and stored
in aliquots at −20°C. Prior to use, the compounds were diluted in
cell culture medium. The effects of 0.1–100 nM everolimus or
temsirolimus were determined on cell growth to evaluate dose
dependency. All further experiments were conducted with 1 nM
everolimus or temsirolimus. Cells treated with culture medium alone
(supplemented with DMSO, diluted 1:105-1:108)
served as controls. To evaluate the toxic effects of the drugs,
following a 72 h incubation at 37°C in a humidified incubator
containing 5% CO2, cell viability was determined by
trypan blue staining (Gibco; Thermo Fisher Scientific GmbH). A
Zeiss ID 03 light microscope (Zeiss AG, Oberkochen, Germany) was
used.
Measurement of tumor cell growth
Cell growth was assessed using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
dye reduction assay (Roche Diagnostics GmbH, Penzberg, Germany).
HepG2, Hep3B and HuH6 cells, (50 µl, 1×105 cells/ml)
were seeded onto 96-well tissue culture plates. After 24, 48 and 72
h incubation at 37°C MTT (0.5 mg/ml) was added for an additional 4
h. Thereafter, cells were lysed in a buffer containing 10% SDS in
0.01 M HCl. The plates were incubated overnight at 37°C in an
atmosphere containing 5% CO2. Absorbance was measured at
550 nm using a microplate ELISA reader. Each experiment was
conducted in triplicate. After subtracting background absorbance,
results were expressed as percentage difference, related to a
control set to 100%.
Clonogenic growth
HepG2, HuH7, or Hep3B cells, treated with 1 nM
everolimus or temsirolimus, were transferred to 6-well plates at
2,000 cells/well. Following 10 days of incubation, without changing
the cell culture medium (everolimus and temsirolimus remained in
culture), colonies were fixed with 1% glutaraldehyde for 10 min at
room temperature and counted. Colonies containing ≥50 cells were
counted using a Zeiss ID 03 light microscope (Zeiss AG).
Non-treated cells served as controls.
Cell cycle analysis
Cell cycle analysis was performed once the tumor
cell cultures had grown to sub-confluency, and after 24 h of drug
treatment. Tumor cell populations were stained with propidium
iodide using a Cycleyest Plus DNA Reagent kit (BD Biosciences,
Heidelberg, Germany) and were then subjected to flow cytometry
using a FACScan flow cytometer (BD Biosciences). A total of 10,000
events were collected for each sample. Data acquisition was
conducted using CellQuest version 6.0 software (BD Biosciences) and
cell cycle distribution was calculated using ModFit version 3.0
software (BD Biosciences). The number of gated cells in the
G1-, S-, or G2/M-phases was expressed as a
percentage of the total cell population.
Western blot analysis
To investigate the expression levels of cell
cycle-regulating proteins, tumor cell lysates (50 µg; protein
concentration was quantified by Bradford assay) were loaded onto a
7% polyacrylamide gel and were electrophoresed for 90 min at 100 V.
The lysis buffer consisted of Tris-NaCl, 10% Tergitol, 0.25%
Na-deoxycholate, 1 mM EDTA, 1 mg/ml aprotinin, 1 mg/ml leupeptin, 1
mg/ml pepstatin, 2 mM NaF, 2 mM Na3VO4 and 2
mM PMSF. Protein was then transferred to nitrocellulose membranes
for 1 h at 100 V. After blocking with non-fat dry milk for 1 h at
room temperature, the membranes were incubated overnight at 4°C
with monoclonal antibodies directed against the following cell
cycle-associated proteins: Phosphorylated (p)-Akt [clone 104A282,
mouse immunoglobulin (Ig)G1; dilution 1:500; cat. no. 550747; BD
Biosciences], p-mTOR (clone D9C2; IgG, Ser2448; dilution 1:1,000;
cat. no. 5536S) and p-Raptor (IgG, Ser792; dilution 1:1,000; cat.
no. 2083S; all Cell Signaling Technology Europe, B.V., Leiden, The
Netherlands). Horseradish peroxidase-conjugated goat-anti-mouse IgG
(dilution, 1:5,000; cat. no. 161-0380; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) served as the secondary antibody (30 min
incubation at room temperature). The membranes were briefly
incubated at room temperature with an enhanced chemiluminescence
(ECL) detection reagent (ECL™; Merck KGaA, Darmstadt, Germany; cat.
no. WBKLS0100) to visualize the proteins and were then analyzed
using the Fusion FX7 system (Peqlab Biotechnologie GmbH, Erlangen,
Germany). β-actin (1:1,000; cat. no. A5441; Sigma-Aldrich; Merck
KGaA) served as an internal control.
Tumor cell adhesion assay
For the tumor cell adhesion assay, 6-well plates
were coated with collagen G (extracted from calfskin, consisting of
90% collagen type I and 10% collagen type III; diluted to 400 µg/ml
in PBS; Thermo Fisher Scientific GmbH) overnight. Plastic dishes
served as the background control. Plates were washed with 1% bovine
serum albumin (BSA; Sigma Aldrich; Merck KGaA) in PBS to block
nonspecific cell adhesion. HCC tumor cells (0.5×106)
were then added to each well for 60 min. Subsequently, non-adherent
tumor cells were washed off, and the remaining adherent cells were
fixed for 10 min at room temperature with 1% glutaraldehyde and
counted under a Zeiss ID 03 light microscope (Zeiss AG). Mean
cellular adhesion, defined as adherent cellscoated
well-adherent cellsbackground, was calculated from
five different observation fields (5×0.25 mm2).
Tumor cell chemotaxis
Serum-induced chemotaxis was investigated using
6-well Transwell chambers (Greiner Bio-One GmbH, Frickenhausen,
Germany) with 8-µm pores. HCC tumor cells (0.5×106/ml)
were placed in the upper chamber in serum-free medium. The lower
chamber contained complete cell culture medium including 10% serum
(Sigma Aldrich; Merck KGaA). After 20 h at 37°C, the upper surface
of the Transwell membrane was gently wiped with a cotton swab to
remove cells that had not migrated. Cells that had moved to the
lower surface of the membrane were stained using hematoxylin and
counted under a light microscope (Zeiss ID 03; Zeiss AG). Mean
chemotaxis was calculated from five different observation fields
(5×0.25 mm2).
Integrin surface expression
HepG2, HuH7 or Hep3B cells were detached from their
culture flasks by Accutase (PAA Laboratories; GE Healthcare) and
washed in blocking solution (PBS, 0.5% BSA). Cells were then
incubated for 60 min at 4°C with phycoerythrin (PE)-conjugated
monoclonal antibodies (mAbs) directed against the following
integrin subtypes: Anti-α1 (mouse IgG1, clone SR84; cat. no.
742362), anti-α2 (mouse IgG2a, clone 12 F1; cat. no. 555669),
anti-α3 (mouse IgG1, clone C3 II.1; cat. no. 746157), anti-α4
(mouse IgG1, clone 9F10; cat. no. 658332), anti-α5 (mouse IgG1,
clone IIA1; cat. no. 555615), anti-α6 (rat IgG2a, clone GoH3; cat.
no. 555734), anti-β1 (mouse IgG1, clone MAR4; cat. no. 557332),
anti-β3 (mouse IgG1, clone VI-PL2; cat. no. 555752), and anti-β4
(rat IgG2a; clone 439-9B; cat. no. 555720; all 20 µl/test; using
the manufacturing dilution; all BD Biosciences). Tumor cell
integrin expression was then measured using a FACScan [BD
Biosciences; FL-2H (log) channel histogram analysis;
1×104 cells per scan] and was expressed as mean
fluorescence units. A mouse IgG1-PE (MOPC-21) or IgG2a-PE
(G155-178) (BD Biosciences) was used as an isotype control.
Integrin suppression
To determine whether integrin α1 impacts tumor
growth, HepG2 cells were incubated for 60 min at 37°C with 10 µg/ml
function-blocking anti-integrin α1 (clone FB12) mAb (Merck KGaA;
cat. no. MAB1973Z). Controls were incubated with cell culture
medium alone. Subsequently, tumor cell growth was analyzed using
the MTT assay, as aforementioned. To evaluate whether integrin α1
acts on tumor cell motility, adhesion and migration experiments
were conducted using HepG2 cells following integrin α1 suppression.
To evaluate tumor cell binding to matrix proteins, 24-well plates
coated with fibronectin (BD Biosciences) were used. Plastic dishes
served as the background control. Plates were washed with 1% BSA in
PBS to block nonspecific cell adhesion. Subsequently,
0.5×106 tumor cells were added to each well and
incubated for 60 min at 37°C. Non-adherent tumor cells were washed
off and the remaining adherent cells were fixed with 1%
glutaraldehyde for 10 min at room temperature and counted by a
light microscope (Zeiss ID 03; Zeiss AG). The mean cellular
adhesion rate, defined as adherent cellscoated
well-adherent cellsbackground, was calculated from
five different observation fields. To evaluate tumor cell
migration, serum-induced chemotaxis was examined using 6-well
Transwell chambers (Greiner Bio-One GmbH) with 8-µm pores preformed
as described above (Tumor cell chemotaxis subsection),
however, only 0.5×106 HepG2 cells were applied.
Statistical analysis
Statistical analysis was performed using BiAS
software version 11.06 (http://www.bias-online.de/). All experiments were
performed between three and six times. Data are presented as the
mean ± standard deviation. Statistical significance between groups
was determined using the Mann-Whitney U test. P<0.05 was
considered to indicate a statistically significant difference.
Results
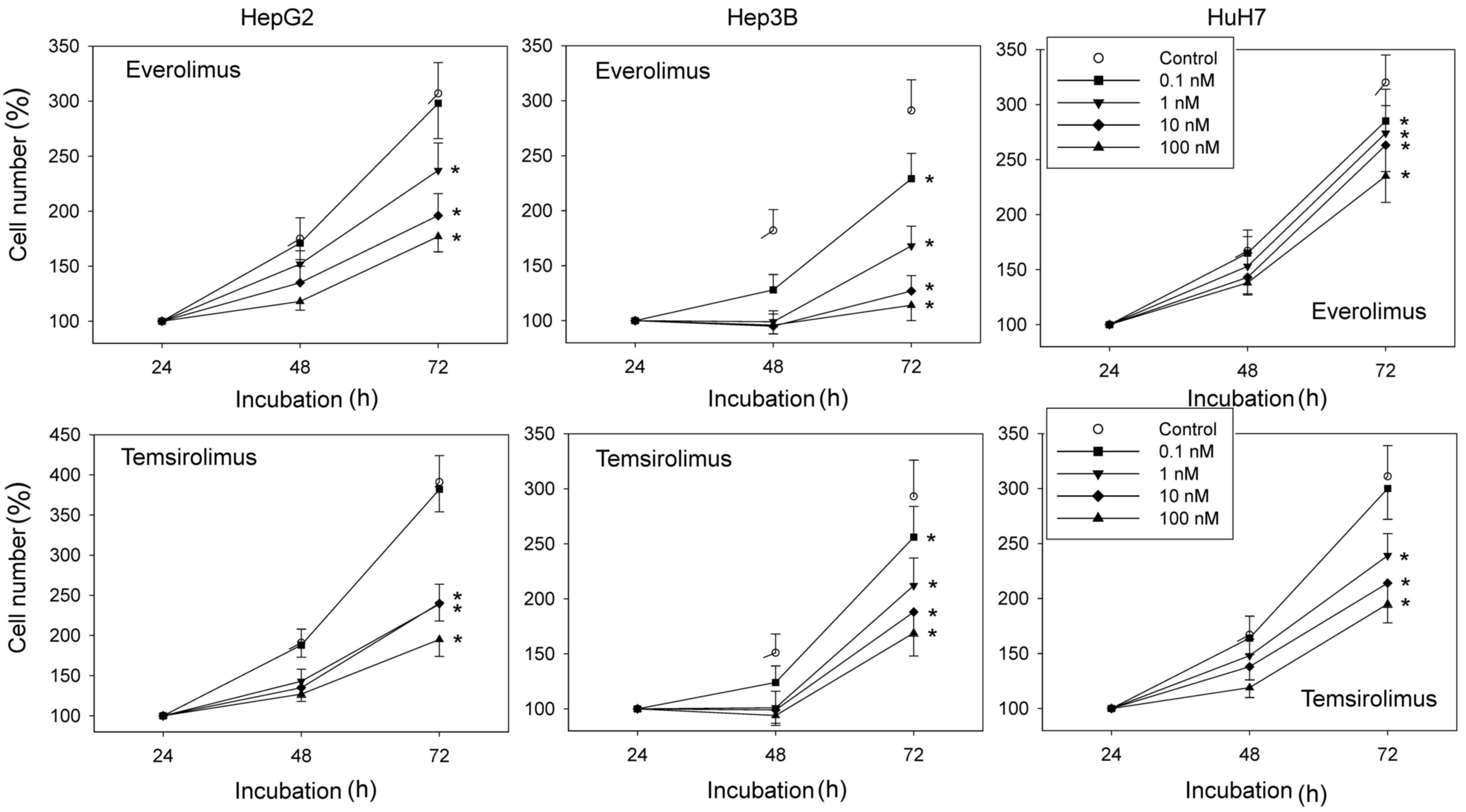
Exposure to everolimus or temsirolimus
reduces HCC cell growth
Ascending concentrations of everolimus or
temsirolimus induced a dose-dependent significant reduction in the
number of HCC cells. Everolimus exerted a growth inhibitory effect
at 0.1 nM in Hep3B and HuH7 cells, where temsirolimus exerted an
inhibitory effect at 1 nM in HepG2 and HuH7 cells; the most obvious
effect was apparent at 100 nM compared with the untreated controls
(Fig. 1). No signs of toxicity
were apparent, as determined by a trypan blue exclusion assay (data
not shown).
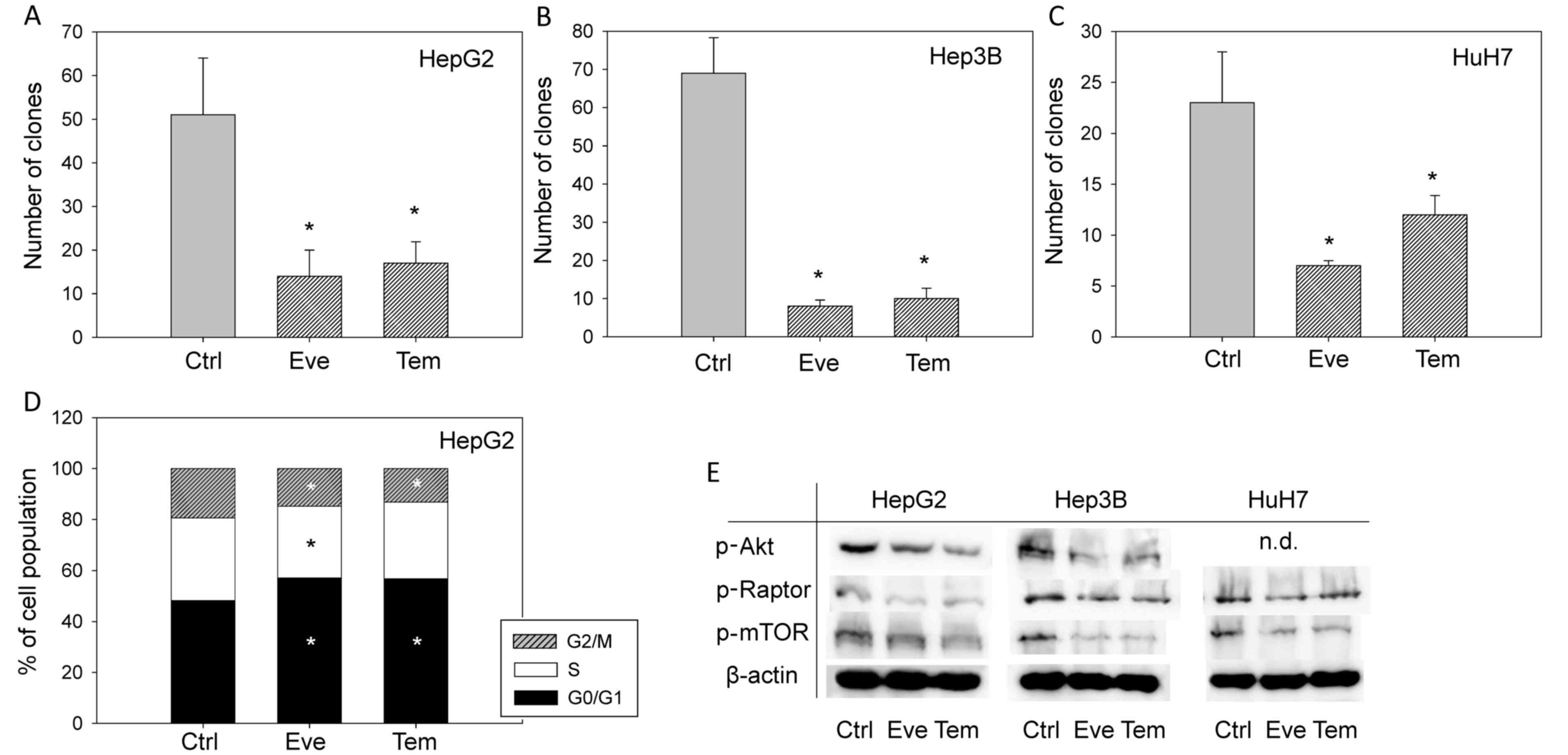
Clonogenic growth was significantly reduced when all
three tumor cell lines were treated with 1 nM everolimus or
temsirolimus (Fig. 2A-C). In
addition, everolimus (1 nM) and temsirolimus (1 nM) modulated cell
cycle progression. The number of HepG2 cells in the
G2/M- and S-phases was reduced, whereas the number of
tumor cells in the G0/G1-phase was increased compared
with the controls (Fig. 2D).
Since everolimus and temsirolimus target the mTOR
signaling pathway, the Akt-mTOR axis was also evaluated. p-mTOR and
p-Raptor expression was suppressed following treatment of all cell
lines with both compounds (Fig.
2E). However, as total protein content was not detected in the
present study, the activation status of the proteins cannot be
confirmed. In addition, p-Akt expression was reduced in the HepG2
and Hep3B cell lines, but was undetectable in HuH7 cells.
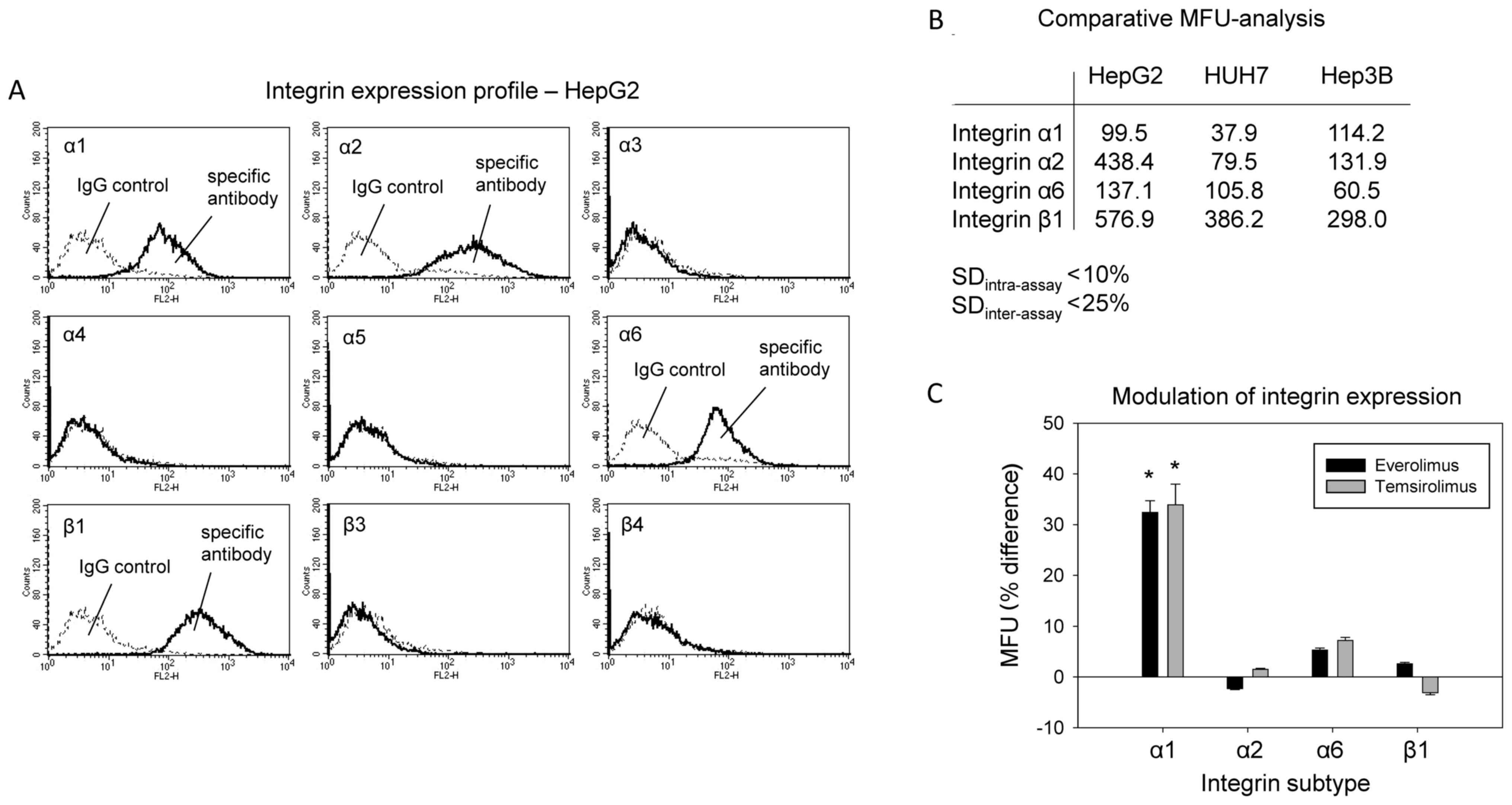
Everolimus and temsirolimus modify
integrin α1 expression
The integrin subtypes α1, α2, α6 and β1 were
strongly expressed on HepG2 cells, whereas α3, α4, α5, β3 and β4
were not detected (Fig. 3A).
Integrin expression was also detected on Hep3B and HuH7 cells; it
was demonstrated that α1, α2, α6 and β1 were expressed on these
cell lines (Fig. 3B). Treatment of
HepG2 cells with everolimus or temsirolimus significantly elevated
the expression of integrin α1 on the tumor cell surface, without
acting on α2, α6 or β1 expression (Fig. 3C).
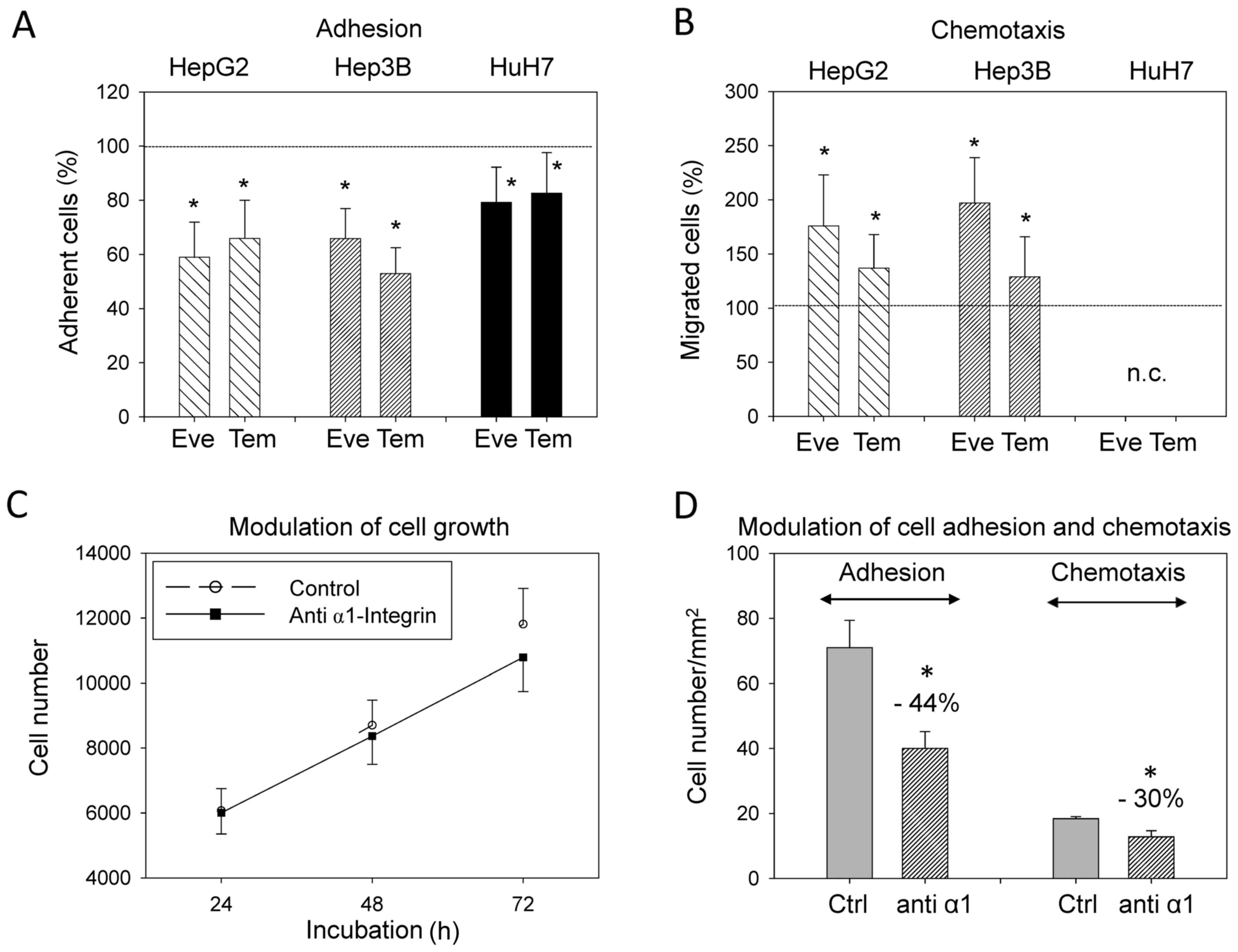
Everolimus and temsirolimus suppress
HCC cell adhesion but not migration
Treatment with everolimus or temsirolimus (1 nM)
significantly downregulated adhesion of HepG2, HuH7 and Hep3B cells
to immobilized collagen (Fig. 4A).
The most prominent effects were evoked in HepG2 and Hep3B cells.
Control values were 61.4±11.2 cells/mm2 (HepG2),
66.8±14.2 cells/mm2 (HuH7) and 79.4±14.8
cells/mm2 (Hep3B). A chemotaxis assay revealed that
HepG2 and Hep3B motility was enhanced following exposure to
temsirolimus or everolimus exposure (Fig. 4B). However, the control values were
low (HepG2: 16.2±4.2 cells/mm2, Hep3B: 6.8±3.5
cells/mm2). HuH7 cells did not migrate through the
filter membrane.
Integrin α1 suppression
Since everolimus and temsirolimus induced a distinct
upregulation in integrin α1 on HepG2 cells, subsequent experiments
aimed to determine the functional relevance of this receptor.
Suppression of integrin α1 slightly decreased HepG2 cell growth;
however, this was not significant (Fig. 4C). The strongest effects were
observed with respect to tumor cell adhesion, which was reduced by
>40% following integrin α1 suppression (Fig. 4D). Chemotactic activity was also
significantly decreased in the presence of anti-integrin α1
compared with the control group (30% decrease; Fig. 4D).
Discussion
Better understanding of tumor cell biology has led
to the development of numerous targeted therapeutic agents. The
PI3K/Akt/mTOR pathway has been identified as a pivotal key
regulator of cell growth, cell proliferation and cell survival.
Targeting the mTOR pathway has, therefore, been proposed as an
innovative strategy to treat cancer, and mTOR inhibitors have been
approved for RCC treatment. However, the clinical relevance of mTOR
inhibition in treating HCC remains unclear. Everolimus has been
reported to delay tumor progression in patients with
sorafenib-refractory HCC (9);
however, a recent investigation did not demonstrate improved
patient survival in a second-line setting or in
sorafenib-intolerant patients (10). Yeo et al (11) and Cho et al (12) hypothesized that only selected
patients, depending on the expression of relevant target proteins,
may benefit from an mTOR-based treatment regimen. The benefit of an
everolimus-sorafenib combination is controversial, since although a
dose-finding study on patients with advanced HCC yielded
encouraging results (13), a
randomized multicenter, multinational phase II trial did not
indicate a synergistic effect (14).
The present study demonstrated the growth inhibitory
effects of everolimus and temsirolimus on numerous HCC cell lines;
these agents exhibited effects at 0.1 or 1 nM, respectively. The
effects observed may, at least in part, be induced by the
downregulation of p-mTOR and particularly p-Raptor. Notably, p-Akt
was not detected in HUH7, which indicates that this cell type may
be associated with an Akt independent pathway with very low
(undetectable) Akt activity or a less sensitive antibody may have
been used in the present study.
In accordance with the present study, HepG2 cells
have been reported to respond to 1 nM everolimus in vitro
(15). In addition, the half
maximal inhibitory concentration values of everolimus or
temsirolimus in HepG2 cells have been reported to range between 0.9
nM (16) and 9 µM (17). Similar sensitivity to mTOR
inhibition has been reported for Hep3B and HuH7 cells (18,19),
indicating that mTOR inhibition may be of clinical relevance in
treating HCC. When discussing the behavior of the cell lines used
in the present study, it must be emphasized that HepG2 cells were
isolated from a liver biopsy with primary hepatoblastoma and HCC
characteristics (20). Although
HepG2 cells possess tumor-specific characteristics, they do not
form tumors when injected into athymic mice and are histologically
similar to the hepatoblastoma from which the cell line was derived
(20). Therefore, HepG2 should be
considered a hepatoblastoma, rather than a carcinoma, cell
line.
Based on the results of a cell cycle analysis, cell
growth inhibition in the presence of everolimus and temsirolimus
may be caused by driving the tumor cells from G2/M- and
S-phases into G0/G1. The differences between
the number of G2/M- and S-phase cells in the treated and
non-treated tumor cells were moderate; however, similar moderate
differences have also recently been observed, when HepG2 cells were
exposed to the mTOR inhibitor sirolimus. Suppression of tumor cell
proliferation by this drug was also associated with only a moderate
G0/G1 increase and S-phase decrease (21). In addition, it has been speculated
that a G0/G1 phase arrest, but not apoptosis,
may be the predominant mechanism responsible for the
antiproliferative activity of sirolimus (22). Everolimus has been reported to
suppress cyclins A, B1 and D1 in an in vivo HCC xenograft
model (22). Although the role of
these cyclins in cell cycle progression has yet to be elucidated in
detail, there is evidence that loss of cyclin A, B1 and/or D1 may
at least partially contribute to HCC cell accumulation in
G0/G1-phase (23,24).
Notably, exposing Hep3B and HuH7 cells to everolimus also induced a
G0/G1-phase arrest, indicating a common
mechanism for this class of drugs (15).
Suppression of adhesion by everolimus and
temsirolimus was determined in the present study. Maximum effects
were exerted on HepG2 and Hep3B cells, with a >40 and 50%
reduction, respectively. This is important, since tumor cell-matrix
interaction is critical for malignant tumor progression and
metastatic spreading. The results of the chemotaxis assay are
difficult to interpret in this context, since the percentage of
migrating cells increased in the presence of mTOR inhibitors.
However, it must be noted that only a few cells migrated across the
filter membrane in the control group. Therefore, slight differences
in the absolute number of motile cells following everolimus or
temsirolimus treatment are associated with large percentage
changes. An elevation of just 3 single Hep3B cells following
treatment was associated with a percentage difference >40%
compared with the control group. Whether the moderate elevation in
cell migration, based on absolute cell number count, is clinically
relevant or unspecific remains to be elucidated. Based on a
wound-healing assay, the motility of HepG2, HuH7 and Hep3B cells
has recently been reported to be significantly reduced by
everolimus, whereas a Transwell migration assay, which was
conducted in parallel, demonstrated a slight elevation in HepG2 and
HuH7 migration (18). These data
suggested a partial assay-dependent effect. To determine the
effects of mTOR inhibitors on tumor cell migration, further
investigation is required. Since everolimus and temsirolimus
markedly suppressed HCC cell adhesion in the present in
vitro model, it is hypothesized that this property is the
dominant mechanism underlying the effects of these compounds.
Integrins serve a significant role in tumorigenesis
and progression, including tumor growth, adhesion and invasion. The
integrin profile analysis conducted in the present study detected a
strong surface expression of the α1, α2, α6, and β1 integrin
subtypes. Integrin α5 was not detected on HepG2 cells, which is
contradictory to the findings of an earlier study, which revealed a
moderate integrin α5 surface level on HepG2 cells (24). However, in the previous
investigation, integrin α5 was not detected by western blotting,
thus suggesting that integrin α5 does not serve a crucial role in
HepG2 cell behavior. In the present study, integrin α1 was the only
integrin significantly increased in response to everolimus or
temsirolimus treatment. The relevance of this increase is not yet
clear. Based on integrin blocking studies, integrin α1 does not
appear involved in cell growth regulation. However, suppression of
integrin α1 was strongly associated with reduced adhesion and
migration, with a stronger effect on adhesion.
The role of integrin α1 in HCC metastasis is
currently unclear. Liu et al (25) detected a positive correlation
between integrin α1 and migration and invasion, which is in
accordance with the present study. However, compared with the
present investigation, adhesion assays were not conducted. The
integrin-blocking model used in the present study explored the
mechanical contact of α1 to its substrate. Since everolimus- or
temsirolimus-induced suppression of HCC cell adhesion was
accompanied by elevated integrin α1 expression, the positive
association between α1 and migration cannot be transferred to the
adhesion process. Another mode of action must, therefore, be
assumed, which is not associated with mechanical contact inhibition
of α1 and its receptor in the collagen matrix. Notably, previous
studies have indicated that integrin α1 may indirectly influence
HCC progression by deactivating oncogenic signaling (26,27).
Chen et al (28)
hypothesized that integrin α1 may initiate mesenchymal to
epithelial transition, with a high expression level associated with
a low tumor cell adhesion capacity. This scenario may also hold
true in the present model system, where this particular integrin
may control cellular re-differentiation, leading to a less adhesive
phenotype. However, this hypothesis is speculative and requires
further confirmation. In addition, integrin α1 expression has been
reported to be reduced in patients with metastatic breast cancer
(29), and a high integrin α1
level has been revealed to correlate with a beneficial therapeutic
response in a patient with melanoma (30).
In conclusion, in the present study, the in
vitro malignancy of HCC was reduced following treatment with
the mTOR inhibitors, everolimus and temsirolimus, thus indicating
that the mTOR pathway may be a potential target in the treatment of
HCC. Both drugs suppressed HCC cell growth and adhesion, and were
demonstrated to regulate integrin α1 expression, which is a novel
finding. In addition, tumor-matrix interaction was blocked and,
consequently, migration may decrease. However, the effects of mTOR
inhibitors on invasive processes require further evaluation. At
present, clinical reports remain ambivalent regarding the value of
mTOR inhibition (31,32). Possibly, identification of a
predictive biomarker may aid in selecting patients most likely to
respond to everolimus or temsirolimus. In addition to optimizing
the mTOR inhibitor-based protocol, which includes dual targeting of
the mTOR complexes (mTORCs), mTORC1 and mTORC2 (18,33),
further studies should focus on combination strategies that
interfere not only with the mTOR signaling cascade, but with
different tumor targets.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: Sorafenib in advanced hepatocellular carcinoma. N Engl J
Med. 359:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S,
Kim JS, Luo R, Feng J, Ye S, Yang TS, et al: Efficacy and safety of
sorafenib in patients in the Asia-Pacific region with advanced
hepatocellular carcinoma: A phase III randomised, double-blind,
placebo-controlled trial. Lancet Oncol. 10:25–34. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Abou-Alfa GK, Schwartz L, Ricci S, Amadori
D, Santoro A, Figer A, De Greve J, Douillard JY, Lathia C, Schwartz
B, et al: Phase II study of sorafenib in patients with advanced
hepatocellular carcinoma. J Clin Oncol. 24:4293–4300. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hu J, Che L, Li L, Pilo MG, Cigliano A,
Ribback S, Li X, Latte G, Mela M, Evert M, et al: Co-activation of
AKT and c-Met triggers rapid hepatocellular carcinoma development
via the mTORC1/FASN pathway in mice. Sci Rep. 6:204842016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ewald F, Nörz D, Grottke A, Bach J,
Herzberger C, Hofmann BT, Nashan B and Jücker M: Vertical targeting
of AKT and mTOR as well as dual targeting of AKT and MEK signaling
is synergistic in hepatocellular carcinoma. J Cancer. 6:1195–1205.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou L, Huang Y, Li J and Wang Z: The mTOR
pathway is associated with the poor prognosis of human
hepatocellular carcinoma. Med Oncol. 27:255–261. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Najjar YG and Rini BI: Novel agents in
renal carcinoma: A reality check. Ther Adv Med Oncol. 4:183–194.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu AX, Abrams TA, Miksad R, Blaszkowsky
LS, Meyerhardt JA, Zheng H, Muzikansky A, Clark JW, Kwak EL, Schrag
D, et al: Phase 1/2 study of everolimus in advanced hepatocellular
carcinoma. Cancer. 117:5094–5102. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhu AX, Kudo M, Assenat E, Cattan S, Kang
YK, Lim HY, Poon RT, Blanc JF, Vogel A, Chen CL, et al: Effect of
everolimus on survival in advanced hepatocellular carcinoma after
failure of sorafenib: The EVOLVE-1 randomized clinical trial. JAMA.
312:57–67. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yeo W, Chan SL, Mo FK, Chu CM, Hui JW,
Tong JH, Chan AW, Koh J, Hui EP, Loong H, et al: Phase I/II study
of temsirolimus for patients with unresectable Hepatocellular
Carcinoma (HCC)- a correlative study to explore potential
biomarkers for response. BMC Cancer. 15:3952015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cho J, Lee J, Kim J, Kim ST, Lee S, Kim
SY, Ha SY, Park CK and Lim HY: Loss of tuberous sclerosis complex 2
(TSC2) as a predictive biomarker of response to mTOR inhibitor
treatment in patients with hepatocellular carcinoma. Transl Oncol.
9:466–471. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kelley RK, Nimeiri HS, Munster PN, Vergo
MT, Huang Y, Li CM, Hwang J, Mulcahy MF, Yeh BM, Kuhn P, et al:
Temsirolimus combined with sorafenib in hepatocellular carcinoma: A
phase I dose-finding trial with pharmacokinetic and biomarker
correlates. Ann Oncol. 24:1900–1907. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Koeberle D, Dufour JF, Demeter G, Li Q,
Ribi K, Samaras P, Saletti P, Roth AD, Horber D, Buehlmann M, et
al: Sorafenib with or without everolimus in patients with advanced
hepatocellular carcinoma (HCC): A randomized multicenter,
multinational phase II trial (SAKK 77/08 and SASL 29). Ann Oncol.
27:856–861. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Grabinski N, Ewald F, Hofmann BT, Staufer
K, Schumacher U, Nashan B and Jücker M: Combined targeting of AKT
and mTOR synergistically inhibits proliferation of hepatocellular
carcinoma cells. Mol Cancer. 11:852012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou Q, Wong CH, Lau CP, Hui CW, Lui VW,
Chan SL and Yeo W: Enhanced antitumor activity with combining
effect of mTOR inhibition and microtubule stabilization in
hepatocellular carcinoma. Int J Hepatol. 2013:1038302013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou Q, Lui VW, Lau CP, Cheng SH, Ng MH,
Cai Y, Chan SL and Yeo W: Sustained antitumor activity by
co-targeting mTOR and the microtubule with temsirolimus/vinblastine
combination in hepatocellular carcinoma. Biochem Pharmacol.
83:1146–1158. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim JO, Kim KH, Song IS, Cheon KS, Kim OH,
Lee SC, Lee SK and Kim SJ: Potentiation of the anticancer effects
of everolimus using a dual mTORC1/2 inhibitor in hepatocellular
carcinoma cells. Oncotarget. 8:2936–2948. 2017.PubMed/NCBI
|
|
19
|
Chan SL, Wong CH, Lau CP, Zhou Q, Hui CW,
Lui VW, Ma BB, Chan AT and Yeo W: Preclinical evaluation of
combined TKI-258 and RAD001 in hepatocellular carcinoma. Cancer
Chemother Pharmacol. 71:1417–1425. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Aden DP, Fogel A, Plotkin S, Damjanov I
and Knowles BB: Controlled synthesis of HBsAg in a differentiated
human liver carcinoma-derived cell line. Nature. 282:615–616. 1979.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pivonello C, Negri M, De Martino MC,
Napolitano M, de Angelis C, Provvisiero DP, Cuomo G, Auriemma RS,
Simeoli C, Izzo F, et al: The dual targeting of insulin and
insulin-like growth factor 1 receptor enhances the mTOR
inhibitor-mediated antitumor efficacy in hepatocellular carcinoma.
Oncotarget. 7:9718–9731. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kirstein MM, Boukouris AE, Pothiraju D,
Buitrago-Molina LE, Marhenke S, Schütt J, Orlik J, Kühnel F,
Hegermann J, Manns MP, et al: Activity of the mTOR inhibitor
RAD001, the dual mTOR and PI3-kinase inhibitor BEZ235 and the
PI3-kinase inhibitor BKM120 in hepatocellular carcinoma. Liver Int.
33:780–793. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gao Y, Lin LP, Zhu CH, Chen Y, Hou YT and
Ding J: Growth arrest induced by C75, A fatty acid synthase
inhibitor, was partially modulated by p38 MAPK but not by p53 in
human hepatocellular carcinoma. Cancer Biol Ther. 5:978–985. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gnainsky Y, Spira G, Paizi M, Bruck R,
Nagler A, Genina O, Taub R, Halevy O and Pines M: Involvement of
the tyrosine phosphatase early gene of liver regeneration (PRL-1)
in cell cycle and in liver regeneration and fibrosis effect of
halofuginone. Cell Tissue Res. 324:385–394. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu X, Tian H, Li H, Ge C, Zhao F, Yao M
and Li J: Derivate isocorydine (d-ICD) suppresses migration and
invasion of hepatocellular carcinoma cell by downregulating ITGA1
expression. Int J Mol Sci. 18:5142017. View Article : Google Scholar :
|
|
26
|
Chen X, Wang H, Liao HJ, Hu W, Gewin L,
Mernaugh G, Zhang S, Zhang ZY, Vega-Montoto L, Vanacore RM, et al:
Integrin-mediated type II TGF-β receptor tyrosine dephosphorylation
controls SMAD-dependent profibrotic signaling. J Clin Invest.
124:3295–3310. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xia W, Lo CM, Poon RYC, Cheung TT, Chan
ACY, Chen L, Yang S, Tsao GSW and Wang XQ: Smad inhibitor induces
CSC differentiation for effective chemosensitization in cyclin D1-
and TGF-β/Smad-regulated liver cancer stem cell-like cells.
Oncotarget. 8:38811–38824. 2017.PubMed/NCBI
|
|
28
|
Chen A, Beetham H, Black MA, Priya R,
Telford BJ, Guest J, Wiggins GA, Godwin TD, Yap AS and Guilford PJ:
E-cadherin loss alters cytoskeletal organization and adhesion in
non-malignant breast cells but is insufficient to induce an
epithelial-mesenchymal transition. BMC Cancer. 14:5522014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gui GP, Wells CA, Browne PD, Yeomans P,
Jordan S, Puddefoot JR, Vinson GP and Carpenter R: Integrin
expression in primary breast cancer and its relation to axillary
nodal status. Surgery. 117:102–108. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gilhar A, Ullmann Y, Kalish RS, Berkutski
T, Azizi E and Bank I: Favourable melanoma prognosis associated
with the expression of the tumour necrosis factor receptor and the
alpha1beta1 integrin: A preliminary report. Melanoma Res.
7:486–495. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Finn RS: Current and future treatment
strategies for patients with advanced hepatocellular carcinoma:
Role of mTOR inhibition. Liver Cancer. 1:247–256. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ashworth RE and Wu J: Mammalian target of
rapamycin inhibition in hepatocellular carcinoma. World J Hepatol.
6:776–782. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen BW, Chen W, Liang H, Liu H, Liang C,
Zhi X, Hu LQ, Yu XZ, Wei T, Ma T, et al: Inhibition of mTORC2
induces cell-cycle arrest and enhances the cytotoxicity of
doxorubicin by suppressing MDR1 expression in HCC cells. Mol Cancer
Ther. 14:1805–1815. 2015. View Article : Google Scholar : PubMed/NCBI
|