Introduction
Type 2 diabetes mellitus (T2DM) is a primary health
concern, reaching epidemic proportions worldwide. The International
Diabetes Federation has predicted that the number of patients with
diabetes will increase from 240 million in 2007 to 380 million in
2025 (1). Current treatments for
T2DM involve attenuating the increased glucose level by insulin, or
by agents which increase the sensitivity of tissue to insulin, or
inhibit glucose absorption by small intestine. Useful therapeutic
targets for T2DM remain to be identified. Resistance to the
pleiotropic effects of insulin in the primary insulin target organs
is a critical process in the development of the disease; however,
the molecular mechanisms underlying insulin resistance remain to be
fully elucidated. MicroRNAs (miRNAs) are a class of small,
non-coding RNAs widely expressed in all multicellular organisms
that post-transcriptionally regulate gene expression (2). Previous studies have highlighted the
significance of miRNAs in maintaining metabolic homeostasis.
miR-130-3p and miR-108a, for example, have been demonstrated to
regulate insulin sensitivity (3,4).
Muscle tissue is an important site of postprandial
glucose uptake; ~75% of insulin-dependent glucose removal from the
plasma takes place in the skeletal muscle. Therefore, muscle is
critical for systemic glucose homeostasis. miR-24 and miR-126
contribute to the adaptation of muscle tissue to increased glucose
levels; however, they do not appear to be involved in the
pathogenesis of insulin resistance (5). Previous studies have demonstrated
that miR-106b is highly expressed in the skeletal muscle of
diabetes patients (6) and insulin
resistant mice (7). Our previous
study revealed that miR-106b induced mitochondrial dysfunction and
insulin resistance in C2C12 myotubes by targeting mitofusin-2
(Mfn2) (8); silencing miR-106b
improved the tumor necrosis factor (TNF)-α- and palmitic acid
(PA)-induced insulin resistance and mitochondrial dysfunction in
these cells (8,9). The present study aimed to further
investigate the contribution of miR-106b to skeletal muscle insulin
sensitivity and glucose homeostasis in vivo and discuss the
role of Mfn2 in miR-106b-induced insulin resistance in
vitro.
Materials and methods
Plasmids and lentiviruses construction
and packaging
Lentiviruses with miR-106b for overexpression and
miR-106b sponge for downregulation were constructed and packaged as
previously described (8). The
pre-miR-106b fragment was cloned from the genomic DNA of mouse
skeletal muscle tissue. The miR-106b inhibitor sponge was
synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China).
Plasmids of miR-106b and miR-106b sponge were inserted into the
lentiviral vector pGIPZ (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). The DNA fragment encoding Mfn2 was
amplified using genomic DNA of mouse skeletal muscle as the
polymerase chain reaction (PCR) template and inserted into the
expression vector pc3.1-puro (Invitrogen; Thermo Fisher Scientific,
Inc.).
Animals and treatment
A total of 24 male C57BL/6J mice (age, 6–8 weeks)
were obtained from the Laboratory Animal Center of Jinling Hospital
(Nanjing, China). Mice were housed under a 12-h light/dark cycle at
25°C with ad libitum access to commercial rodent chow and
tap water prior to the initiation of the experiments. The present
study was approved by, and conducted in accordance with the
guidelines of, the Institutional Animal Care and Use Committee of
Nanjing General Hospital of Nanjing Military Command (Nanjing,
China). Mice were randomly divided into 4 groups and infected with
lentivirus expressing miR-106b (n=6) or miR-106b sponge (n=6) and
corresponding empty vectors (n=6 in each group). Briefly,
1×107 transduction units of lentivirus in
phosphate-buffered saline at a volume of 10 ml/kg were
intravenously injected into the tail vein of mice one week prior to
a glucose tolerance test (GTT). Blood glucose levels were measured
using an Elite glucometer (Bayer AG, Leverkusen, Germany). GTT was
performed by intraperitoneal injection of 2 g/kg glucose following
12 h of fasting. Blood glucose levels were measured 15, 60 and 90
min following GTT. Subsequently, mice were anesthetized by an
intraperitoneal injection of 1% sodium pentobarbital (40 mg/kg;
Invitrogen; Thermo Fisher Scientific, Inc.). The quadricep muscles
were collected following perfusion with saline.
Cell culture and treatment
C2C12 mouse myoblasts (American Type Culture
Collection, Manassas, VA, USA) were cultured at 37°C in Dulbecco's
modified Eagle's medium (DMEM) containing 10% fetal bovine serum
(Wisent, Inc., St. Bruno, QC, Canada). C2C12 myoblasts seeded in
12-well plates (4×104 cells/well) were transfected with
the aforementioned Mfn2 plasmid (1.6 µg/well), 20 µM Mfn2 small
interfering (si)RNA or control siRNA (Santa Cruz Biotechnology,
Inc., Dallas, TX, USA), and lentiviruses with miR-106b or miR-106b
sponge. The stable C2C12 cells lines with miR-106b overexpression
and downregulation were obtained by selecting with puromycin for
two weeks. When C2C12 myoblasts reached confluence, the medium was
replaced with differentiation medium containing DMEM and 2% horse
serum (Gibco; Thermo Fisher Scientific, Inc.), which was replaced
every other day. Following five additional days, the differentiated
C2C12 myoblasts had fused into myotubes.
2-Deoxyglucose uptake assay
The uptake of 2-deoxy-D-[3H]
glucose (Beijing CIC Technology Co., Ltd., Beijing, China) was
assayed as previously described (8). Following 4 h of serum starvation in
DMEM, C2C12 myotubes were rinsed twice with
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered
saline (20 mM HEPES, 140 mM NaCl, 5 mM KCl, 2.5 mM
MgSO4, 1 mM CaCl2; pH 7.4), and stimulated
with 100 nM insulin (Peptide Institute, Inc., Osaka, Japan) for 30
min at 37°C. Glucose uptake was assessed by the addition of 10 µM
2-deoxyglucose containing 0.2 µCi [3H]-2-deoxyglucose in
HEPES-buffered saline for 10 min. The uptake of
[3H]-2-deoxyglucose was terminated by ice-cold
phosphate-buffered saline (PBS; 8% glucose), and the cells were
immediately washed with ice-cold PBS. Subsequently, cells were
lysed with 0.25 N NaOH and the cell lysates were transferred to
scintillation vials (Beckman Coulter, Inc., Brea, CA, USA) to
measure the radioactivity using an LS6500 liquid scintillation
counter (Beckman Coulter).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
miRNAs were extracted using an miRNeasy Mini kit
(Qiagen China Co., Ltd., Shanghai, China). miR-106b cDNA was
generated with a RT primer provided with the TaqMan®
MicroRNA Reverse Transcription kit (Thermo Fisher Scientific,
Inc.). qPCR was performed on an ABI7500 RT-PCR system using TaqMan
Gene Expression assays for miR-106b (Mm03306675_pri; Thermo Fisher
Scientific, Inc.) and U6 (Mm01164115_g1; Thermo Fisher Scientific,
Inc.). The cycling conditions were as follows: Initial denaturation
for 10 min at 95°C, followed by 40 cycles of 15 sec of denaturation
at 95°C, 30 sec annealing at the optimal primer temperature and 36
sec extension at 72°C. Each sample was assayed in duplicate.
Negative controls (no template) were run to ensure the absence of
contamination. Analysis was performed using the 2−ΔΔCq
method (10) and the data was
normalized to U6.
Western blot analysis
Western blot analysis was performed as previously
described (8). Cells and tissue
were lysed with radioimmunoprecipitation assay lysis buffer
(Beyotime Institute of Biotechnology, Haimen, China) for 20 min on
ice. Protein levels were quantified using a bicinchoninic acid
protein assay kit (Pierce; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Proteins from muscle
tissue (60 µg) or cells (30 µg) were loaded onto 10% SDS-PAGE gels.
Following electrophoresis, proteins were transferred to
nitrocellulose membranes (EMD Millipore, Billerica, MA, USA).
Membranes were blocked with 5% bovine serum albumin (BSA) in
Tris-buffered saline with Tween-20 [TBST; 50 mM Tris (pH 7.5), 150
mM NaCl, 0.05% Tween-20] for 12 h. Following this, membranes were
incubated at 4°C overnight with one of the following primary
antibodies at the indicated dilutions in TBS with 5% BSA: Rabbit
anti-Mfn2 (1:500; catalog no. ab50838), rabbit anti-glucose
transporter-4 (Glut4; 1:500; catalog no. ab654), and rabbit
anti-β-actin (1:5,000; catalog no. ab8227), purchased from Abcam
(Cambridge, MA, USA). The membranes were washed five times for 5
min with TBST. Subsequently, the membranes were incubated with a
horseradish peroxidase-conjugated goat anti-rabbit IgG (1:5,000;
catalog no. ab6721; Abcam) for 1 h at room temperature, washed with
TBST and developed using an Enhanced Chemiluminescence kit (GE
Healthcare Life Sciences, Chalfont, UK). Densitometry was performed
using Quantity One software version 4.6.4 (Bio-Rad Laboratories,
Inc., Hercules, CA, USA).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Differences between groups were assessed by two-tailed Student's
t-test or one-way analysis of variance followed by the
Student-Newman-Keuls test. P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-106b regulates skeletal muscle
insulin sensitivity and glucose homeostasis in vivo
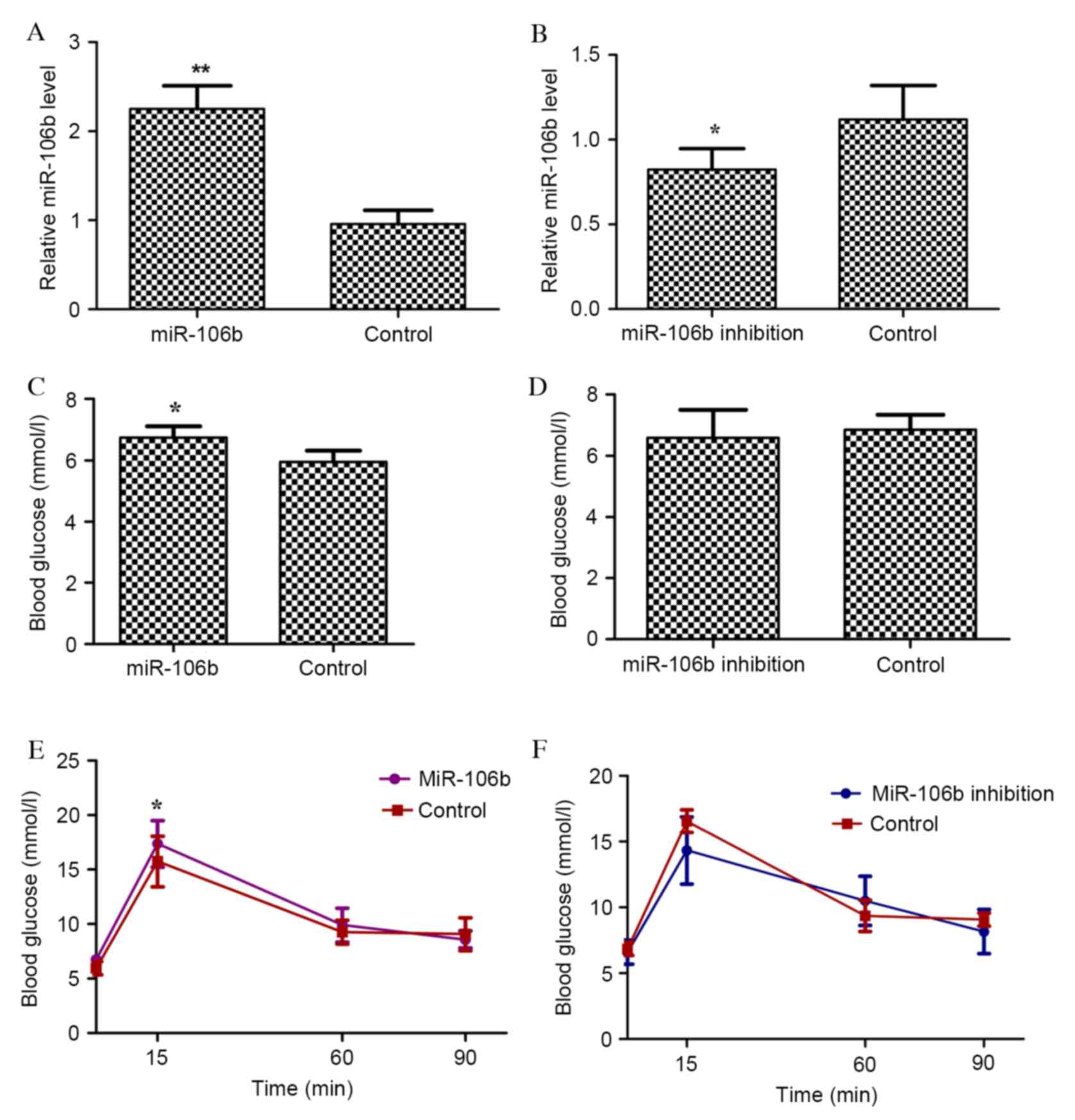
To investigate whether miR-106b regulated glucose
homeostasis in vivo, lentiviruses with miR-106b for
overexpression, with miR-106b sponge for downregulation or empty
lentiviral vectors were used to infect C57BL/6 mice by tail vein
injection. The expression level of miR-106b was markedly up and
downregulated in the muscle of mice infected with lentiviruses
expressing miR-106b precursors (designated as miR-106b mice;
P=0.009; Fig. 1A) and miR-106b
sponge (designated as miR-106b inhibition mice; P=0.007; Fig. 1B), respectively. miR-106b mice had
significantly greater blood glucose levels following 16 h of
fasting (P=0.005; Fig. 1C),
whereas miR-106b inhibition mice had no significant alteration in
blood glucose (Fig. 1D).
Similarly, miR-106b mice had impaired glucose tolerance
(P<0.001; Fig. 1E), whereas
miR-106b inhibition mice did not (Fig.
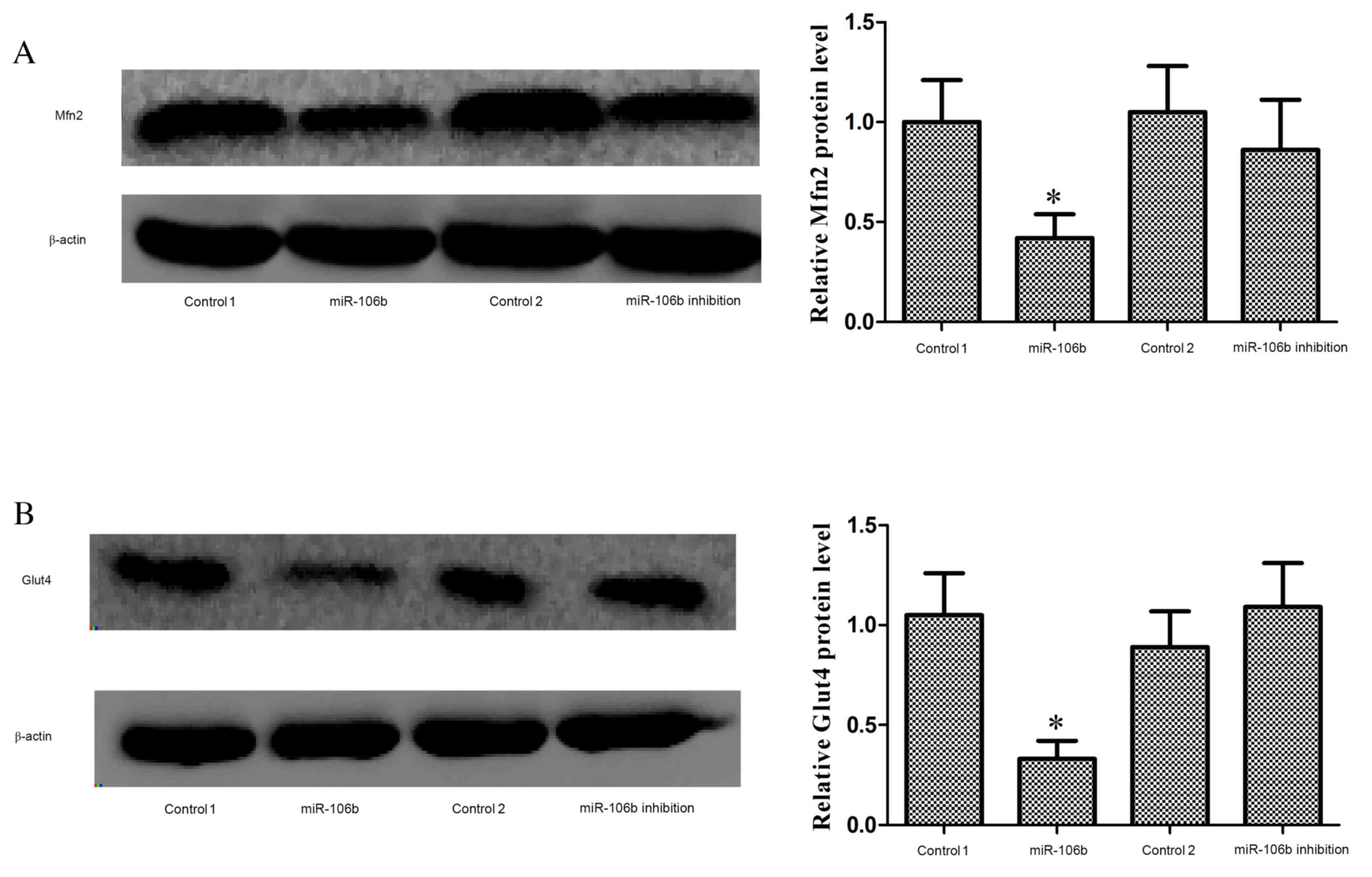
1F). Mfn2 (P=0.027; Fig. 2A)
and plasmalemma Glut4 (P=0.034; Fig.
2B) protein expression levels were significantly reduced in the
muscle of miR-106b mice, whereas these protein levels were
unaffected in miR-106b inhibition mice. The decrease of Glut4
translocation to cell membrane in the muscle indicated reduced
insulin sensitivity.
Mfn2 is involved in the regulation of
C2C12 myotubes insulin sensitivity by miR-106b in vitro
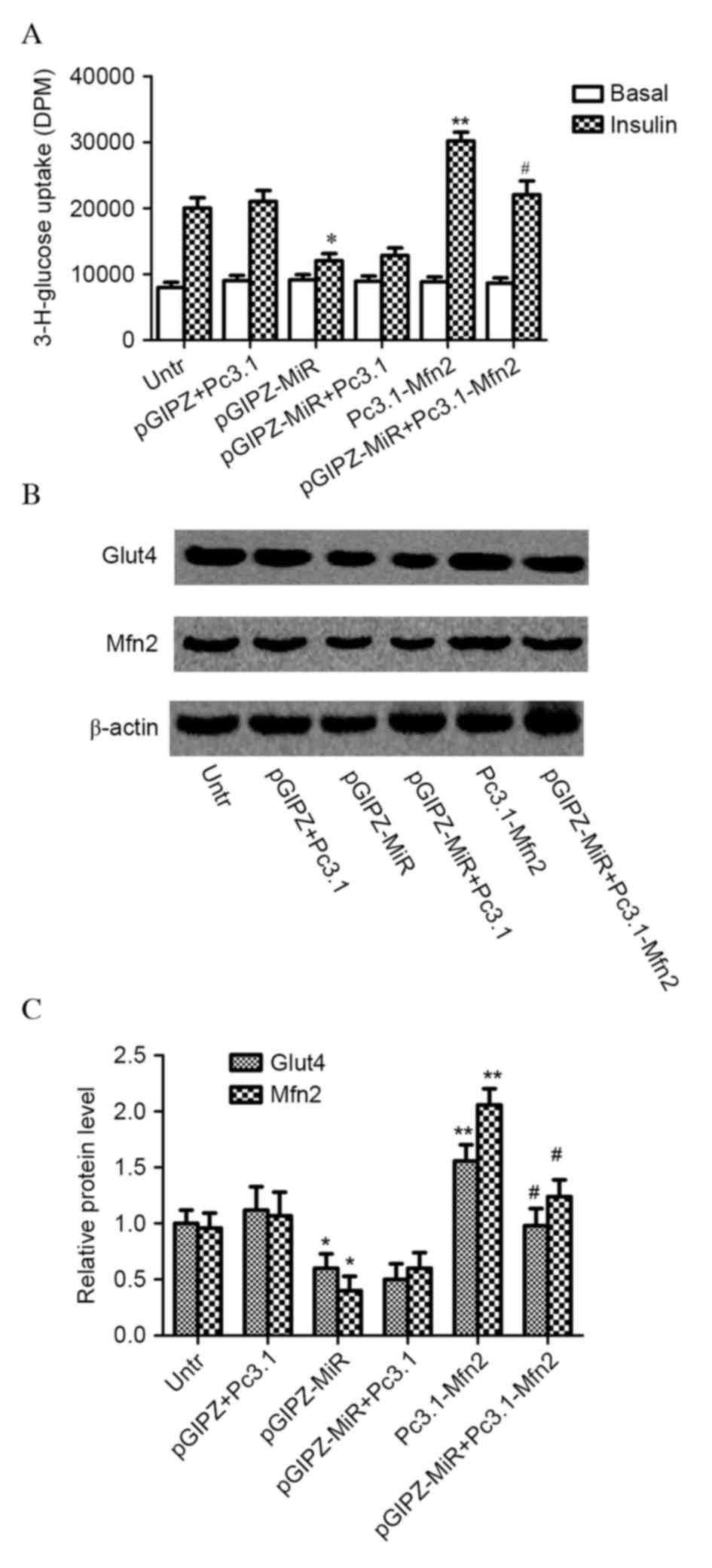
To investigate whether Mfn2 is involved in the
regulation of muscle insulin sensitivity by miR-106b, C2C12
myotubes were transfected with miR-106b, miR-106b sponge, Mfn2
plasmid or Mfn2 siRNA. miR-106b successfully decreased
insulin-stimulated glucose uptake in C2C12 myotubes overexpressing
miR-106b (pGIPZ-MiR; P=0.042; Fig.
3A). However, the repressive effect of miR-106b on glucose
uptake was completely inhibited in C2C12 myoblasts with miR-106b
overexpression and Mfn2 plasmid infection (Pc3.1-Mfn2; P=0.032),
but not by control plasmid (Pc3.1). Mfn2 protein expression levels
(P=0.043) and translocation of Glut4 (P=0.034) were significantly
decreased in C2C12 myotubes with miR-106b overexpression; however,
Mfn2 plasmid infection significantly increased Mfn2 (P=0.023) and
Glut4 protein expression levels (P=0.011) in C2C12 myotubes with
miR-106b overexpression compared with control plasmids (Fig. 3B and C).
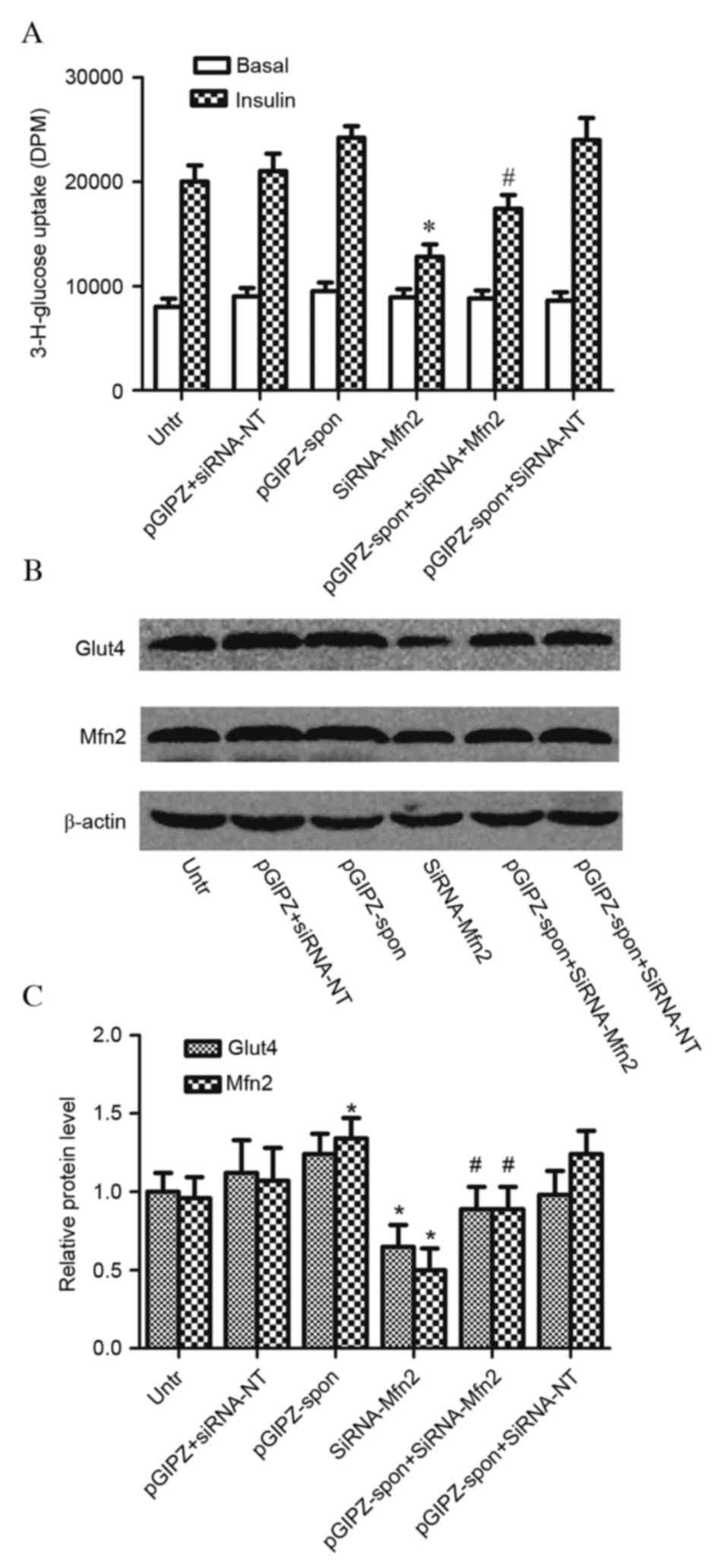
Inhibition of miR-106b (pGIPZ-spon) did not
significantly affect glucose uptake in C2C12 myotubes infected with
control siRNA [siRNA-no target (NT); P=0.322]. C2C12 myotubes
treated with Mfn2 siRNA (siRNA-Mfn2) had decreased glucose uptake
(P=0.012); however, this was increased by the concomitant
inhibition of miR-106b (P=0.032; Fig.
4A). Glut4 translocation was unaffected by inhibition of
miR-106b (P=0.123); however, Mfn2 protein expression levels were
significantly increased in C2C12 myotubes with miR-106b inhibition
(P<0.001; Fig. 4B and C). Mfn2
siRNA infection significantly decreased Mfn2 expression levels and
Glut4 translocation in C2C12 myotubes; miR-106b inhibition
partially reversed these effects. These results indicated that
miR-106b regulated skeletal muscle insulin sensitivity by targeting
Mfn2.
Discussion
Gallagher et al (6) identified, using an miRNA microarray,
that miR-106b was highly expressed in the skeletal muscle of T2DM
patients. A separate study revealed, using microarrays and RT-qPCR,
that miR-106b expression in the skeletal muscle of mice fed a
high-fat diet was 4.19-fold greater than mice on a normal diet
(7). However, the role of miR-106b
in regulating skeletal muscle insulin sensitivity and glucose
homeostasis remains to be fully elucidated.
In our previous study, miR-106b overexpression was
demonstrated to decrease glucose uptake and Glut4 translocation in
C2C12 myotubes, indicating insulin resistance (8). In addition, it was revealed that
miR-106b targeted Mfn2, an important protein mediating
mitochondrial fusion and dynamics, inducing mitochondrial
dysfunction, which may contribute to muscle insulin resistance
(8). The inflammatory factor TNF-α
and the saturated fatty acid PA increased miR-106b expression in
C2C12 myotubes in a time- and dose-dependent manner (7,8).
miR-106b loss of function attenuated TNF-α- and PA-induced
mitochondrial dysfunction and insulin resistance (8,9).
In the present study miR-106b mice and miR-106b
inhibition mice were created to increase or reduce miR-106b
expression levels in the muscle, through tail-vein injection of
lentiviruses with miR-106b for overexpression or miR-106b sponge
for downregulation. miR-106b mice had decreased Mfn2 and Glut4
expression levels in the muscle, and impaired glucose tolerance at
the first phase by GTT. miR-106b inhibition failed to regulate
systemic glucose homeostasis in vivo. This may be due to the
decrease of glucose uptake by the muscle being compensated for by
the increase of glucose uptake by other insulin target organs,
including the liver and adipose tissue. Furthermore, energy
metabolism is a complex and integrated process, and gluconeogenesis
and glycolysis additionally contribute to systemic glucose
homeostasis. The present study indicated that miR-106b contributed
to glucose homeostasis in certain pathological processes.
In our previous study, it was demonstrated that Mfn2
was a direct target of miR-106b (8). In the present study, it was further
revealed, by overexpression or knockdown of Mfn2 expression in
C2C12 myotubes, that Mfn2 was the direct target through which
miR-106b exerted its effects. Mfn2 protein, a dynamin-related
protein with GTPase activity, is anchored in the external
mitochondrial membrane to mediate mitochondrial fusion (11). In addition, Mfn2 localizes to the
endoplasmic reticulum (ER) membrane, serving to tether the ER to
mitochondria (12). Mfn2 is highly
expressed in skeletal muscle, contributes to the maintenance of
mitochondrial morphology and regulates mitochondrial metabolism and
intracellular signaling (13).
Mfn2 suppression has been detected in skeletal muscle of T2DM
patients (14). In addition, a
positive correlation between Mfn2 expression in skeletal muscle and
insulin sensitivity has been detected, as assessed by glucose
disposal rates in healthy controls, obese and T2DM patients
(14). Furthermore, this positive
correlation has been demonstrated in morbidly obese subjects and
bariatric surgery patients (14,15).
Recently, it was reported that Mfn2 deficiency induced oxidative
stress, which contributed to insulin resistance in skeletal muscle
cells (16). Mfn2 reduced the
accumulation of lipid intermediates in skeletal muscle and
alleviated insulin resistance (17). The offspring of rats fed a high-fat
diet had reduced Mfn2 mRNA expression levels at postnatal day 35
and 130 (18).
In conclusion, the results of the present study
indicated that miR-106b regulated skeletal muscle insulin
sensitivity and glucose homeostasis, with Mfn2 contributing to this
process. The finding that miR-106b overexpression induced skeletal
muscle insulin resistance and impaired glucose homeostasis may
suggest a potential mechanism underlying insulin resistance and
T2DM.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81100592
and 81270800).
References
|
1
|
International Diabetes Federation:
Diabetes Atlas. 3rd edition. International Diabetes Federation;
Brussels, Belgium: 2006
|
|
2
|
Guo H, Ingolia NT, Weissman JS and Bartel
DP: Mammalian microRNAs predominantly act to decrease target mRNA
levels. Nature. 466:835–840. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xiao F, Yu J, Liu B, Guo Y, Li K, Deng J,
Zhang J, Wang C, Chen S, Du Y, et al: A novel function of microRNA
130a-3p in hepatic insulin sensitivity and liver steatosis.
Diabetes. 63:2631–2642. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou B, Li C, Qi W, Zhang Y, Zhang F, Wu
JX, Hu YN, Wu DM, Liu Y, Yan TT, et al: Downregulation of miR-181a
upregulates sirtuin-1 (SIRT1) and improves hepatic insulin
sensitivity. Diabetologia. 55:2032–2043. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ferland-McCollough D, Ozanne SE, Siddle K,
Willis AE and Bushell M: The involvement of microRNAs in Type 2
diabetes. Biochem Soc Trans. 38:1565–1570. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gallagher IJ, Scheele C, Keller P, Nielsen
AR, Remenyi J, Fischer CP, Roder K, Babraj J, Wahlestedt C,
Hutvagner G, et al: Integration of microRNA changes in vivo
identifies novel molecular features of muscle insulin resistance in
type 2 diabetes. Genome Med. 2:92010. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen GQ, Lian WJ, Wang GM, Wang S, Yang YQ
and Zhao ZW: Altered microRNA expression in skeletal muscle results
from high-fat diet-induced insulin resistance in mice. Mol Med Rep.
5:1362–1368. 2012.PubMed/NCBI
|
|
8
|
Zhang Y, Yang L, Gao YF, Fan ZM, Cai XY,
Liu MY, Guo XR, Gao CL and Xia ZK: MicroRNA-106b induces
mitochondrial dysfunction and insulin resistance in C2C12 myotubes
by targeting mitofusin-2. Mol Cell Endocrinol. 381:230–240. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang Y, Zhao YP, Gao YF, Fan ZM, Liu MY,
Cai XY, Xia ZK and Gao CL: Silencing miR-106b improves palmitic
acid-induced mitochondrial dysfunction and insulin resistance in
skeletal myocytes. Mol Med Rep. 11:3834–3841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liesa M, Palacin M and Zorzano A:
Mitochondrial dynamics in mammalian health and disease. Physiol
Rev. 89:799–845. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
de Brito OM and Scorrano L: Mitofusin 2
tethers endoplasmic reticulum to mitochondria. Nature. 456:605–610.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zorzano A: Regulation of mitofusin-2
expression in skeletal muscle. Appl Physiol Nutr Metab. 34:433–449.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bach D, Naon D, Pich S, Soriano FX, Vega
N, Rieusset J, Laville M, Guillet C, Boirie Y, Wallberg-Henriksson
H, et al: Expression of Mfn2, the Charcot-Marie-Tooth neuropathy
type 2A gene, in human skeletal muscle: Effects of type 2 diabetes,
obesity, weight loss, and the regulatory role of tumor necrosis
factor alpha and interleukin-6. Diabetes. 54:2685–2693. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cartoni R, Léger B, Hock MB, Praz M,
Crettenand A, Pich S, Ziltener JL, Luthi F, Dériaz O, Zorzano A, et
al: Mitofusins 1/2 and ERRalpha expression are increased in human
skeletal muscle after physical exercise. J Physiol. 567:349–358.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nie Q, Wang C, Song G, Ma H, Kong D, Zhang
X, Gan K and Tang Y: Mitofusin 2 deficiency leads to oxidative
stress that contributes to insulin resistance in rat skeletal
muscle cells. Mol Biol Rep. 41:6975–6983. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang X, Wang C, Song G, Gan K, Kong D,
Nie Q and Ren L: Mitofusion-2-mediated alleviation of insulin
resistance in rats through reduction in lipid intermediate
accumulation in skeletal muscle. J Biomed Sci. 20:452013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Borengasser SJ, Faske J, Kang P, Blackburn
ML, Badger TM and Shankar K: In utero exposure to prepregnancy
maternal obesity and postweaning high-fat diet impair regulators of
mitochondrial dynamics in rat placenta and offspring. Physiol
Genomics. 46:841–850. 2014. View Article : Google Scholar : PubMed/NCBI
|