Introduction
Kisspeptin is a protein encoded by the KISS1
gene in humans, which has been reported to serve a diverse role in
various physiological and pathological conditions (1–5).
Kisspeptin has been reported to suppress the metastatic
capabilities of various types of cancer cells, through the
activation of its G-protein coupled receptor (GPR) 54 (5–7).
Kisspeptin has been demonstrated to activate various cytosolic
protein kinases, including B, C and D in several types of cells
in vitro (8–10). However, kisspeptin-mediated
signaling pathways are complex, and the molecular mechanisms
underlying the involvement of kisspeptin-induced intracellular
signaling in the regulation of the migratory and invasive
capabilities of cancer cells have yet to be fully elucidated
(9,11–13).
The eukaryotic translation initiation factor 2α
kinase 2 (EIF2AK2; also known as double-stranded RNA-activated
protein kinase, PKR), has been revealed to be ubiquitously
expressed across numerous cell types, and may be activated under
various types of cellular stress, including viral infections,
hypoxia and nutritional depletion (14). EIF2AK2 activation has been reported
to suppress tumor growth (15–18);
however, its expression and activity levels have been demonstrated
to be increased in various types of cancer cells, including breast,
lung and colorectal cancer cells (19–21).
It has previously been reported that invasive ductal carcinoma
cells exhibit increased EIF2AK2 expression levels compared with
adjacent normal breast tissue (21); however, the expression and activity
levels of EIF2AK2 have been negatively correlated with the
metastatic capabilities of breast cancer cells (22). Furthermore, the double-stranded
RNA-mediated activation of EIF2AK2 has been implicated in the
regulation of breast cancer cell motility (23). These results suggest that EIF2AK2
activity may be critical for the suppression of cancer metastasis.
Conversely, EIF2AK2 has been reported to be implicated in
hepatocellular carcinoma growth and migration, as well as gastric
cancer metastasis (24,25). Therefore, it may be hypothesized
that its anti-tumor or oncogenic role is determined by the type of
cancer cell.
Previous studies have proposed a role for EIF2AK2 in
the regulation of cytoskeletal dynamics (23,26),
which appear to be similar to the roles of kisspeptin in the
regulation of cellular architecture (7,8,13).
However, whether kisspeptin may inhibit cancer cell migration and
invasion through the modulation of EIF2AK2 has yet to be
elucidated. Therefore, the present study aimed to investigate the
role of EIF2AK2 in the mechanisms underlying the inhibitory effects
of kisspeptin on cancer metastasis. The results of the present
study demonstrated that kisspeptin activated EIF2AK2 in several
highly metastatic types of cancer cells. Furthermore, kisspeptin
failed to suppress the migratory and invasive capabilities of mouse
embryonic fibroblasts (MEFs) following the deletion of the
EIF2AK2 gene. An in vivo experimental metastasis
assay revealed that kisspeptin-induced EIF2AK2 activation
attenuated cancer cell migration and invasion, thus suppressing
distant metastasis to the lungs. In addition, kisspeptin was
demonstrated to promote the interaction between the Ras homolog
gene family member A (RhoA) and EIF2AK2, resulting in the
autophosphorylation of EIF2AK2. The present results suggest that
kisspeptin may suppress the metastatic capabilities of cancer cells
via activation of EIF2AK2. Therefore, kisspeptin treatment may be
one of options in preventing cancer metastases.
Materials and methods
Cell lines and reagents
The following cell lines used in the present study
were kindly provided by Professor Barat B. Aggarwal (University of
Texas, Anderson Cancer Center, Houston, Texas): wild-type MEFs
(MEF-EIF2AK2+/+), MEFs where the EIF2AK2
gene was deleted (MEF-EIF2AK2−/−), human breast
SK-BR-3, prostatic PC-3 and colorectal LoVo adenocarcinoma cells.
Cells were cultured in Dulbecco's modified Eagle's medium or
RPMI-1640 (Cellgro; Corning Incorporated, Corning, NY, USA),
supplemented with 10% fetal bovine serum (Cellgro; Corning
Incorporated) and 1% penicillin/streptomycin (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and 5% CO2
humidified air at 37°C. Kisspeptin (C-terminal amidated
kisspeptin-10) was purchased from Tocris Bioscience (Bristol, UK).
The Rho-associated protein kinase (ROCK) inhibitor Y-27632 was
purchased from Selleck Chemicals (Houston, TX, USA). The EIF2AK2
inhibitor C16 was purchased from Santa Cruz Biotechnology, Inc.
(sc-204200; Dallas, TX, USA).
Short interfering (si)RNA
experiment
3×106 LoVo cells were transfected with
EIF2AK2 siRNA or negative control siRNA using Lipofectamine 2000
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). In brief,
1×106 LoVo cells were cultured in DMEM medium without
antibiotics a day before transfection. Then, 20 µl of Lipofectamine
2000 reagent in 500 µl of serum-free DMEM medium and 10 µl of each
siRNA in 500 µl of serum-free DMEM medium were gently mixed and
incubated for 30 min at room temperature, and the mixed solution
was added in the cells. The cells were then incubated at 37°C in a
CO2 incubator for 24 h and then used for the assays such
as the migration and invasion assay. EIF2AK2 siRNA and negative
control siRNA were purchased from Santa Cruz Biotechnology, Inc.
These siRNA products consist of pools of three to five
target-specific 19 to 25 nucleotide siRNAs designed to knock down
gene expression. EIF2AK2 silencing was confirmed by western blot
analysis.
Western blot analysis
A total of 3×106 cells (all cell types
used in this study) were lyzed using radioimmunoprecipitation lysis
buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM
Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate,
2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM
Na3VO4, 1 µg/ml leupeptin, 1 mM PMSF) for 30
min on ice and centrifuged at 20,000 × g for 10 min at 4°C. Protein
concentrations were measured using Pierce BCA protein assay kit
(Pierce; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Equal amounts of extracted protein samples
(30 µg) were separated by 6–12% SDS-PAGE and transferred onto
polyvinylidene difluoride membranes. Subsequent to blocking with 5%
milk for 1 h at room temperature, membranes were probed for 1 h at
room temperature with the following primary antibodies:
Anti-phosphorylated (p)-cofilin (sc-271921; 1:250; Santa Cruz
Biotechnology, Inc.), anti-cofilin (sc-376476; 1:250; Santa Cruz
Biotechnology, Inc.), anti-p-myosin light chain (MLC; 3674;
1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA),
anti-MLC (8505; 1:1,000; Cell Signaling Technology, Inc.),
anti-RhoA (sc-166399; 1:250; Santa Cruz Biotechnology, Inc.),
anti-EIF2AK2 (sc-6282; 1:250; Santa Cruz Biotechnology, Inc.)
anti-p-EIF2AK2 (3075; 1:1,000; Cell Signaling Technology, Inc.) and
anti-β-actin (4967; 1:1,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA). The membranes were then incubated in secondary
antibodies conjugated to horseradish peroxidase (anti-rabbit; 7074;
1:10,000, anti-mouse; 7076; 1:10,000; Cell Signaling Technology,
Inc.) for 1 h at room temperature. Bands were detected using
LumiGLO chemiluminescent reagent and peroxidase (7003; Cell
Signaling Technology, Inc.). Active GTP-bound RhoA was detected by
a glutathione-S-transferase (GST) pull down assay using a human
Rhotekin Rho binding domain (RBD)-GST-fusion protein.
Scratch wound assay
An in vitro scratch-wound healing assay was
used to evaluate cellular migration; 3×105 cells (from
all cell lines tested in the present study) were cultured in 6-well
plates containing DMEM medium at 37°C in CO2 incubator
for 24 h and scratched when cell confluence reached about 80%. A
‘scratch’ was created by scraping the cell culture in a straight
line with a 200 µl pipette tip and cells were washed with PBS three
times to remove debris. The cells were then treated with kisspeptin
at 100 nM for 24 h at 37°C and then random cell migration was
evaluated via counting the number of cells that had migrated in the
scratched region. Experiments were performed in triplicate.
Non-treated cells were used as a control in the migration assay.
Cell migration was observed using a Zeiss Axiovert inverted
microscope, and images were analyzed using Zen software version
3.00 (Carl Zeiss AG, Oberkochen, Germany).
Cell invasion analysis
An invasion assay was used to evaluate the invasive
capabilities of cancer cells; 3×105 cells (from all cell
lines tested in this study) were seeded into the upper chambers of
Transwell inserts (Corning Incorporated, Corning, NY, USA)
pre-coated with Matrigel (BD Biosciences, San Jose, CA, USA). The
upper chambers were filled with DMEM medium supplemented with 1%
serum, whereas the lower chambers were filled with DMEM medium
supplemented with 10% serum as a chemoattractant. The cells in the
upper chamber were treated with kisspeptin at 100 nM for 24 h.
Non-treated cells were used as a control. Following incubation at
37°C for 24 h, cells on the top of the membrane were carefully
removed with cotton swabs. Cells that had invaded the lower
membrane were fixed with 4% formaldehyde at room temperature for 5
min, stained with 1% crystal violet at room temperature for another
5 min, and then counted. Experiments were performed in triplicate.
Cell invasion was observed using a Zeiss Axiovert inverted
microscope, and images were analyzed using Zen software version
3.00 (Carl Zeiss AG). A total of 4 fields were randomly selected
and the invaded cells were counted.
In vivo studies
Cultured LoVo cells (3×105) were
subcutaneously injected into the back skin of 6-week old male nude
mice (Orient Bio Inc., Seongnam, Korea). A day after tumor cell
injection, kisspeptin (100 nM) alone (n=6), or in combination with
C16 (200 nM) (n=6) or control (saline, n=6) was subcutaneously
injected every other day into the tumor site for 14 days. Tumor
volume was measured every other day, according to the following
formula: Tumor volume (mm3) = 0.5 × (tumor length, mm) ×
(tumor width, mm)2. Mice were sacrificed 14 days after
treatment, and tumors were removed for immunohistochemical
analysis. For the experimental metastasis assay, LoVo cells
(1×106) were treated with kisspeptin (100 nM) alone or
in combination with C16 (200 nM) for 1 h at 37°C, and then injected
into the tail vein of the mice. A total of 2 weeks following the
injection, mice were sacrificed, the lungs were isolated and
metastatic foci were counted. Mice were maintained under a 12-h
light/dark cycle with free access to food and water and diet at
24±1°C and a relative humidity of 40±5%. All experimental
procedures were approved by the Institutional Animal Care and Use
Committee of the Kyung Hee University (Seoul, Republic of
Korea).
Immunohistochemistry
Tumor xenografts were removed, cut and put on
cassettes, fixed with 4% formaldehyde at room temperature for 24 h
and embedded in paraffin wax. Tumor tissue was sectioned at 10 µm,
deparaffinized, and antigen-retrieval performed with 8 M Urea,
H2O2 was used to block endogenous peroxidase.
The sections were washed with TBST (0.1% Tween-20) twice, and
blocked in 10% normal serum with 1% BSA for 1 h at room
temperature, stained with an anti-p-EIF2AK2 antibody (ab32036;
Abcam, Cambridge, UK) for 1 h at room temperature, incubated with
anti-rabbit secondary antibody (ab205718; Abcam) for another 1 h at
room temperature and then incubated with DAB for 2 min at room
temperature using DAB substrate kit (ab64238; Abcam). Stained
sections were observed under ×20 magnification. Cell invasion was
observed using a Zeiss Axiovert inverted microscope, and images
were analyzed using Zen software version 3.00 (Carl Zeiss AG).
Statistical analysis
The statistical significance of the differences
between groups was assessed using unpaired Student's t-test or
one-way analysis of variance with a post-hoc Tukey's test using
SPSS version 22 (IBM Corp., Armonk, NY, USA). All in vitro
experiments were conducted at least three times. Data are expressed
as the mean ± standard deviation. P<0.05 was considered to
indicate a statistically significant difference.
Results
EIF2AK2 is involved in
kisspeptin-mediated inhibition of cellular migration and
invasion
To investigate whether EIF2AK2 may be involved in
the mechanism underlying the inhibitory effects of kisspeptin on
cellular migration, wild-type MEFs
(MEF-EIF2AK2+/+) and MEFs where the
EIF2AK2 gene was deleted (MEF-EIF2AK2−/−)
received a scratch wound and were subsequently treated with
kisspeptin for 24 h. EIF2AK2 expression in
MEF-EIF2AK2+/+, and its absence in
MEF-EIF2AK2−/− cells were confirmed using western
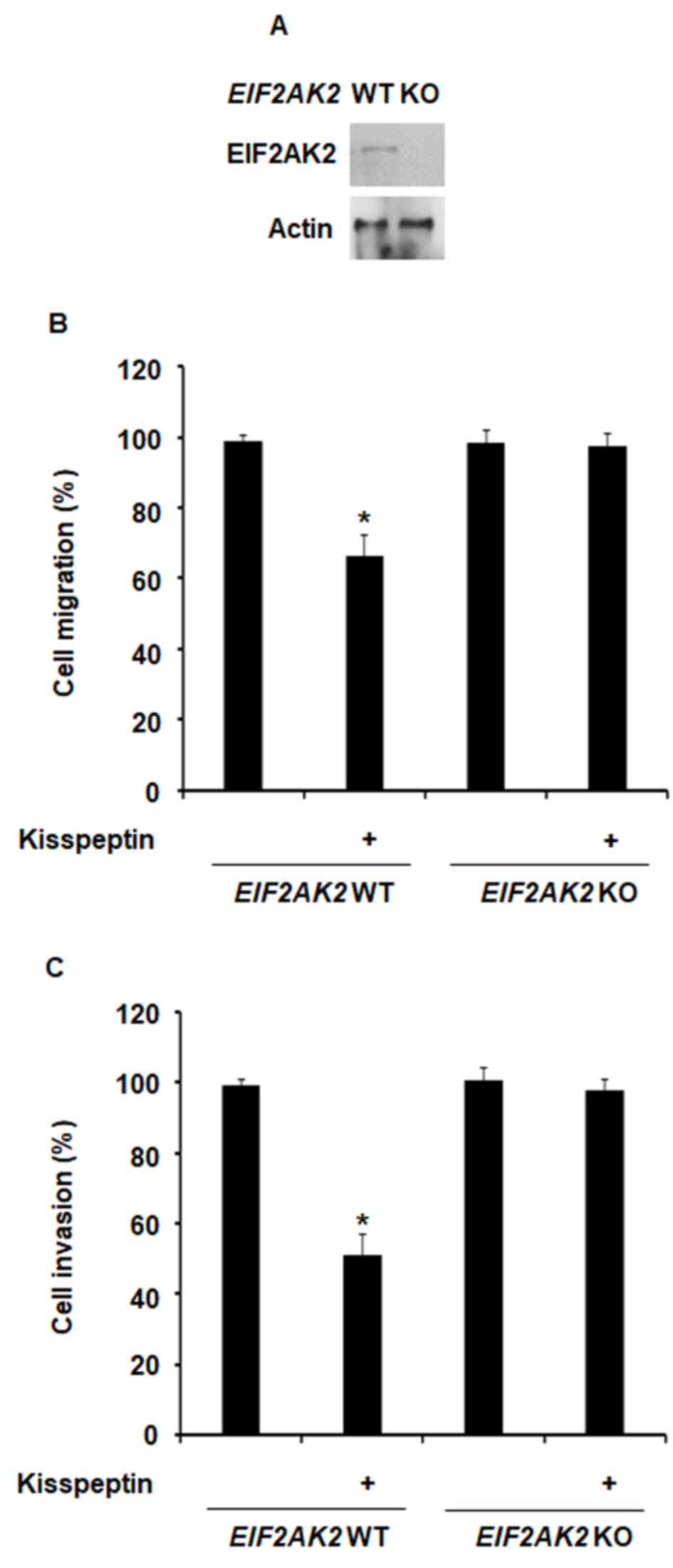
blot analysis (Fig. 1A). Treatment
with kisspeptin was revealed to inhibit the migratory capabilities
of normal MEFs; however, in MEFs where EIF2AK2 was knocked
out, the inhibitory effects of kisspeptin on cellular migration
were abolished (Fig. 1B).
Similarly, kisspeptin failed to suppress the invasive capabilities
of MEF-EIF2AK2−/− cells (Fig. 1C). These results suggested that
EIF2AK2 may be involved in the mechanisms underlying the inhibitory
effects of kisspeptin on cellular migration and invasion.
Kisspeptin-mediated EIF2AK2 activation
is crucial for the inhibition of cancer cell migration and
invasion
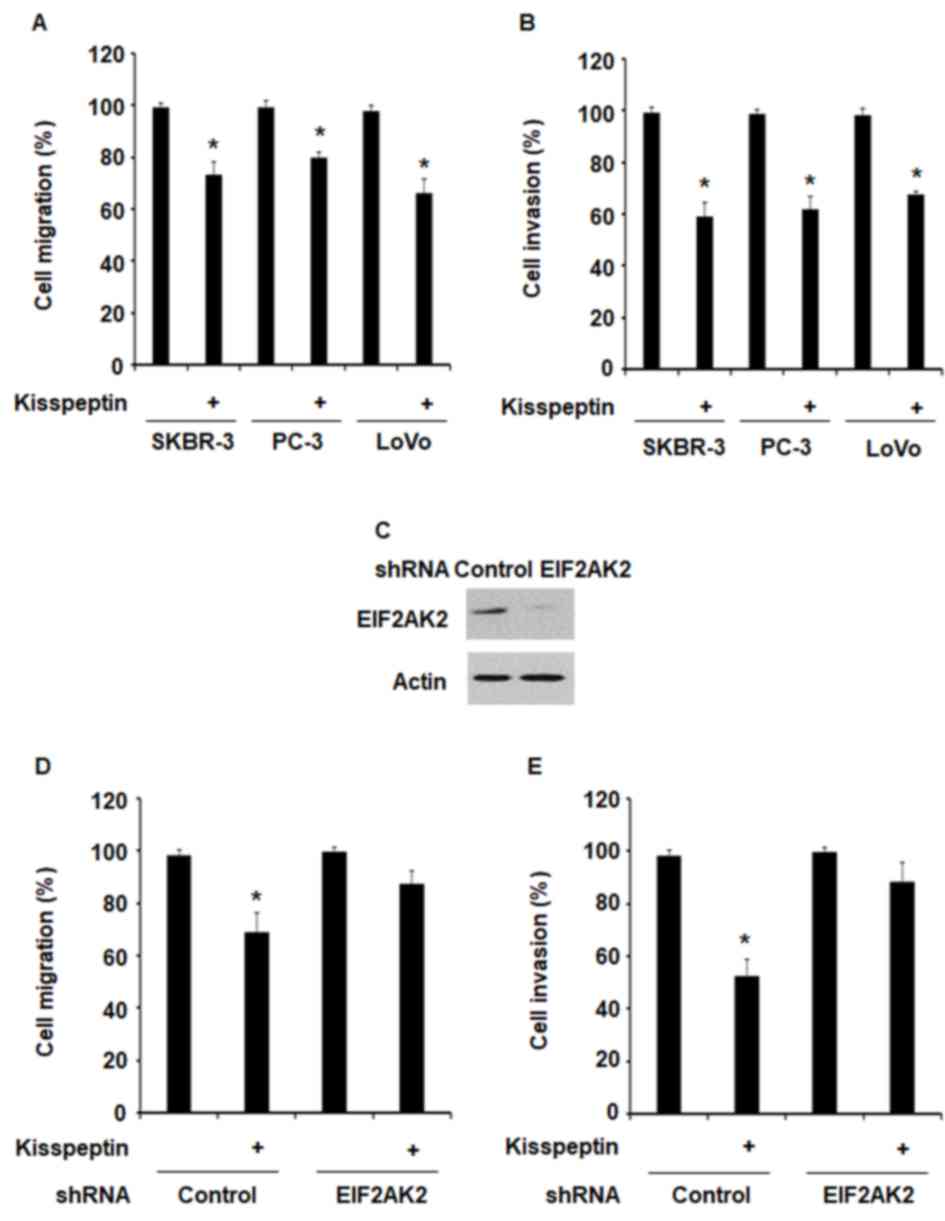
To investigate the effects of kisspeptin on the
migratory and invasive capabilities of cancer cells, SK-BR-3, PC-3
and LoVo cells were treated with kisspeptin for 24 h and
subsequently evaluated in scratch wound and invasion assays. The
present results revealed that kisspeptin significantly suppressed
cancer cell migration and invasion (Fig. 2A and B).
RNA interference was used to silence the expression
of EIF2AK2 in LoVo cells (Fig.
2C). Following EIF2AK2 knockdown, the inhibitory effects of
kisspeptin on cancer cell migration and invasion were abolished
(Fig. 2D and E). The results of
the present study suggested that kisspeptin may exert its
suppressive effects on the metastatic capabilities of cancer cells
via an EIF2AK2-mediated pathway.
Kisspeptin-mediated EIF2AK2
phosphorylation requires RhoA activation
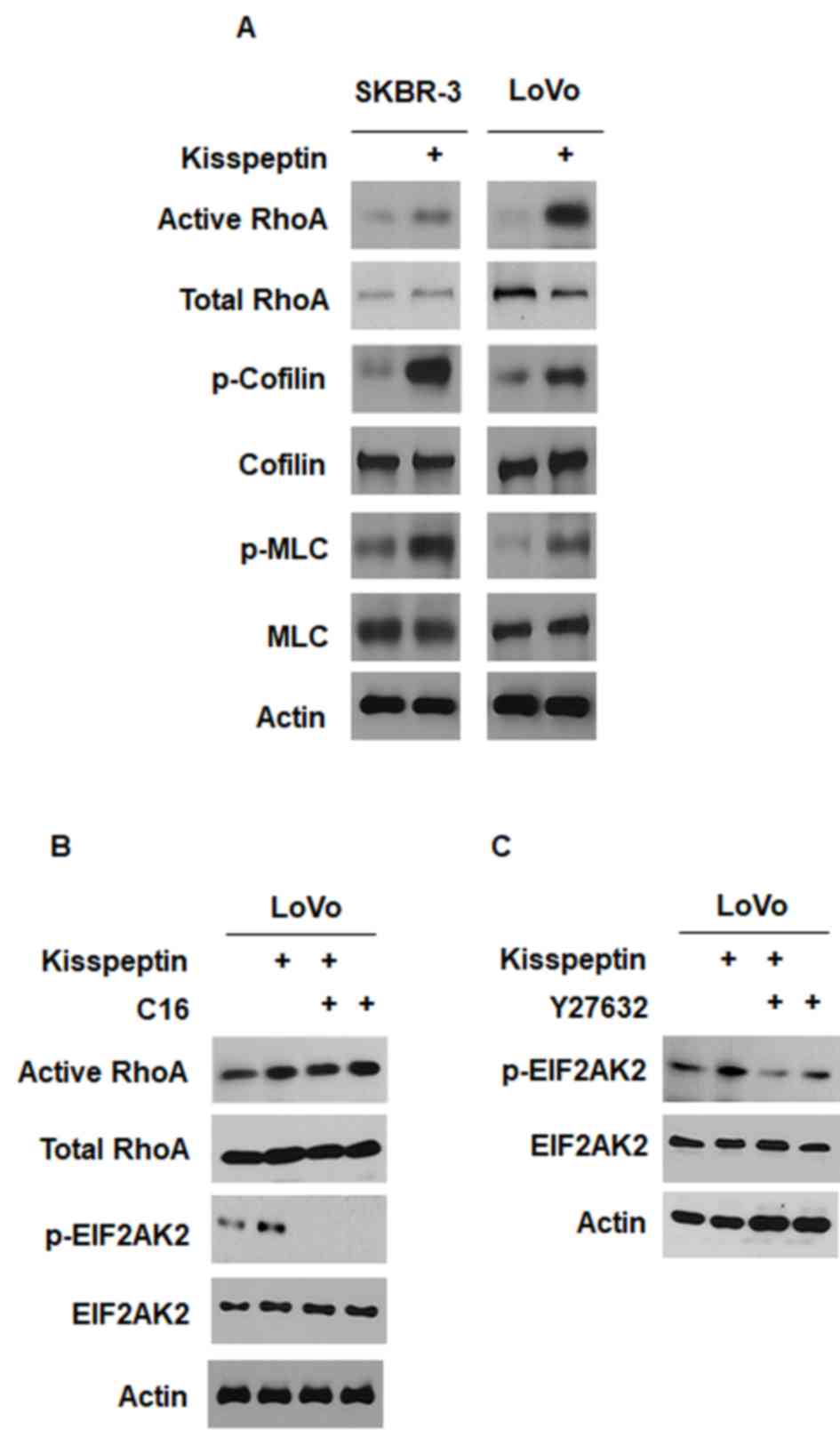
Kisspeptin has been reported to modulate the RhoA
intracellular signaling pathway (8,12,27).
In LoVo and SK-BR-3 cells, a GST pull-down assay demonstrated that
kisspeptin activated RhoA (Fig.
3A). Western blot analysis revealed that treatment with
kisspeptin increased the phosphorylation of the downstream
mediators of RhoA, MLC and cofilin (Fig. 3A). Notably, the inhibition of
EIF2AK2 with C16 (100 nM) did not appear to prevent the activation
of RhoA in LoVo cells (Fig. 3B).
However, the inhibition of the downstream effector of RhoA ROCK
with Y-27632 (100 nM) appeared to prevent the phosphorylation of
EIF2AK2 in LoVo cells (Fig. 3C).
These results suggested that RhoA-mediated signaling may be
implicated in the mechanisms underlying the kisspeptin-induced
phosphorylation of EIF2AK2.
Kisspeptin-induced EIF2AK2 activation
inhibits cancer metastasis in vivo
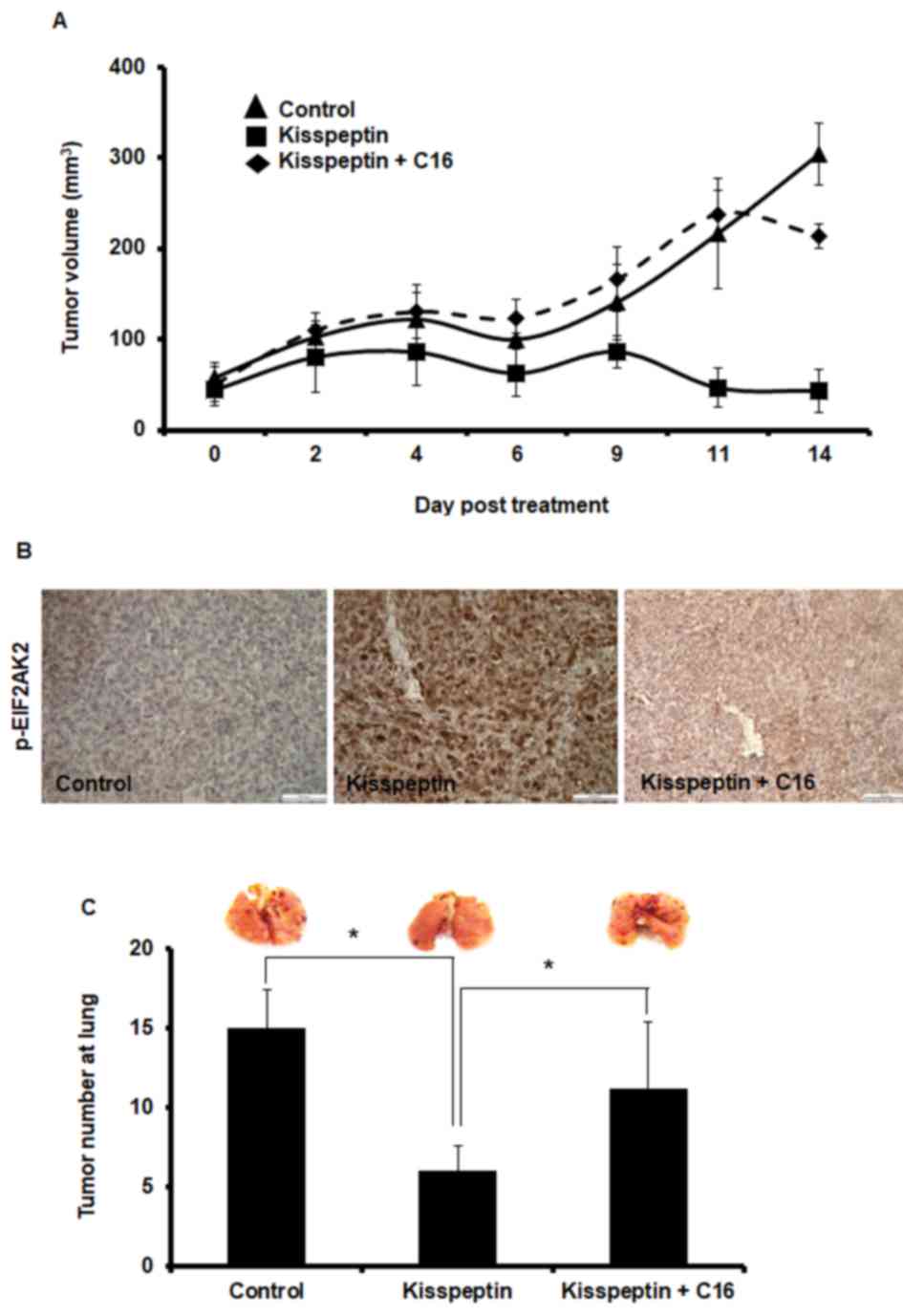
Kisspeptin has been reported to suppress cancer
metastasis (6,7). The aforementioned results suggested
that kisspeptin-induced EIF2AK2 activation may be involved in the
inhibitory effects on cancer cell motility. Therefore, the
implication of EIF2AK2 in the mechanism of action of kisspeptin was
investigated in cancer metastasis in vivo. LoVo-derived
tumor xenografts were implanted into nude mice, and kisspeptin
administration was revealed to suppress tumor growth (Fig. 4A). Notably, the inhibition of
EIF2AK2 following C16 co-administration appeared to abolish the
inhibitory effects of kisspeptin in vivo (Fig. 4A). Similarly, following treatment
with kisspeptin, phosphorylation levels of EIF2AK2 in xenografted
tumor tissue appeared to be increased, whereas C16 effectively
prevented EIF2AK2 phosphorylation (Fig. 4B).
An experimental metastasis assay was used to
investigate the effects of kisspeptin on the metastatic
capabilities of cancer cells in vivo. The present results
demonstrated that kisspeptin prevented the development of
LoVo-derived lung metastases in nude mice, whereas EIF2AK2
inhibition with C16 abolished the inhibitory effects of kisspeptin
on the metastatic capabilities of LoVo cells (Fig. 4C). These results suggested that
kisspeptin may inhibit cancer growth and metastasis in vivo
through the activation of EIF2AK2-mediated signaling pathways.
Discussion
Kisspeptin is a protein encoded by the KISS1
gene, which has been reported to suppress the metastatic
capabilities of various types of cancer cells (6,7), and
KISS1 expression levels have been revealed to be
downregulated in metastatic compared with non-metastatic cancer
tissue (28). However, those
findings inversely indicate that KISS1 expression level is
higher in tumor tissues than in normal tissues. Conversely, the
G-protein coupled receptor for kisspeptin, GPR54, has been
implicated in breast cancer development (27). These contradictory results suggest
that the effects of kisspeptin during cancer development and
metastasis may differ across various types of cancer cells
(5). Kisspeptin-mediated
intracellular signaling has been reported to serve diverse roles in
physiological and pathological conditions; however, the implication
of kisspeptin-mediated pathways in the regulation of cancer cell
migration and invasion has yet to be elucidated. The results of the
present study suggested that kisspeptin-mediated signaling
suppressed the metastatic capabilities of cancer cells, and EIF2AK2
may be implicated in the molecular mechanisms underlying these
effects.
In the present study, kisspeptin-mediated inhibition
of MEF migration and invasion was abolished following the deletion
of the EIF2AK2 gene, thus suggesting that EIF2AK2-mediated
signaling pathways may be involved in kisspeptin-induced inhibition
of cellular migration and invasion. Similarly, kisspeptin-mediated
EIF2AK2 activation was demonstrated to be required for the
kisspeptin-induced suppression of the migratory and invasive
capabilities of cancer cells. The present results are in accordance
with a previous study reporting the implication of EIF2AK2 in the
regulation of breast cancer cell migration (23). Therefore, it may be hypothesized
that EIF2AK2 can be activated by kisspeptin, and kisspeptin
regulates cancer cell motility via EIF2AK2-mediated signaling
pathways.
Kisspeptin has been reported to activate RhoA via a
GPR54/Gq/11/p63 RhoA-specific guanine nucleotide
exchange factor (p63RhoGEF)-mediated signaling pathway (27). The present study suggested that
RhoA may be required for the kisspeptin-induced activation of
EIF2AK2. Therefore, it may be hypothesized that the signaling
pathway underlying the effects of kisspeptin may be represented by
the following scheme:
GPR54/Gq/11/p63RhoGEF/RhoA/EIF2AK2.
The results of the present study suggested that
autophagy may be linked to cytoskeletal dynamics (29), since the roles of EIF2AK2 in the
regulation of autophagy have been previously demonstrated (30). However, the effects of kisspeptin
in the regulation of autophagy have yet to be elucidated, and
further studies are required to investigate whether
kisspeptin-mediated EIF2AK2 activation may modulate autophagy, as
well as the association between autophagy and cytoskeletal
dynamics. Vesicle trafficking has been associated with cytoskeletal
dynamics (31). Therefore, it may
be hypothesized that the trafficking of autophagosomes by
cytoskeletal components serves a role in cancer cell motility, as
well as in their metastatic capabilities. However, further studies
are required to investigate the putative relationship between
autophagy and cell motility. In conclusion, the present study
suggested that kisspeptin may regulate the metastatic capabilities
of cancer cells via EIF2AK2-associated signaling pathways in
vitro and in vivo. The present study, therefore, may
improve our knowledge of the role of kisspeptin as a metastasis
suppressor and address its pharmaceutical application in cancer
treatment.
Acknowledgements
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
funded by the Ministry of Science, ICT and Future Planning (grant
no. NRF-2014R1A1A1035831).
References
|
1
|
Oakley AE, Clifton DK and Steiner RA:
Kisspeptin signaling in the brain. Endocr Rev. 30:713–743. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Colledge WH, Doran J and Mei H: Model
systems for studying kisspeptin signalling: Mice and cells. Adv Exp
Med Biol. 784:481–503. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Silveira LG, Latronico AC and Seminara SB:
Kisspeptin and clinical disorders. Adv Exp Med Biol. 784:187–199.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Castellano JM and Tena-Sempere M:
Metabolic regulation of kisspeptin. Adv Exp Med Biol. 784:363–383.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cho SG, Li D, Tan K, Siwko SK and Liu M:
KiSS1 and its G-protein-coupled receptor GPR54 in cancer
development and metastasis. Cancer Metastasis Rev. 31:585–591.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee JH and Welch DR: Suppression of
metastasis in human breast carcinoma MDA-MB-435 cells after
transfection with the metastasis suppressor gene, KiSS-1. Cancer
Res. 57:2384–2387. 1997.PubMed/NCBI
|
|
7
|
Ohtaki T, Shintani Y, Honda S, Matsumoto
H, Hori A, Kanehashi K, Terao Y, Kumano S, Takatsu Y, Masuda Y, et
al: Metastasis suppressor gene KiSS-1 encodes peptide ligand of a
G-protein-coupled receptor. Nature. 411:613–617. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Navenot JM, Fujii N and Peiper SC:
Activation of Rho and Rho-associated kinase by GPR54 and KiSS1
metastasis suppressor gene product induces changes of cell
morphology and contributes to apoptosis. Mol Pharmacol.
75:1300–1306. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tan K, Cho SG, Luo W, Yi T, Wu X, Siwko S,
Liu M and Yuan W: KiSS1-induced GPR54 signaling inhibits breast
cancer cell migration and epithelial-mesenchymal transition via
protein kinase D1. Curr Mol Med. 14:652–662. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiang Y, Berk M, Singh LS, Tan H, Yin L,
Powell CT and Xu Y: KiSS1 suppresses metastasis in human ovarian
cancer via inhibition of protein kinase C alpha. Clin Exp
Metastasis. 22:369–376. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roseweir AK, Katz AA and Millar RP:
Kisspeptin-10 inhibits cell migration in vitro via a receptor-GSK3
beta-FAK feedback loop in HTR8SVneo cells. Placenta. 33:408–415.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cho SG, Li D, Stafford LJ, Luo J,
Rodriguez-Villanueva M, Wang Y and Liu M: KiSS1 suppresses
TNFalpha-induced breast cancer cell invasion via an inhibition of
RhoA-mediated NF-kappaB activation. J Cell Biochem. 107:1139–1149.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stafford LJ, Xia C, Ma W, Cai Y and Liu M:
Identification and characterization of mouse metastasis-suppressor
KiSS1 and its G-protein-coupled receptor. Cancer Res. 62:5399–5404.
2002.PubMed/NCBI
|
|
14
|
Williams BR: PKR; a sentinel kinase for
cellular stress. Oncogene. 18:6112–6120. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meurs EF, Galabru J, Barber GN, Katze MG
and Hovanessian AG: Tumor suppressor function of the
interferon-induced double-stranded RNA-activated protein kinase.
Proc Natl Acad Sci USA. 90:232–236. 1993; View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hii SI, Hardy L, Crough T, Payne EJ,
Grimmett K, Gill D and McMillan NA: Loss of PKR activity in chronic
lymphocytic leukemia. Int J Cancer. 109:329–335. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shir A and Levitzki A: Inhibition of
glioma growth by tumor-specific activation of double-stranded
RNA-dependent protein kinase PKR. Nat Biotechnol. 20:895–900. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Watanabe MA, Souza L Rodrigues, Murad JM
and De Lucca FL: Antitumor activity induced by regulatory RNA:
Possible role of RNA-dependent protein kinase and nuclear
factor-kappaB. Eur J Pharmacol. 465:205–210. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim SH, Gunnery S, Choe JK and Mathews MB:
Neoplastic progression in melanoma and colon cancer is associated
with increased expression and activity of the interferon-inducible
protein kinase, PKR. Oncogene. 21:8741–8748. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim SH, Forman AP, Mathews MB and Gunnery
S: Human breast cancer cells contain elevated levels and activity
of the protein kinase, PKR. Oncogene. 19:3086–3094. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Haines GK, Cajulis R, Hayden R, Duda R,
Talamonti M and Radosevich JA: Expression of the double-stranded
RNA-dependent protein kinase (p68) in human breast tissues. Tumour
Biol. 17:5–12. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Savinova O, Joshi B and Jagus R: Abnormal
levels and minimal activity of the dsRNA-activated protein kinase,
PKR, in breast carcinoma cells. Int J Biochem Cell Biol.
31:175–189. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu M, Chen G, Wang S, Liao M, Frank JA,
Bower KA, Zhang Z, Shi X and Luo J: Double-stranded RNA-dependent
protein kinase regulates the motility of breast cancer cells. PLoS
One. 7:e477212012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yuan X, Wang W, Li J, Zheng P, Dong P,
Chen L, Zhou Y, Xie G, Xu D, Liu Y and Shen L: Gelsolin suppresses
gastric cancer metastasis through inhibition of PKR-p38 signaling.
Oncotarget. 7:53459–53470. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang X, Dong JH, Zhang WZ, Leng JJ, Cai
SW, Chen MY and Yang X: Double stranded RNA-dependent protein
kinase promotes the tumorigenic phenotype in HepG2 hepatocellular
carcinoma cells by activating STAT3. Oncol Lett. 8:2762–2768.
2014.PubMed/NCBI
|
|
26
|
Irving AT, Wang D, Vasilevski O,
Latchoumanin O, Kozer N, Clayton AH, Szczepny A, Morimoto H, Xu D,
Williams BR and Sadler AJ: Regulation of actin dynamics by protein
kinase R control of gelsolin enforces basal innate immune defense.
Immunity. 36:795–806. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cho SG, Wang Y, Rodriguez M, Tan K, Zhang
W, Luo J, Li D and Liu M: Haploinsufficiency in the prometastasis
Kiss1 receptor Gpr54 delays breast tumor initiation, progression,
and lung metastasis. Cancer Res. 71:6535–6546. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shengbing Z, Feng LJ, Bin W, Lingyun G and
Aimin H: Expression of KiSS-1 gene and its role in invasion and
metastasis of human hepatocellular carcinoma. Anat Rec (Hoboken).
292:1128–1134. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Caino MC, Chae YC, Vaira V, Ferrero S,
Nosotti M, Martin NM, Weeraratna A, O'Connell M, Jernigan D,
Fatatis A, et al: Metabolic stress regulates cytoskeletal dynamics
and metastasis of cancer cells. J Clin Invest. 123:2907–2920. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pietrocola F, Izzo V, Niso-Santano M,
Vacchelli E, Galluzzi L, Maiuri MC and Kroemer G: Regulation of
autophagy by stress-responsive transcription factors. Semin Cancer
Biol. 23:310–322. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mackeh R, Perdiz D, Lorin S, Codogno P and
Poüs C: Autophagy and microtubules-new story, old players. J Cell
Sci. 126:1071–1080. 2013. View Article : Google Scholar : PubMed/NCBI
|