Introduction
Among 278 million suffering from deafness worldwide,
half are induced by hereditary factors, with 200 to 300 genes
having been identified (1). Based
on the presence of other symptoms, hearing impairment may be
divided into syndromic (SHL, ~30%) and nonsyndromic hearing loss
(NSHL) (2,3). SHL has >400 types of symptoms in
the skin, outer ears, eyes and endocrine metabolism. The present
study examined a Chinese patient originally suspected to have
certain types of SHL. The proband exhibited deafness along with
various additional symptoms, including dry loose skin, tooth decay
and hypotonia. These phenotypes partially conform to the symptoms
of some diseases and syndromes, such as Hutchinson-Gilford progeria
syndrome (HGPS) and congenital ectodermal dysplasia syndrome
(4–7). HGPS is a rare syndrome characterized
by slow growth, prominent eyes, protruding ears, a small chin, hair
loss, ageing skin and loss of subcutaneous fat tissue. (8,9)
Generally, patients were born normal; however, ageing occurs
rapidly, leading to alterations in various organs (10). Some of the proband's symptoms also
fall within those of ectodermal dysplasia syndrome (skin and tooth
abnormality), whereas other diseases are also possible due to the
multiple symptoms of the patient, such as hypogonadism.
Genetically, HGPS occurs due to an autosomal dominant inheritance
of a mutant lamin A/C (LMNA) gene (11). Ectodermal dysplasia syndrome is
attributed to copy number variations (CNVs) or mutations in
ectodysplasin A (EDA) gene family members, including
EDA, EDA receptor (EDAR) and EDAR-associated death
domain. (12,13) Therefore, genetic screening is of
the utmost importance for diagnosis and treatment.
Given the possibility of HGPS and ectodermal
dysplasia syndromes for the patient, sequencing of LMNA
exons was performed, followed by CNV examination of EDA gene
family members; however, no pathogenic clues were identified.
Subsequently, 438 deafness-associated genes were sequenced, in
order to identify if these mutations in these genes are associated
with the patient's phenotypes. Whole-genome sequencing (WGS) was
performed in order to identify potential pathogenic genes that may
account for the symptoms observed. The obtained gene list with CNVs
or single nucleotide variations (SNVs) was compared with the
Human/Mouse Disease Connection database, other SNV-related
databases and previous studies in order to identify potential
candidate genes. Multiple genes were selected for further
analysis.
Case report
Patient, ethics, consent and
permission
Clinical information about the patient, aged 4,
male, and the parents was collected in June 2014, followed by a
systemic health check up, under signed informed consent forms to
participate and publish the data in accordance with the Ethics
Committee of the Chinese PLA General Hospital (Beijing, China).
Peripheral blood (5 ml) was collected for genomic DNA (gDNA)
isolation.
Sequencing of the HGPS-associated LMNA
gene
Primers were based on the NCBI reference sequence of
LMNA (Genbank, NM_001282625) and were designed by Shanghai
Genesky Biotech Co., Ltd. (Shanghai, China) to amplify exons with
~50 bp of flanking introns. Primer sequences are presented in
Table I. Polymerase chain reaction
(PCR) products were purified for sequencing. PCR was performed by
Shanghai Genesky Biotech Co., Ltd.
 | Table I.Amplification primers of LMNA
gene. |
Table I.
Amplification primers of LMNA
gene.
| Exons | Forward
(5′-3′) | Reverse
(5′-3′) | Length (bp) |
|---|
| 1 |
CCAGGAGCAAGCCGAGAG |
ACAATTCCCCTTGACACTGC | 990 |
| 2 |
GGATGCCCTCTCCTGGTAAT |
GGCTCTGAAATCAGGTGACAG | 700 |
| 3 |
CCTGGACCTGTTTCCACAT |
TAACCTGGGAGCTGAGTGCT | 680 |
| 4 |
CCTAGTGGACAGGGAGTTGG |
CCTGAGTTGGGCATCACTG | 640 |
| 5 |
TAGCAGTGATGCCCAACTCA |
GCCATCTGACTCCACATCCT | 640 |
| 6,7 |
CTCTGGGGAAGCTCTGATTG |
TCTCACAGCCAAAGAGTCCA | 1,130 |
| 8,9 |
CAGGGGTGTGTGTAGATGGA |
GTTTGCCTACTGGGTGGAGA | 860 |
| 10 |
AAGTTGCAGGTGGTCACTGG |
GAAAGTTCCCACTCCCTTCC | 600 |
| 11 |
GCACAGAACCACACCTTCCT |
GGTGGGCTGTCTAGGACTCA | 800 |
| 12 |
CATCCTGCCCCTCTTGTCT |
TTTTGCTTGTGTTTTTCCTTCA | 520 |
Multiplex ligation-dependent probe
amplification (MLPA) for EDA testing
MLPA was used for genetic testing of EDA
using P183 kit according to the manufacturer's instructions
(MRC-Holland, Amsterdam, Netherlands). DNA was denatured and
hybridized with SALSA probe mix, followed by ligation and
polymerase chain reaction amplification. Capillary electrophoresis
was performed to generate fragment length and peak area using
Genemapper software, version 3.0 (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Copy number ratio was denoted as peak area ratio
of patient vs. references. A ratio between 0.7 and 1.3 indicated a
normal individual, whereas the subject may be diagnosed with
ectodermal dysplasia syndrome when the value falls between 1.7 and
2.3. Tests were repeated when the value was near the aforementioned
boundaries.
Sequencing of hearing
impairment-associated genes
A total of 2 µg gDNA was sheared with
NEBNext® dsDNA Fragmentase (New England Biolabs, Inc.,
Ipswich, MA, USA) and end repaired with DNA End Repair Mix (Thermo
Fisher Scientific, Inc.), followed by 3′-end adenylation (A-tailing
kit; Generay Biotech Co., Ltd., Shanghai, China) and adaptor
ligation (NEBNext® Multiplex Oligos for
Illumina®; New England BioLabs, Inc.) according to the
respective manufacturer's instructions. PCR was performed with a
primer cocktail using NEBNext® Ultra™ II DNA Library
Prep kit (New England BioLabs, Inc.), followed by purification. The
thermocycling conditions were as follows: 98°C for 30 sec, 10
cycles of 98°C for 10 sec, 60°C for 30 sec and 72°C for 30 sec,
followed by 72°C for 5 min. Subsequently, the library was pooled,
hybridized and purified prior to another round of PCR amplification
with the same thermocycling conditions as the ones stated above.
Finally, samples were mounted on the Illumina HiSeq 2000 loading
unit for sequencing.
WGS procedure
WGS was performed using Illumina TruSeq Nano DNA HT
Sample prep kit (Illumina, Inc., San Diego, CA, USA) and HiSeq X
system (Illumina, Inc.), according to the manufacturer's protocol.
For CNV analysis, sequences of 6 unrelated individuals were used as
references. For SNV analysis, the criteria for gene selection were
as follows: i) Existing mutations in HGMD; ii) conservation
analysis; iii) frequency <0.001 in 1,000 genomes or <0.01 in
its own genome; iv) frequency in ESP6500 <0.01; v) single
nucleotide polymorphism (SNP) calling quality not L (L meaning that
the SNP cannot be called by either the GAKT or varscan programs),
and the ratio of genotyping quality L <50%; vi) homology is 1;
and vii) zero occurrence in the Genesky database. SNP calling was
performed using Genome Analysis Toolkit (version 3.7, Broad
Institute; software.broadinstitute.org/gatk) and VarScan (version
2.4.0; Genome Institute at Washington University; genome.wustl.edu). An SNP was labeled as ‘H’ if it was
identified by both programs, whereas the quality was ‘M’ when it
was detected by only one. The quality was further downgraded if
there were short tandem repeats, indel or homologous sequences
flanking the SNP. SNPs labeled as ‘H’ were selected in priority.
The overall quality of SNP genotyping was ‘L’ or ‘M’; therefore, if
one sample was graded as ‘L’ or ‘M’ those graded as ‘L’ were
selected. Genes with sorting intolerant from tolerant (SIFT) values
<0.05 were selected, as SIFT values indicate the impact of the
mutation on protein functions. Additionally, sites with mutation
taster scores have a higher probability of mutation. The original
gene list was narrowed down according to the aforementioned
criteria and genes associated with hearing impairment and HGPS were
selected as priority. The list of genes carrying CNV or SNV was
matched with the patient's symptoms, including progeria, either in
the gene list or the Human-Mouse Disease Connection database
(www.informatics.jax.org/mgihome/homepages/humanDisease.shtml).
Genes of interest were selected for further analysis. An extensive
search in existing literature was performed for the refined genes,
in order to identify whether the detected mutations in the current
patient had been reported to be the pathogenic cause of the
phenotypes observed.
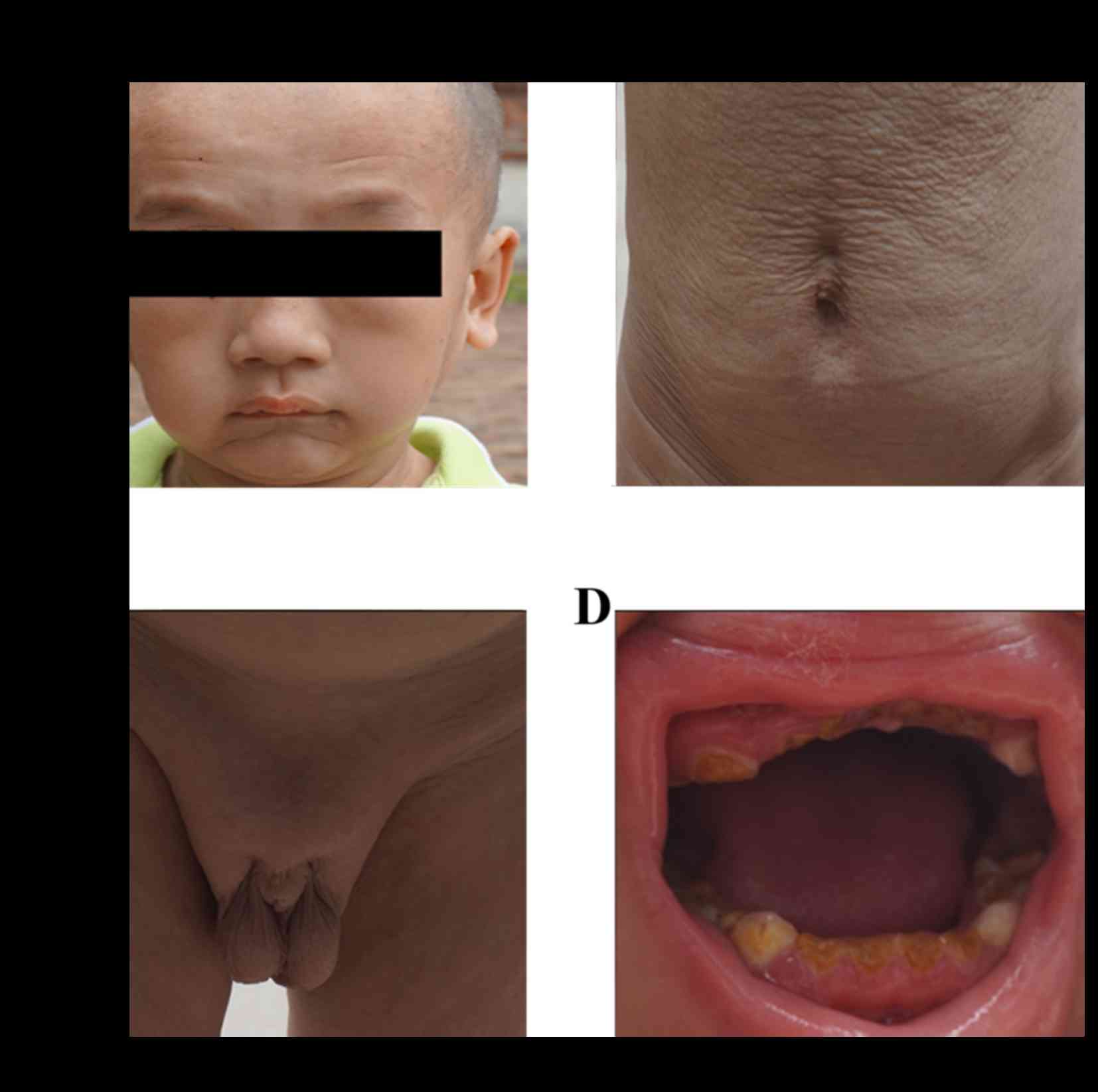
Clinical check up of the family
The patient exhibited ageing skin loosening
following birth, with limited skin elasticity and subcutaneous fat
storage. He exhibited progeroid symptoms without age pigment
deposition. Facial skin loosening was moderate; however, there was
abnormal development in hair and teeth, with evident tooth decay.
The patient had slow reaction to external forces and low muscle
tension; however, no abnormality in eyesight, intelligence and
overall skeletal development (no bone integration or bone loss) was
observed. Additionally, no obvious abnormalities were detected in
the nervous system check up. However, the development of the gonads
was limited (Fig. 1).
The patient was not treated after birth. At age 1,
the child was diagnosed with severe deafness. No abnormality was
observed in skull/temporal bone computed tomography and
skull/internal auditory canal magnetic resonance imaging scan prior
to artificial cochlea implantation. The patient exhibited high
aminotransferase levels; however, no chromosomal abnormality was
identified. Following artificial cochlea implantation in 2013,
hearing and skin resilience improved. His parents and sister were
also examined and were determined to be in healthy condition. The
patient was initially suspected to be deaf and have HGPS;
therefore, genetic screening was used for diagnosis.
LMNA gene mutation
No mutation was identified in the exons of the
HGPS-associated gene LMNA. Exon sequencing of LMNA
for the patient and his parents revealed no mutation of LMNA
in the family.
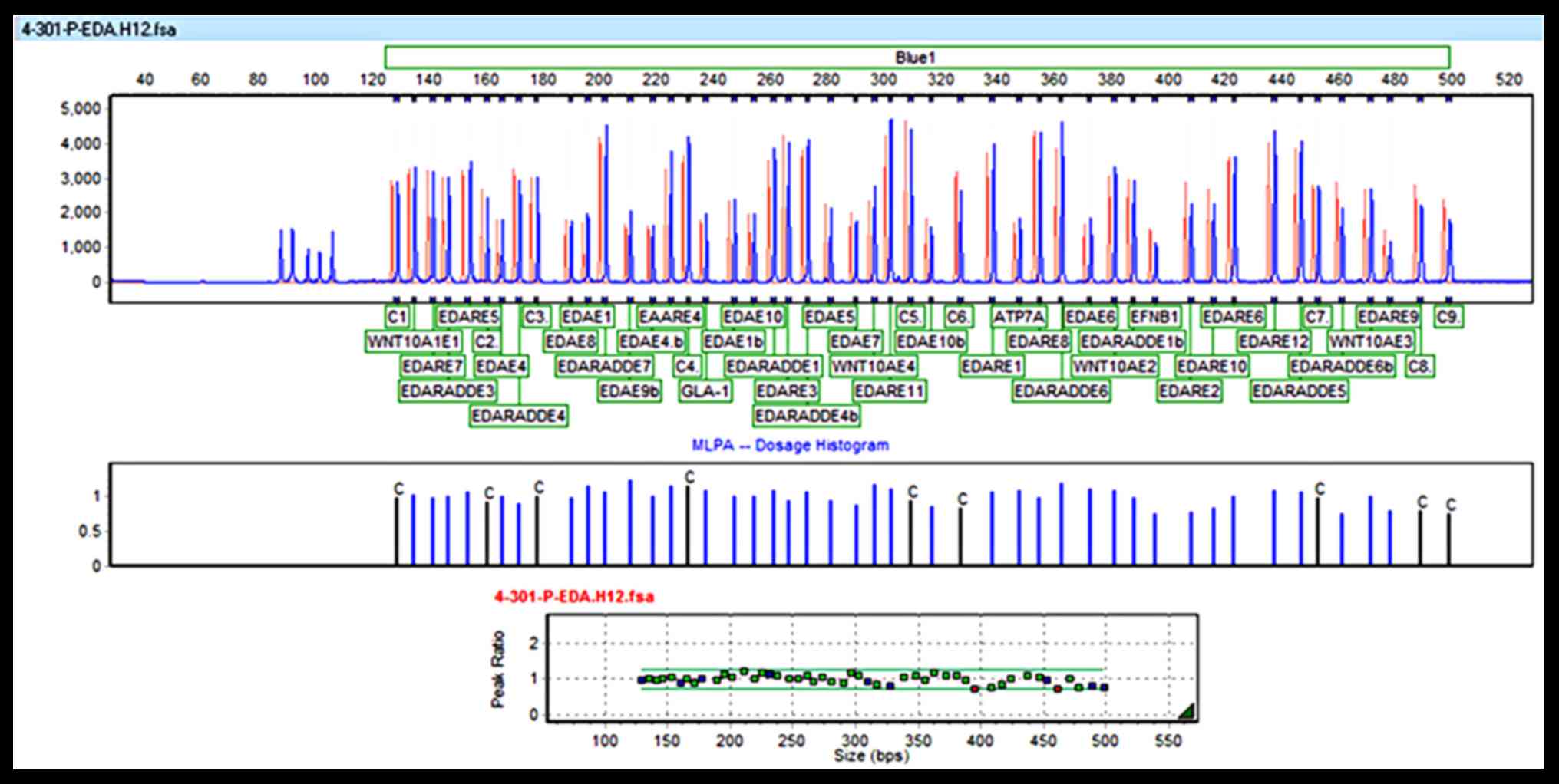
MLPA analysis excluded EDA
syndrome
EDA peaks for the patient and reference
samples are presented in the MLPA histogram of Fig. 2 along with their ratios. Copy
number ratios were ~1, suggesting no change in copy number.
Therefore, ectodermal dysplasia syndrome due to dysfunction of EDA
was excluded for this patient.
Mutations of deafness-associated
genes
Mono-allelic heterozygous mutations were identified
in some genes, including coenzyme Q6 monooxygenase, FGFR3,
melanogenesis associated transcription factor (MITF),
otoferlin, DNA polymerase γ catalytic subunit (POLG) and
usherin (USH2A). Mutation of these genes may contribute to
other unrelated symptoms. For instance, a POLG mutation may
lead to a mitochondrial DNA depletion syndrome, which is
characterized by tubulopathy, seizures, respiratory distress,
diarrhea and lactic acidosis, which were inconsistent with the
majority of the phenotypes observed in the proband. Mutations in
some of these genes were also revealed in WGS, and therefore will
be discussed further in the following section.
WGS revealed multiple genes that may
account for specific symptoms
WGS identified CNV and SNV. From the CNV analysis,
2,653 genes were determined to have half or less copy numbers.
Conversely, the remaining 720 genes had copy numbers ~2. For the
SNV analysis, WGS confirmed the mutations of the aforementioned
deafness-associated gene sequencing. Additionally, a full list of
genes with autosomal dominant and recessive inheritance patterns
were identified.
The genes of interest were identified by searching
the lists against the symptoms. Briefly, their mutations were
checked in the literature to see what symptoms they may cause. They
were categorized according to the phenotypes of the patient in
Table II. Some of these genes or
their SNVs have been well-characterized and have been identified to
be responsible for various diseases. For instance, USH2A and
CDH23 were determined to be involved in the pathogenesis of
Usher syndrome (14,15). However, none of the identified
genes were capable of simultaneously explaining the majority of the
symptoms present in the proband, suggesting that the disease may
not be attributed to a monogene and multiple genetic lesions may
cooperatively contribute to the presentations in the patient.
Additionally, the exact SNVs in a number of the listed genes were
never reported to be pathogenic in any of the literature, let alone
the symptoms presented by the proband in our study. In some cases,
the diseases associated with the genes were reported to present
major symptoms that were not observed in the present case, such as
the Donnai-Barrow syndrome which occurs due to LRP2 mutation
(16). Therefore, the majority of
the listed genes were filtered out and only SNVs in the
LMNA, DNA polymerase δ 1, catalytic subunit (POLD1),
crystallin mu (CRYM), RAB3 GTPase activating protein
catalytic subunit 1 (RAB3GAP1) and Wnt family member 10A
(WNT10A) genes were further discussed.
| Table II.Patient phenotypes, associated genes,
mutations and diseases attributed to alterations in the genes. |
Table II.
Patient phenotypes, associated genes,
mutations and diseases attributed to alterations in the genes.
| A,
Aging/progeria |
|---|
|
|---|
| Gene name | Disease | RefSeq mRNA | CNV/SNV |
|---|
| LRP1 | NA | NM_002332 |
c.12161A>T:p.Y4054F |
| EDNRA | Mandibulofacial
dysostosis with alopecia | NM_001166055 |
c.503T>C:p.L168P |
| POLG | Mitochondrial DNA
depletion syndrome | NM_001126131 |
c.1840T>C:p.Y614H |
| SREBF1 | NA | NM_001005291 |
c.547G>A:p.A183T |
| CANX | NA | NM_001024649 |
c.418C>A:p.L140M |
| BAK1 | NA | NM_001188 | Splicing |
| POLD1 | Mandibular
hypoplasia, deafness, progeroid features and lipodystrophy
syndrome | NM_001256849 |
c.1932C>G:p.D644E |
| LRP2 | Donnai-barrow
syndrome | NM_004525 | CNV ratio=0.54 |
| CISD2 | Wolfram
syndrome | NM_001008388 | CNV ratio=2.03 |
| VCAM1 | NA | NM_001078 | CNV ratio=0.52 |
| CASP7 | NA | NM_033338 | CNV ratio=0.52 |
| SLC18A2 | NA | NM_003054 | CNV ratio=0.60 |
| IL15 | NA | NM_000585 | CNV ratio=0.60 |
| ADH5 | NA | NM_000671 | CNV ratio=0.58 |
| SLC6A3 | NA | NM_001044 | CNV ratio=0.53 |
| HMGCR | NA | NM_000859 | CNV ratio=0.58 |
| DLD | NA | NM_000108 | CNV ratio=0.58 |
| DDC | NA | NM_001082971 | CNV ratio=0.59 |
| MSRA | NA | NM_001135670 | CNV ratio=1.84 |
| GSN | NA | NM_000177 | CNV ratio=0.52 |
| MLIP | NA | NM_001281746 | CNV ratio=0.54 |
|
| B, Deafness |
|
| Gene name | Disease | RefSeq mRNA | CNV/SNV |
|
| USH2A | Usher syndrome
II | NM_206933 |
c.5608C>T:p.R1870W
c.9340C>T:p.P3114S |
| CDH23 | Usher syndrome | NM_022124 |
c.5418C>G:p.D1806E |
| CRYM |
| NM_001888.4 | Splicing |
| COQ6 | Nephrotic
syndrome | NM_182476 |
c.186C>A:p.D62E |
| FBXO2 | NA | NM_012168 | Splicing |
| STRC | NA | NM_153700 |
c.179T>C:p.F60S |
| VPS13B | Cohen syndrome | NM_152564 |
c.11884C>G:p.P3962A |
| OTOF |
| NM_194248 |
c.2123G>A:p.R708Q |
| CISD2 | Wolfram
syndrome |
| CNV ratio=2.03 |
| LRP2 | Donnai-barrow
syndrome | NM_004525 | CNV ratio=0.54 |
| SMCHD1 | Facioscapulohumeal
muscular dystrophy | NM_015295 |
c.4071T>G:p.I1357M |
| POLD1 | Mandibular
hypoplasia, deafness, Progeroid | NM_001256849 |
c.1932C>G:p.D644E |
|
| features, and
lipodystrophy syndrome | NM_001256849 |
c.1932C>G:p.D644E |
| POLG | Mitochondrial DNA
depletion syndrome | NM_001126131 |
c.1840T>C:p.Y614H |
|
| C,
Hypogonadism |
|
| Gene name | Disease | RefSeq mRNA | CNV/SNV |
|
| RAB3GAP | Warburg syndrome,
martsolf syndrome | NM_012233 |
c.1175G>A:p.R392Q |
| MITF | Warburg
syndrome | NM_198158 |
c.1235C>T:p.T412I |
| MAGEL2 | Prader-willi
syndrome | NM_019066 |
c.1425_1445del:p.475_482del |
| KISS1 |
| NM_002256 |
c.417delA:p.X139W |
| NRP2 |
| NM_201266 |
c.1333A>C:p.I445L |
| CISD2 | Wolfram
syndrome | NM_001008388 | CNV
ratio=2.031485778 |
| POLD1 | Mandibular
hypoplasia, deafness, progeroid features and lipodystrophy
syndrome | NM_001256849 |
c.1932C>G:p.D644E |
| Fras1 | Fraser
syndrome | NM_025074 | CNV ratio=0.59 |
|
|
| NM_025074 |
c.9356A>G:p.N3119S |
|
| D, Hypotonia |
|
| Gene name | Disease | RefSeq mRNA | CNV/SNV |
|
| MAGEL2 | Prader-willi
syndrome | NM_019066 |
c.1425_1445del:p.475_482del |
| POLG | Mitochondrial DNA
depletion syndrome | NM_001126131 |
c.1840T>C:p.Y614H |
| SMCHD1 | Facioscapulohumeral
muscular dystrophy | NM_015295 |
c.4071T>G:p.I1357M |
|
| E, Tooth
development |
|
| Gene name | Disease | RefSeq mRNA | CNV/SNV |
|
| EDNRA | Mandibulofacial
dysostosis with alopecia | NM_001166055 |
c.503T>C:p.L168P |
| POLD1 | Mandibular
hypoplasia, deafness, progeroid features and lipodystrophy
syndrome | NM_001256849 |
c.1932C>G:p.D644E |
| WNT10A |
Odonto-onycho-dermal dysplasia | NM_025216 |
c.637G>A:p.G213S |
Discussion
The proband exhibited multiple symptoms, including
deafness, ageing and hypogonadism, which partially conform to the
symptoms of HGPS and ectodermal dysplasia syndrome. HGPS is a rare
hereditary disease which occurs due to autosomal dominant
inheritance of mutant LMNA, the product of which promotes
nuclear membrane deformation and reduces cellular lifespan
(17). Previous studies identified
the following pathogenic mutations c.1824C>T (p.Gly608Gly),
c.1822G>A (p.Gly608Ser), c.1821G>A (p.Val607Val),
c.1968+1G>A (18,19). Pathogenic mutations such as
c.1824C>T (p.Gly608Gly), do not alter the amino acid sequence;
however, they activate a hidden cleavage site, which leads to the
deletion of 50 amino acids in the resulting protein (20,21).
The present study also identified a synonymous SNV of c.1698C>T,
p.His566His (chr1:156107534, NM_001282626, rs4641) at the splice
region. However, the allele frequency carrying this SNV is 26.55%
according to ExAC Browser database; therefore, excluding the
possibility of rare HGPS in the current patient.
Another gene of interest is POLD1, the
defects of which (serine 605 loss or R507C substitution at exon 13)
lead to loss of δ DNA polymerase activity and impairment of
proof-reading exonuclease activity (22–24).
This may lead to mandibular dysplasia with deafness and progeroid
features (MDP), which is characterized by lipodystrophy, deafness,
a small lower jaw, low testosterone levels, claw toes, joint
stiffness and hypogonadism (25).
However, the nonsynonymous SNV detected in this patient is
1932C>G at exon 16 in POLD1 gene (rs80214209), which has
been reported in >90 individuals, particularly in East Asia
according to ExAC Browser database. Given that the MDP syndrome is
an extremely rare syndrome with only 5 reported cases exhibiting a
different POLD1 SNV (24),
it is unlikely that the SNV in POLD1 observed in the present
study leads to MDP syndrome. The molecular pathogenesis that
results in progeria-like features remains to be further
elucidated.
Ectodermal dysplasia syndrome occurs partially due
to mutations and CNVs in EDAs (12,13).
The protein products of these genes participate in signaling
pathways that regulate interactions between the ectoderm and
mesoderm, critical for the formation of skin, hair, teeth and sweat
glands. However, no CNV or SNV were detected in the EDAs;
therefore, this case is unlikely to be EDA-caused ectodermal
dysplasia. However, it is possible that ectodermal dysplasia may be
induced by other gene mutations; for example, c.637G>A, p.G213S
of WNT10A.
Deafness may be induced by mutations in multiple
genes, such as USH2A, CDH23 as listed in Table II. A dominant splicing alteration
(rs189371585) in the CRYM gene was identified as a candidate
etiological gene for deafness, with an allele frequency of 0.46% in
the 1,000 genome phase 1 population according to the SNP database
(dbSNP). Alterations of CRYM (X315Y and K314T) have been
determined to lead to autosomal dominant NSHL (26–28).
However, the pathogenic changes previously observed occurred due to
an amino acid substitution (26),
whereas the present case exhibited alterations in splicing. At
present, no existing literature is available to link this
alteration to any pathogenic consequences. Further functional
analysis is required to confirm the biological effects of this
splicing mutation.
Mutations in several genes may give rise to
hypogonadism, including MITF and KiSS-1
metastasis-suppressor. Additionally, diseases associated with
mutations in RAB3GAP1 include Warburg Micro syndrome and
Martsolf syndrome (29–33). Leiden open source variation
database archived nonsense mutations at c.1174 that led to the
production of truncated protein terminating at p.R392 and
contributed to Warburg Micro Syndrome. The novel SNV of
c.1175G>A in RAB3GAP1 identified in the present study led
to a p.R392Q amino acid substitution. Albeit at the same position,
no previous studies have reported the pathogenic role of the SNV
(p.R392Q) detected in the present study in Warburg Micro syndrome.
Additionally, Warburg Micro syndrome is an autosomal recessive
disease; however, only the symptom of hypogonadism was consistent
with the present case. Therefore, the function of the
RAB3GAP1 mutation in the current proband remains
unclear.
The present study was unable to identify definitive
gene lesions that may account for the aforementioned symptoms.
However, an SNV in WNT10A was confirmed to be etiological of
tooth agenesis and ectodermal dysplasia. WNT10A produces a
protein that triggers the Wnt pathway, which is important for
development and oncogenesis. Mutations in the WNT10A gene
may lead to aberrant development, such as odonto-onycho-dermal
dysplasia, featured by tooth agenesis and ectodermal dysplasia
(34–40). Previous studies also demonstrated
some overlapping functions of WNT10A with EDAs in
inducing hypodontia and ectodermal dysplasia (41–43).
Ectodermal dysplasia syndrome exhibits a broad range of symptoms,
including but not confined to abnormality of hair growth, absence
or malformation of some or all teeth, inability to perspire,
impairment or loss of hearing or vision and irregular skin
pigmentation. The case presented here conforms to these symptoms in
terms of tooth agenesis and hearing loss. Using WGS, a
non-synonymous SNV of c.637G>A, p.G213S in WNT10A was
detected. This mutation has been previously reported to be the
etiological variant leading to tooth agenesis (35,43).
In the absence of alterations in EDA members, this variant may also
give rise to ectodermal dysplasia (34). Therefore, the gene screening
performed in the present study identified the function of the
WNT10A mutation p.G213S in the induction of tooth agenesis,
skin abnormalities and hearing loss.
In conclusion, although further investigation is
required to confirm the pathogenic role of some of the SNVs
identified in inducing the phenotypes observed, the present study
provides an example of the use of genetic screening tools for the
diagnosis of a patient with putative syndromic deafness. However,
the symptoms were ultimately determined to be attributed to
multiple genetic lesions as opposed to a single gene, the present
study determined that a WNT10A mutation contributes to the
tooth agenesis and ectodermal dysplasia observed in the patient.
Additionally, a novel SNV of CRYM and an existing SNV of
RAB3GAP1 were identified, their function in inducing
deafness and hypogonadism require further exploration. The present
study provided an example of the use gene screening tools in the
diagnosis of a patient with complicated symptoms.
Acknowledgements
The authors would like to thank the family members
for their participation and support in the present study. The
present study was supported by the National High Technology
Research and Development Program of China (863 Program) (grant no.
2007AA02E466) and the National Natural Science Foundation of China
both to Dr. Huijun Yuan (grant no. 30571018).
Glossary
Abbreviations
Abbreviations:
|
HGPS
|
Hutchinson-Gilford progeria
syndrome
|
|
NSHL
|
nonsyndromic hearing loss
|
|
SHL
|
syndromic hearing loss
|
|
EDA
|
ectodermal dysplasia
|
|
WGS
|
whole-genome sequencing
|
|
CNV
|
copy number variation
|
|
SNV
|
single nucleotide variations
|
References
|
1
|
Shearer AE, Eppsteiner RW, Booth KT,
Ephraim SS, Gurrola J II, Simpson A, Black-Ziegelbein EA, Joshi S,
Ravi H, Giuffre AC, et al: Utilizing ethnic-specific differences in
minor allele frequency to recategorize reported pathogenic deafness
variants. Am J Hum Genet. 95:445–453. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yamamoto N, Okuyama H, Hiraumi H, Sakamoto
T, Matsuura H and Ito J: The outcome of cochlear implantation for
mitochondrial disease patients with syndromic hearing loss. Otol
Neurotol. 36:e129–e133. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dai ZY, Sun BC, Huang SS, Yuan YY, Zhu YH,
Su Y and Dai P: Correlation analysis of phenotype and genotype of
GJB2 in patients with non-syndromic hearing loss in China. Gene.
570:272–276. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lamartine J: Towards a new classification
of ectodermal dysplasias. Clin Exp Dermatol. 28:351–355. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yavuz I, Baskan Z, Ulku R, Dulgergil TC,
Dari O, Ece A, Yavuz Y and Dari KO: Ectodermal dysplasia:
Retrospective study of fifteen cases. Arch Med Res. 37:403–409.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guler N, Cildir S, Iseri U, Sandalli N and
Dilek O: Hypohidrotic ectodermal dysplasia with bilateral impacted
teeth at the coronoid process: A case rehabilitated with mini
dental implants. Oral Surg Oral Med Oral Pathol Oral Radiol Endod.
99:E34–E38. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yavuz I, Kiralp S and Baskan Z:
Hypohidrotic ectodermal dysplasia: A case report. Quintessence Int.
39:81–86. 2008.PubMed/NCBI
|
|
8
|
Fukuo K and Ogihara T: Hutchinson-Gilford
progeria syndrome. Ryoikibetsu Shokogun Shirizu. 318–320. 2000.(In
Japanese). PubMed/NCBI
|
|
9
|
Sinha JK, Ghosh S and Raghunath M:
Progeria: A rare genetic premature ageing disorder. Indian J Med
Res. 139:667–674. 2014.PubMed/NCBI
|
|
10
|
Mazereeuw-Hautier J, Wilson LC, Mohammed
S, Smallwood D, Shackleton S, Atherton DJ and Harper JI:
Hutchinson-Gilford progeria syndrome: Clinical findings in three
patients carrying the G608G mutation in LMNA and review of the
literature. Br J Dermatol. 156:1308–1314. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vetter U, Pontz B, Zauner E, Brenner RE
and Spranger J: Osteogenesis imperfecta: A clinical study of the
first ten years of life. Calcif Tissue Int. 50:36–41. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yasuda M, Kishi C, Yokoyama Y, Amano H and
Ishikawa O: Case of X-linked hypohidrotic ectodermal dysplasia with
a novel EDA missense mutation. J Dermatol. 42:907–908. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Salas-Alanis JC, Wozniak E, Mein CA,
Mckinster CC Duran, Ocampo-Candiani J, Kelsell DP, Hua R,
Garza-Rodriguez ML, Choate KA and Saldaña HA Barrera: Mutations in
EDA and EDAR genes in a large mexican hispanic cohort with
hypohidrotic ectodermal dysplasia. Ann Dermatol. 27:474–477. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang L, Liang X, Li Y, Wang J, Zaneveld
JE, Wang H, Xu S, Wang K, Wang B, Chen R and Sui R: Comprehensive
molecular diagnosis of 67 Chinese Usher syndrome probands: High
rate of ethnicity specific mutations in Chinese USH patients.
Orphanet J Rare Dis. 10:1102015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sodi A, Mariottini A, Passerini I, Murro
V, Tachyla I, Bianchi B, Menchini U and Torricelli F: MYO7A and
USH2A gene sequence variants in Italian patients with Usher
syndrome. Mol Vis. 20:1717–1731. 2014.PubMed/NCBI
|
|
16
|
Pober BR, Longoni M and Noonan KM: A
review of Donnai-Barrow and facio-oculo-acoustico-renal (DB/FOAR)
syndrome: Clinical features and differential diagnosis. Birth
Defects Res A Clin Mol Teratol. 85:76–81. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
De S, andre-Giovannoli A, Bernard R, Cau
P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich
A, Le Merrer M and Lévy N: Lamin a truncation in hutchinson-gilford
progeria. Science. 300:20552003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kieran MW, Gordon L and Kleinman M: New
approaches to progeria. Pediatrics. 120:834–841. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Moulson CL, Fong LG, Gardner JM, Farber
EA, Go G, Passariello A, Grange DK, Young SG and Miner JH:
Increased progerin expression associated with unusual LMNA
mutations causes severe progeroid syndromes. Hum Mutat. 28:882–889.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chu Y, Xu ZG, Xu Z and Ma L:
Hutchinson-Gilford progeria syndrome caused by an LMNA mutation: A
case report. Pediatr Dermatol. 32:271–275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eriksson M, Brown WT, Gordon LB, Glynn MW,
Singer J, Scott L, Erdos MR, Robbins CM, Moses TY and Berglund P:
Recurrent de novo point mutations in lamin A cause
Hutchinson-Gilford progeria syndrome. Nature. 423:293–298. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pelosini C, Martinelli S, Ceccarini G,
Magno S, Barone I, Basolo A, Fierabracci P, Vitti P, Maffei M,
Santini F, et al: Identification of a novel mutation in the
polymerase delta 1 (POLD1) gene in a lipodystrophic patient
affected by mandibular hypoplasia, deafness, progeroid features
(MDPL) syndrome. Metabolism. 63:1385–1389. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weedon MN, Ellard S, Prindle MJ, Caswell
R, Allen H Lango, Oram R, Godbole K, Yajnik CS, Sbraccia P, Novelli
G, et al: An in-frame deletion at the polymerase active site of
POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet.
45:947–950. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lessel D, Hisama FM, Szakszon K, Saha B,
Sanjuanelo AB, Salbert BA, Steele PD, Baldwin J, Brown WT, Piussan
C, et al: POLD1 germline mutations in patients initially diagnosed
with werner syndrome. Hum Mutat. 36:1070–1079. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shastry S, Simha V, Godbole K, Sbraccia P,
Melancon S, Yajnik CS, Novelli G, Kroiss M, Garg A, et al: A novel
syndrome of mandibular hypoplasia, deafness and progeroid features
associated with lipodystrophy, undescended testes, and male
hypogonadism. J Clin Endocrinol Metab. 95:E192–197. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Abe S, Katagiri T, Saito-Hisaminato A,
Usami S, Inoue Y, Tsunoda T and Nakamura Y: Identification of CRYM
as a candidate responsible for nonsyndromic deafness, through cDNA
microarray analysis of human cochlear and vestibular tissues. Am J
Hum Genet. 72:73–82. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yoshimura H, Takumi Y, Nishio SY, Suzuki
N, Iwasa Y and Usami S: Deafness gene expression patterns in the
mouse cochlea found by microarray analysis. PLoS One. 9:e925472014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Oshima A, Suzuki S, Takumi Y, Hashizume K,
Abe S and Usami S: CRYM mutations cause deafness through thyroid
hormone binding properties in the fibrocytes of the cochlea. J Med
Genet. 43:e252006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Morris-Rosendahl DJ, Segel R, Born AP,
Conrad C, Loeys B, Brooks SS, Müller L, Zeschnigk C, Botti C,
Rabinowitz R, et al: New RAB3GAP1 mutations in patients with
warburg micro syndrome from different ethnic backgrounds and a
possible founder effect in the Danish. Eur J Hum Genet.
18:1100–1106. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aligianis IA, Morgan NV, Mione M, Johnson
CA, Rosser E, Hennekam RC, Adams G, Trembath RC, Pilz DT, Stoodley
N, et al: Mutation in Rab3 GTPase-activating protein (RAB3GAP)
noncatalytic subunit in a kindred with Martsolf syndrome. Am J Hum
Genet. 78:702–707. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Handley MT and Aligianis IA: RAB3GAP1,
RAB3GAP2 and RAB18: Disease genes in Micro and Martsolf syndromes.
Biochem Soc Trans. 40:1394–1397. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Handley MT, Morris-Rosendahl DJ, Brown S,
Macdonald F, Hardy C, Bem D, Carpanini SM, Borck G, Martorell L,
Izzi C, et al: Mutation spectrum in RAB3GAP1, RAB3GAP2 and RAB18
and genotype-phenotype correlations in warburg micro syndrome and
Martsolf syndrome. Hum Mutat. 34:686–696. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Asahina M, Endoh Y, Matsubayashi T, Fukuda
T and Ogata T: Novel RAB3GAP1 compound heterozygous mutations in
Japanese siblings with Warburg Micro syndrome. Brain Dev.
38:337–340. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Clauss F, Waltmann E, Barriere P,
Hadj-Rabia S, Manière MC and Schmittbuhl M: Dento-maxillo-facial
phenotype and implants-based oral rehabilitation in ectodermal
dysplasia with WNT10A gene mutation: Report of a case and
literature review. J Craniomaxillofac Surg. 42:e346–351. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mues G, Bonds J, Xiang L, Vieira AR,
Seymen F, Klein O and D'Souza RN: The WNT10A gene in ectodermal
dysplasias and selective tooth agenesis. Am J Med Genet A.
164A:1–2460. 2014.PubMed/NCBI
|
|
36
|
Adams BB: Odonto-onycho-dermal dysplasia
syndrome. J Am Acad Dermatol. 57:732–733. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang J, Wang SK, Choi M, Reid BM, Hu Y,
Lee YL, Herzog CR, Kim-Berman H, Lee M, Benke PJ, et al:
Taurodontism, variations in tooth number and misshapened crowns in
Wnt10a null mice and human kindreds. Mol Genet Genomic Med.
3:40–58. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kimura R, Watanabe C, Kawaguchi A, Kim YI,
Park SB, Maki K, Ishida H and Yamaguchi T: Common polymorphisms in
WNT10A affect tooth morphology as well as hair shape. Hum Mol
Genet. 24:2673–2680. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mostowska A, Biedziak B, Zadurska M,
Matuszewska-Trojan S and Jagodziński PP: WNT10A coding variants and
maxillary lateral incisor agenesis with associated dental
anomalies. Eur J Oral Sci. 123:1–8. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kantaputra P, Kaewgahya M, Jotikasthira D
and Kantaputra W: Tricho-odonto-onycho-dermal dysplasia and WNT10A
mutations. Am J Med Genet A. 164A:1–1048. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bergendal B, Klar J, Stecksén-Blicks C,
Norderyd J and Dahl N: Isolated oligodontia associated with
mutations in EDARADD AXIN2, MSX1 and PAX9 genes. Am J Med Genet A.
155A:1–1622. 2011.PubMed/NCBI
|
|
42
|
Arzoo PS, Klar J, Bergendal B, Norderyd J
and Dahl N: WNT10A mutations account for (1/4) of population-based
isolated oligodontia and show phenotypic correlations. Am J Med
Genet A. 164A:1–359. 2014.PubMed/NCBI
|
|
43
|
He H, Han D, Feng H, Qu H, Song S, Bai B
and Zhang Z: Involvement of and interaction between WNT10A and EDA
mutations in tooth agenesis cases in the Chinese population. PLoS
One. 8:e803932013. View Article : Google Scholar : PubMed/NCBI
|