Introduction
Psoriasis is a complex skin disease involving
reciprocity between immune cells and keratinocytes, and is
characterized by the infiltration of multiple inflammatory cells,
upregulation dermal vascularity and keratinocyte proliferation
(1–4). Angiogenesis commences with early
psoriatic alterations and disappears when the disease is cured. A
number of pro-angiogenic mediators, including vascular endothelial
growth factor (VEGF), tumor necrosis factor, hypoxia-inducible
factor, interleukin (IL)-8, angiopoietins and IL-17, are
upregulated during the development of psoriasis (5–8).
Initially, psoriasis was considered to be a T-helper
(Th)1-mediated skin disease (9).
However, a number of subsequent studies later demonstrated that
Th17 cells, which are activated upon exposure to IL-23, IL-6 and
transforming growth factor-β and produce IL-17, IL-21 and IL-22
cytokines, serves a key role in the pathogenesis of psoriasis
(9–12). IL-17 is one of the most potent
proinflammatory cytokines, and is secreted by Th17 cells (9). A previous study indicated that IL-17
may induce keratinocytes to produce VEGF, which is an important
angiogenic mediator (13).
Therefore, elucidation of the mechanisms by which IL-17 signaling
in keratinocytes is regulated may facilitate the development of
novel treatments for Th17-mediated angiogenesis.
The Janus kinase/signal transducer and activator of
transcription signaling (JAK/STAT) signaling pathway controls a
number of important biological responses, including cellular
differentiation, immune functions, hematopoiesis and cellular
growth, and it is initiated when a receptor is bound by its
corresponding cytokine and subsequently transmits signals from the
cell surface membrane to the target genes (14). A previous study confirmed that
IL-17 induced upregulation of VEGF expression via activation of the
JAK2/STAT3 signaling pathway (15). However, the precise mechanisms
involved required further investigation. An increasing number of
studies have indicated that microRNAs (miRNAs/miRs) may be involved
in the pathogenesis of psoriasis, and miRNAs associated with
psoriasis have been identified by comparing their expression in
normal and psoriatic skin samples (16,17).
However, whether specific miRNAs influence the process of
IL-17-induced VEGF expression remains unclear.
miR-203 is preferentially expressed in the skin and
is an important regulator of keratinocyte differentiation. miR-203
has been implicated in a number of skin diseases, particularly
psoriasis (18). However, the role
of miR-203 in IL-17-induced VEGF secretion has yet to be
elucidated. The aim of the present study was to investigate the
role of miR-203 in IL-17-induced VEGF secretion to further
elucidate the mechanism of miR-203 in psoriasis.
Materials and methods
Animals and stimulation with
IL-17
A total of 16 BALB/c mice (age, 6–8 weeks; weight,
18–22 g) were obtained from the Center of Experimental Animals of
China Medical University (Shenyang, China). The mice were
maintained in cages (4 mice/cage) under controlled conditions
(temperature, 20–25°C; humidity 40–70%) with daily 12-h light/dark
cycles. Mice were provided with food and water ad libitum.
All animal experiments were approved by the Institutional Animal
Ethics Committee of China Medical University, and were performed in
accordance with the Animal Care Guidelines for Experimental Animals
(19). Mice were divided into the
IL-17 stimulated group and control group (n=8 mice/group), then the
mice were injected intradermally into each ear with 30 µl PBS,
either alone or with 3 µg recombinant mouse IL-17A (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) using a 30-gauge needle once a day
for two consecutive days.
Immunohistochemical staining
Following 1 week, all mice were sacrificed and mouse
ear tissues were fixed in 4% paraformaldehyde for 48 h at 4°C, and
embedded in paraffin wax. Sections were cut at 4 µm and mounted
onto slides. The tissue sections were dewaxed in xylene,
re-hydrated using a descending ethanol series, and subjected to
antigen retrieval in 0.01 M citrate buffer. Following inhibition of
endogenous peroxidase by incubating samples with 3%
H2O2 at 37°C for 30 min, the sections were
subsequently incubated with anti-VEGF (cat no. sc-80442; 1:200
dilution) and cluster of differentiation (CD)34 monoclonal
antibodies (cat no. sc-74499; 1:200 dilution) (both from Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) overnight at 4°C. Following
washing with PBS, tissue sections were incubated with a horseradish
peroxidase-conjugated goat anti-mouse IgG1 secary antibody (cat no.
ab97240; 1:1,000 dilution; Abcam, Cambridge, MA, USA) at 37°C for
30 min. Reaction products were visualized by incubation with
diaminobenzidine for 60 sec at room temperature and then
counterstained with hematoxylin for 5 min at room temperature. As a
negative control, tissue samples were subject to the same staining
procedures without incubation with primary antibodies. Regions of
positive staining were quantified by calculating the pixel density
using analysis LS Research image analysis software v5.0 (Olympus
Soft Imaging Solutions GmbH, Münster, Germany).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The mice were injected intradermally into each ear
with 30 µl PBS, either alone or with 3 µg recombinant mouse IL-17A
once a day for two consecutive days. Following 1 week, the ear
tissues were collected and the expression of miR-203a-3p.1 in ear
tissues was determined by RT-qPCR. Total RNA from ear tissues was
extracted using TRIzol (Takara Biotechnology Co., Ltd., Dalian,
China) following the manufacturer's instructions. Single strand
cDNA was synthesized using the PrimeScript miRNA cDNA Synthesis kit
(Takara Biotechnology Co., Ltd.) using 2 µg of total RNA as a
template according to the manufacturer's instructions. qPCR was
performed using SYBR Premix Ex Taq (Takara Biotechnology Co., Ltd.)
according to the manufacturer's instructions. The thermocycling
conditions were as follows: 95°C for 30 sec, followed by 45 cycles
of 95°C for 5 sec and 53°C for 20 sec. The expression of U6 small
nuclear B non-coding RNA was used as an internal normalization
control. The primers used were as follows: miR-203a-3p.1 primer,
5′-TACGAGTGAAATGTTTAGGACCACTAG-3′; U6,
5′-ATTGGAACGATACAGAGAAGATT-3′.
HaCaT cells were treated with medium only or with
IL-17 for 48 h, in the presence or absence of miR-203 inhibitor for
24 h. The expression of suppressor of cytokine signaling 3 (SOCS3)
in HaCaT cells was then determined by RT-qPCR. cDNA was synthesized
using the PrimeScript RT-PCR kit (Takara Biotechnology Co., Ltd.)
using 2 µg of total RNA as a template according to the
manufacturer's instructions. qPCR was performed using SYBR Premix
Ex Taq (Takara Biotechnology Co., Ltd.) according to the
manufacturer's instructions. The following thermocycling conditions
were used: 95°C for 30 sec, followed by 45 cycles of 95°C for 5 sec
and 60°C for 20 sec. β-actin was used to normalize the expression
levels of SOCS3. The primers used were as follows: SOCS3 forward,
5′-TGGATGGAGCGGGAGGCT-3′, and reverse,
5′-ACGGACATCTTTCACCTCAGGCTCCT-3′; β-actin forward,
5′-GACAGGATGCAGAAGGAGATTACT-3′ and reverse,
5′-TGATCCACATCTGCTGGAAGGT-3′. All RT-qPCR experiments were
performed in triplicate using the Applied Biosystems 7500 Real-Time
PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). All primers were synthesized by Guangzhou
RiboBio Co., Ltd. (Guangzhou, China). Samples were analyzed in
triplicate, and the mean quantification cycle (Cq) was
calculated. Gene expression levels were analyzed by comparing the
ΔCq values of samples [where ΔCq=Cq
(target gene)-Cq (housekeeping gene)] transformed
to a linear scale (2−ΔΔCq) (20).
Cell culture
The human umbilical vein endothelial cells (HUVECs)
cell line was purchased from the American Type Culture Collection
(ATTC; Manassas, VA, USA). Cells were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum (both from Gibco; Thermo
Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml
streptomycin, and maintained in a humidified atmosphere of 5%
CO2 at 37°C. The human keratinocyte cell lines, HaCaT
and HEK293T (both from ATCC), were incubated in Dulbecco's modified
Eagle's medium containing 10% fetal bovine serum (both from Gibco;
Thermo Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml
streptomycin, and maintained in a humidified atmosphere of 5%
CO2 at 37°C.
Cell transfection
HaCaT cells were seeded in 6-well plates
(2×105/well) for 24 h prior to transfection.
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) was
used to transfect HaCaT cells with 50 nmol miR-203 mimic, 150 nmol
miR-203 inhibitor or the same concentration of their respective
negative controls (Guangzhou RiboBio Co., Ltd.), according to the
manufacturer's instructions. The sequence of miR-203 mimic, miR-203
inhibitor and their respective negative controls were as follows:
miR-203a-3p.1 mimic, 5′-GUGAAAUGUUUAGGACCACUAG-3′ and
5′-AGUGGUCCUAAACAUUUCACUU-3′; miR-203a-3p.1 mimic control,
5′-UUCUCCCAACGUGUCACGUTT-3′ and 5′-ACGUGACACGUUCGGAGAATT-3′;
miR-203a-3p.1 inhibitor, 5′-CUAGUGGUCCUAAACAUUUCAC-3′;
miR-203a-3p.1 inhibitor control, 5′-CAGUACUUUUGUGUAGUACAA-3′. HaCaT
cells were co-transfected with 150 nmol SOCS3 small interfering
(si)RNA or scrambled siRNA (Shanghai GenePharma Co., Ltd.,
Shanghai, China) and 150 nmol miR-203 inhibitor using Lipofectamine
2000 according to the manufacturer's instructions (Invitrogen;
Thermo Fisher Scientific, Inc.). At 24 h following transfection,
HaCaT cells were treated with 80 ng/ml IL-17 (PeproTech, Inc.,
Rocky Hill, NJ, USA) for 48 h at 37°C, then cells
(~1.5×106/well) were harvested for analysis. HUVECs were
treated with conditioned HaCaT cell medium only at 37°C for 7
h.
Luciferase reporter assays
For luciferase activity analysis, the 3′-UTR
sequence of SOCS3 or the mutant SOCS3 3′-UTR sequence, which
included a mutation in the miR-203 binding site, was cloned into a
pGL3-promoter vector (Promega Corporation, Madison, WI, USA). Then
HEK293T cells were co-transfected with 500 ng wild-type
pGL3-SOCS3-3′UTR or mutant pGL3-SOCS3-3′UTR and 10 pmol miR-203
mimic or 10 pmol miRNA control using Lipofectamine 2000, according
to the manufacturer's instructions (Invitrogen; Thermo Fisher
Scientific, Inc.). At 24 h following transfection, luciferase
activity was detected using a dual-luciferase reporter assay system
according to the manufacturer's instructions (Promega Corporation).
Luciferase activity was normalized to Renilla luciferase
activity.
Computational prediction
The target gene of miR-203 was predicted using
TargetScan Release 7.1 software (www.targetscan.org/vert_71/).
Western blot analysis
HaCaT cells treated with medium alone or with IL-17
for 48 h, in the presence or absence of miR-203 inhibitor for 24 h.
Total protein was extracted using a protein extraction kit
(Beyotime Institute of Biotechnology, Haimen, China) according to
the manufacturer's protocol. Protein concentration was determined
using a bicinchoninic protein assay kit (Beyotime Institute of
Biotechnology). For western blotting analysis, an equal quantity of
total protein (30 µg/lane) was loaded, and separated by 8%
SDS-PAGE. Following electrophoresis, proteins were transferred to a
polyvinylidene difluoride membrane and blocked for 1 h at room
temperature with 5% milk in TBS with 0.1% Tween-20. The membrane
was incubated with primary antibodies against the IL-17 receptor
(cat no. ab180904; 1:1,000 dilution; Abcam), phosphorylated
(p)-JAK2 (cat no. 4406; 1:1,000 dilution), p-STAT3 (cat no. 9145;
1:1,000 dilution), β-actin (cat no. 4970; 1:1,000 dilution), SOCS1
(cat no. 3950; 1:1,000 dilution) and SOCS3 (cat no. 2932; 1:1,000
dilution) (all from Cell Signaling Technology, Inc., Danvers, MA,
USA) at 4°C overnight. The blots were subsequently incubated with a
with horseradish peroxidase-conjugated secondary antibody (cat no.
E030120-02; 1:2,000 dilution; EarthOx Life Sciences, Millbrae, CA,
USA) for 1 h at room temperature. Proteins were visualized using
Clarity Western ECL Substrate (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) according to the manufacturer's protocol.
Densitometry analysis of the western blots was achieved using
ImageJ v1.48 software (National Institutes of Health, Bethesda, MD,
USA).
Tube formation assay
The tube formation assay was performed as described
previously (21). Briefly, a
96-well plate was coated with 60 µl Matrigel (BD Biosciences,
Franklin Lakes, NJ, USA), which was allowed to polymerize and
solidify at 37°C for 30 min. HUVECs (2×104 cells) were
seeded onto the Matrigel layer in the presence or absence of
conditioned medium from HaCaT cells treated with IL-17 (80 ng/ml
for 48 h), miR-203 inhibitor (150 nmol for 24 h) or inhibitor
control (150 nmol for 24 h), and SOCS3-siRNA (150 nmol for 24 h) or
siRNA-control (150 nmol for 24 h), and cells were incubated at 37°C
for 7 h. The number of blood-vessel-like tubules from six fields of
view selected at random were counted, and images were captured
using an inverted light microscope (Nikon Corporation, Tokyo,
Japan).
Enzyme-linked immunosorbent assay
(ELISA)
HaCaT cells were treated with IL-17 (80 ng/ml for 48
h), miR-203 inhibitor (150 nmol for 24 h) or inhibitor control (150
nmol for 24 h), and SOCS3-siRNA (150 nmol for 24 h) or
siRNA-control (150 nmol for 24 h). The cell culture media were
centrifuged at 1,500 × g for 10 min at 4°C, the supernatants were
collected and and stored at −80°C prior to ELISA analysis. The
level of VEGF secretion in HaCaT cell cultures were measured by an
ELISA kit (cat no. DVE00; R&D Systems, Minneapolis, MN, USA)
according to the manufacturer's instructions.
Statistical analysis
Statistical analysis was performed using SPSS v13.0
software (SPSS, Inc., Chicago, IL, USA). The results were analyzed
by one-way analysis of variance followed by a Student-Newman-Keuls
post hoc test, and presented as the mean ± standard deviation.
P<0.05 was considered to indicate a statistically significant
difference. All experiments were repeated three times.
Results
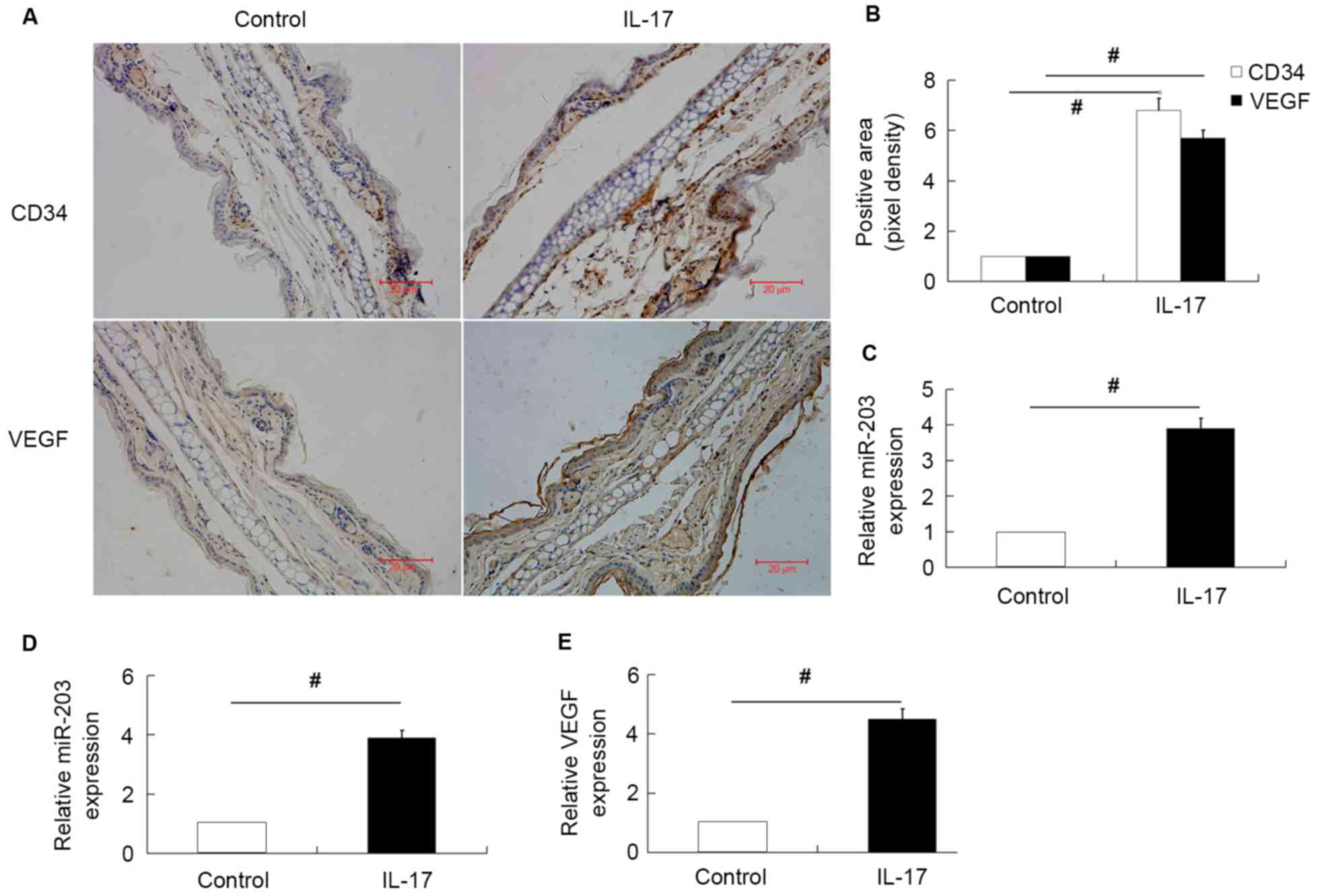
miR-203 is upregulated in the ears of
IL-17-stimulated mice and IL-17-treated HaCaT cells
In order to investigate the effect of miR-203 on
IL-17-induced VEGF expression, miR-203, VEGF and CD34 expression in
the ears of untreated and IL-17-stimulated mice were first
examined. The results demonstrated that the expression of miR-203,
VEGF and CD34 were significantly upregulated in the ears of
IL-17-stimulated mice compared with the normal untreated group
(Fig. 1A-C). The expression of
miR-203 and VEGF secretion in IL-17-stimulated HaCaT cells was then
investigated. Consistent with the in vivo results, miR-203
expression and VEGF secretion levels were significantly increased
in HaCaT cells stimulated with IL-17 (Fig. 1D and E).
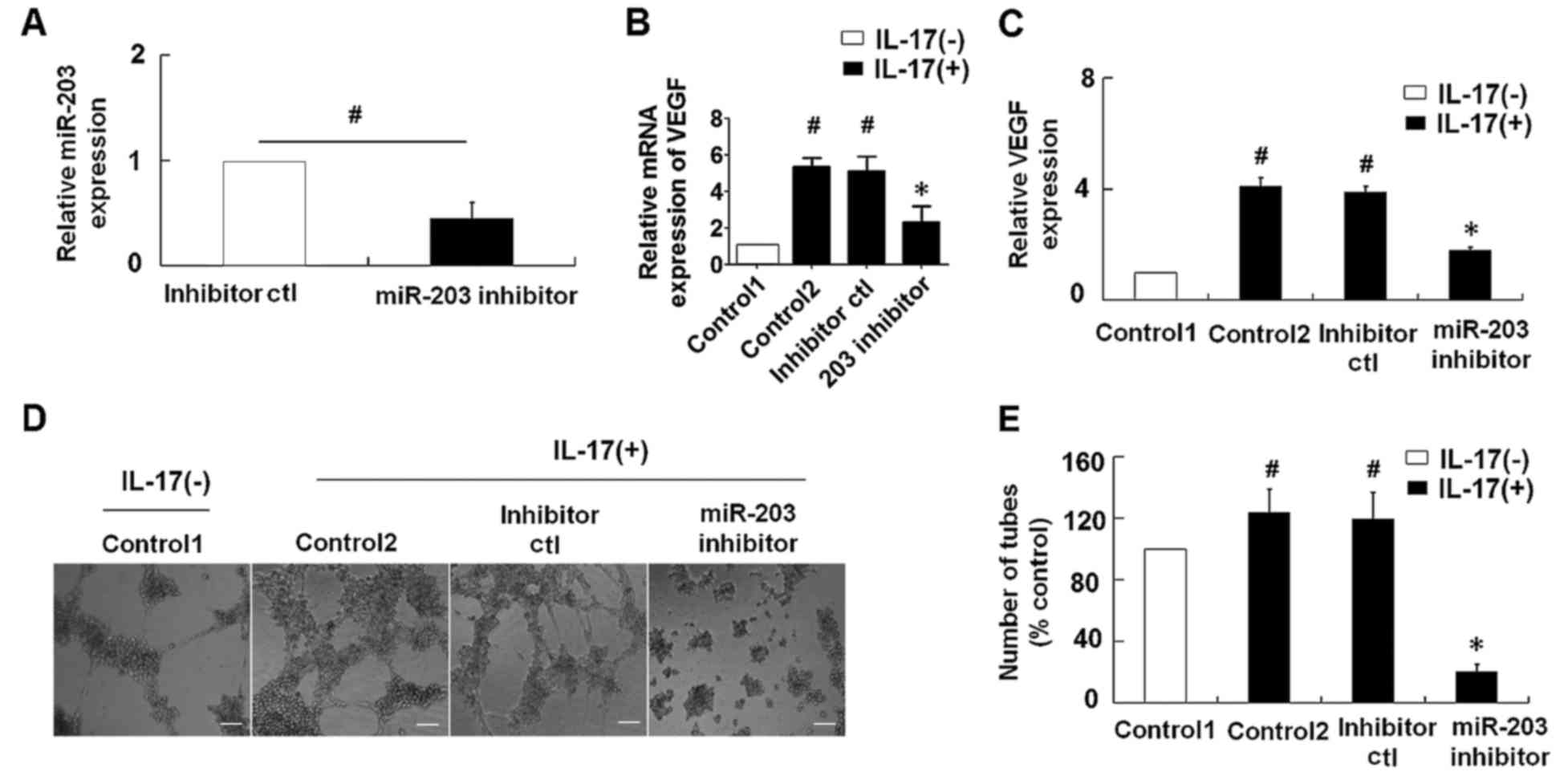
Suppression of miR-203 inhibits
IL-17-induced VEGF secretion in HaCaT cells
To explore the role of miR-203 in IL-17-induced VEGF
secretion, an miR-203 inhibitor was transfected into the HaCaT
cells prior to stimulation with IL-17. As demonstrated in Fig. 2A, transfection of HaCaT cells with
the miR-203 inhibitor was associated with a significant reduction
in relative miR-203 expression levels when compared with cells
transfected with the inhibitor control. IL-17-stimulation was
associated with a significant increase in VEGF mRNA and the levels
of VEGF in the supernatant of HaCaT cells, while repression of
miR-203 significantly inhibited the IL-17-induced upregulation of
VEGF levels (Fig. 2B and C). In
addition, the effect of miR-203 on IL-17-induced VEGF secretion was
assessed using a tube formation assay in HUVECs. Consistent with
the results observed in HaCaT cells, suppression of miR-203
significantly attenuated the IL-17-induced upregulation of VEGF
levels and significantly inhibited tube formation of HUVECs
(Fig. 2B-E).
Repression of miR-203 inhibits
IL-17-induced activation of the JAK2/STAT3 signaling pathway
In order to explore the effect of miR-203 on
IL-17-induced activation of the JAK2/STAT3 signaling pathway, HaCaT
cells were transfected with miR-203 inhibitor or controls for 24 h,
then treated with 80 ng/ml IL-17 for a further 48 h. Protein
expression levels were subsequently detected by western blot
analysis. The results demonstrated that the expression levels of
p-JAK2 and p-STAT3 were significantly reduced in miR-203
inhibitor-transfected cells when compared with IL-17-stimulated and
inhibitor control-transfected cells (Fig. 3A and B). The next aim was to
identify targets of miR-203. As demonstrated in (Fig. 3C-E), the mRNA and protein
expression levels of SOCS3, an inhibitor of the JAK2/STAT3
signaling pathway, was significantly upregulated in miR-203
inhibitor-transfected cells when compared with IL-17-stimulated and
inhibitor control-transfected cells. By contrast, western blot
analysis demonstrated that the protein expression levels of SOCS1,
an additional inhibitor of the JAK2/STAT3 signaling pathway, were
not significantly affected by miR-203 inhibition (Fig. 3D and E). These results suggest that
miR-203 may inhibit JAK2/STAT3 signaling via targeting of SOCS3
expression.
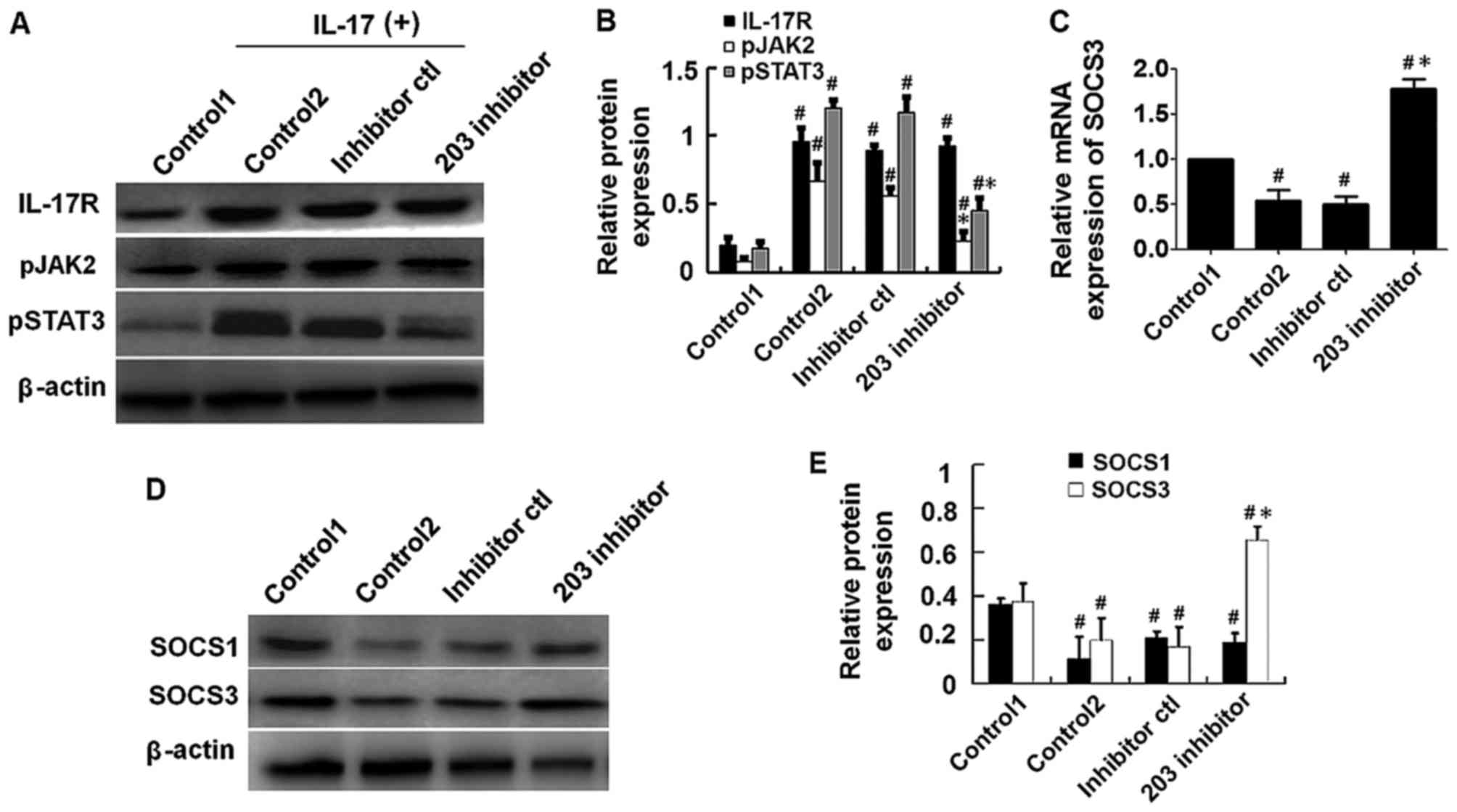 | Figure 3.Repression of miR-203 inhibited
IL-17-induced activation of the JAK2/STAT3 signaling pathway. HaCaT
cells were transfected with miR-203 inhibitor for 24 h and
subsequently treated with 80 ng/ml IL-17 for a further 48 h. (A)
Western blot analysis of the protein expression levels of IL-17R,
pJAK2 and pSTAT3, and (B) quantification of the results by
densitometry analysis using Image J software. (C) The mRNA levels
of SOCS3 were measured by reverse transcription-quantitative
polymerase chain reaction analysis, and β-actin was used as an
endogenous control. (D) Western blot analysis of the protein
expression levels of SOCS1 and SOCS3 inhibitors of the JAK2/STAT3
signaling pathway, and (E) quantification of the results by
densitometry analysis using ImageJ software. Target protein levels
were normalized to β-actin. The control1 group consisted of
untransfected HaCaT cells that were not treated with IL-17, whereas
the control2 group consisted of untransfected HaCaT cells that were
treated with IL-17. The results are presented as the mean ±
standard deviation (n=3). #P<0.05 vs. control1;
*P<0.05 vs. control2. miR, microRNA; IL, interleukin; JAK, Janus
kinase; STAT, signal transducer and activator of transcription;
SOCS, suppressor of cytokine signaling; p-, phosphorylated; R,
receptor. |
miR-203 directly targets SOCS3
To assess the role of miR-203 in regulating the
JAK2/STAT3 signaling pathway, computational analysis (TargetScan
v7.1) was performed to predict the potential target genes of
miR-203. The results indicated that miR-203 may target sequences in
3′-untranslated region (UTR) of SOCS3 (Fig. 4A). The 3′-UTR sequence of SOCS3 was
then cloned into a luciferase reporter plasmid. A mutant SOCS3
3′-UTR sequence, which included a mutation in the miR-203 binding
site, was additionally cloned into the luciferase reporter plasmid
(Fig. 4A). The dual-luciferase
reporter assay was performed using HEK293T cells. The results
demonstrated that the luciferase activity in HEK293T cells
co-transfected with an miR-203 mimic and a luciferase reporter
plasmid containing the wild type SOCS3 3′-UTR sequence was
significantly reduced when compared with the mimic control
(Fig. 4B). By contest, no
significant alterations in luciferase activity were observed
HEK293T cells transfected with miR-203 mimic and a luciferase
reporter plasmid containing the mutant SOCS3 3′-UTR sequence
(Fig. 4B). In addition,
transfection of HaCaT cells with miR-203 mimics was associated with
a significant reduction in the mRNA and protein expression levels
of SOCS3 (Fig. 4C-E). These
results provide evidence to suggest that SOCS3 may be a direct
target of miR-203.
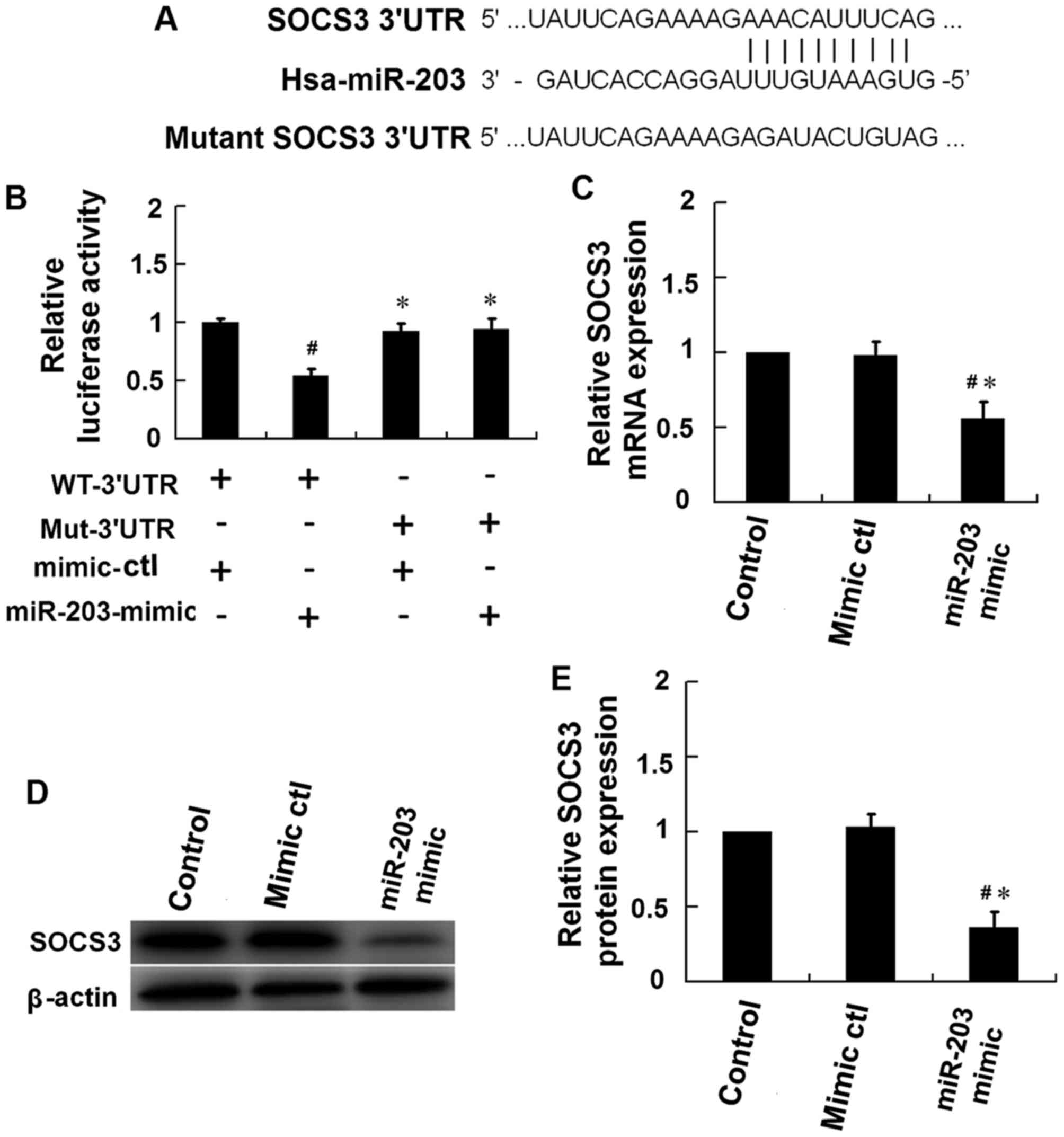 | Figure 4.SOCS3 is the molecular target of
miR-203. (A) Schematic of the predicted miR-203 binding site in the
3′-UTR region of SOCS3, as detected using TargetScan v7.1 software.
The mutant SOCS3 3′-UTR sequence included several mutations in the
miR-203 binding site. (B) A dual-luciferase reporter assay was
performed using HEK293T cells. Cells were co-transfected with a
reporter vector containing the WT-3′UTR or the Mut-3′UTR, in
addition to a miR-203-mimic or a mimic-ctl. The firefly/Renilla
activity ratio was calculated to determine the luciferase activity
(#P<0.05 vs. HaCaT cells transfected with WT-3′UTR
plus mimic-ctl; *P<0.05 vs. HaCaT cells transfected with
WT-3′-UTR plus miR-203 mimic). Following 48 h of IL-17 stimulation,
HaCaT cells were then transfected with an miR-203 mimic or
mimic-ctl, and cells were harvested at 24 h following transfection.
The (C) mRNA and (D) protein levels of SOCS3 were measured by
western blotting and reverse transcription-quantitative polymerase
chain reaction analysis, respectively, and (E) densitometry
analysis of the western blots was performed using ImageJ software.
SOCS3 mRNA and protein expression levels were normalized to that of
β-actin (#P<0.05 vs. control; *P<0.05 vs.
mimic-ctl). The results are presented as the mean ± standard
deviation (n=3). SOCS, suppressor of cytokine signaling; miR,
microRNA; UTR, untranslated region; WT-3′UTR, pGL3 vector
containing the wild-type SOCS3 3′-UTR; Mut-3′UTR, pGL3 vector
containing the mutant SOCS3 3′-UTR; ctl, control. |
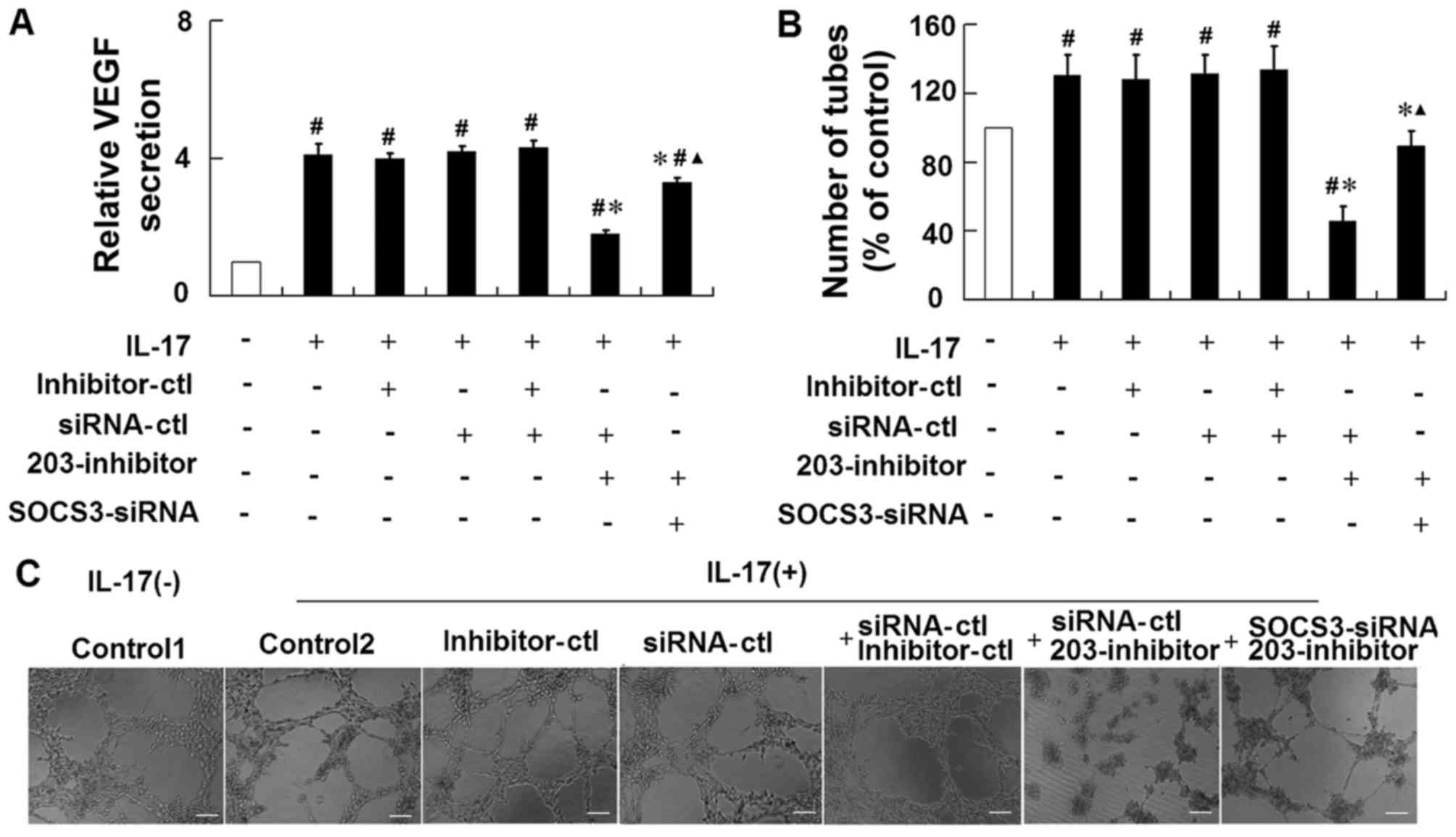
Repression of miR-203 attenuates
IL-17-induced VEGF secretion in HaCaT cells via targeting
SOCS3
To further confirm the role of SOCS3 in
miR-203-mediated VEGF secretion in response to IL-17 stimulation,
HaCaT cells were co-transfected with an miR-203 inhibitor together
with an siRNA targeting SOCS3, and then stimulated with IL-17. The
results demonstrated that treatment of cells with the SOCS3 siRNA
significantly attenuated the effects of the miR-203 inhibitor on
the IL-17-induced upregulation of VEGF levels and increase in tube
formation (Fig. 5). These results
suggest that IL-17 may induce miR-203 expression, which may
subsequently increase VEGF secretion via targeting of SOCS3.
Discussion
The results of the present study demonstrated that
miR-203 expression is significantly upregulated in mice and HaCaT
cells stimulated with IL-17. In addition, VEGF levels were observed
to increase in the ears of IL-17-stimulated mice and in the
supernatant of IL-17-treated HaCaT cells. These results suggest
that IL-17 may induce VEGF expression, and that miR-203 may be
involved in mediating this effect. In addition, the results
indicated that inhibition of miR-203 reversed the IL-17-induced
increase in VEGF secretion and inhibited the IL-17-induced
activation of JAK2/STAT3 signaling. Furthermore, the present study
provided evidence to suggest that SOCS3, a repressor of the
JAK2/STAT3 signaling pathway, may be a direct target of
miR-203.
miRNAs are a class of abundant non-coding RNA
molecules that modulate mRNA degradation or inhibition of
translation by specifically binding to the 3′-UTR of target mRNA
sequences (22–24). miRNAs serve key roles in the
regulation of cell differentiation, growth and death in normal and
malignant tissues (25,26). miR-203 is a miRNA preferentially
expressed in the skin, and is an important regulator of
keratinocyte differentiation (18). miR-203 has been implicated in a
number of skin diseases, particularly psoriasis, via regulation of
pro-inflammatory cytokines (27,28).
However, a complete understanding of the mechanisms underlying the
involvement of miR-203 in psoriasis is lacking. The results of the
present study demonstrated that miR-203 expression is significantly
upregulated in mice and HaCaT cells stimulated with IL-17. In
addition, VEGF levels were increased in the ears of
IL-17-stimulated mice and in the supernatant of IL-17-treated HaCaT
cells. These results implied that miR-203 may be involved in
IL-17-induced VEGF expression. In addition, the results of the
present study demonstrated that miR-203 may function as a positive
effector of IL-17-induced VEGF secretion, as inhibition of miR-203
effectively reversed IL-17-induced VEGF secretion.
Further investigation of the mechanisms by which
miR-203 functions to mediate the IL-17-induced increase in VEGF
secretion demonstrated that SOCS3 may be a direct target of miR-203
via binding to its 3′-UTR, thus inhibiting SOCS3 expression. An
inverse association between miR-203 and SOCS3 expression in HaCaT
cells stimulated by IL-17 was observed. These results suggest that
SOCS3 may be a direct target of miR-203, and potentially mediate
the process of IL-17-induced VEGF expression in HaCaT cells.
In order to prevent the adverse effects of
over-activation, the duration and intensity of JAK/STAT signaling
pathway activation is strictly controlled by negative regulators
(29). SOCS proteins are induced
by growth factors or cytokines, and regulate the duration and
magnitude of inflammatory responses activated by these cytokines
via the inhibition of JAK proteins in a negative feedback loop
(30). Previous studies have
demonstrated that SOCS1 and SOCS3 proteins are the major regulators
of the JAK2/STAT3 signaling pathway (31–33).
The results of the present study demonstrated that SOCS1 and SOCS3
expression was significantly downregulated in HaCaT cells following
stimulation with IL-17. However, only SOCS3 protein expression was
upregulated following inhibition of miR-203 in IL-17-treated HaCaT
cells. This provides additional evidence to suggest that SOCS3 may
be a target of miR-203.
Angiogenesis is an important pathological feature of
psoriasis, and the expression of a number of angiogenic mediators,
including VEGF, increase during the development of psoriasis
(34,35). The present study demonstrated that
IL-17 induces the expression of VEGF in vitro and in
vivo, which is consistent with the results of previous studies
(13,15). Notably, suppression of miR-203
reversed IL-17-induced VEGF expression in HaCaT cells. These
results are consistent with the observed upregulation of SOCS3
expression and the reduction in JAK/STAT signalling pathway
activation in the IL-17 signaling process following inhibition of
miR-203, which indicated that miR-203 may be involved in mediating
IL-17-induced VEGF expression.
In conclusion, the results of the current study
suggest that miR-203 may inhibit the expression of SOCS3 by
directly binding to the 3′-UTR of SOCS3 and promoting the
degradation of the SOCS3 mRNA, thus activating the JAK2/STAT3
signaling pathway and mediating IL-17-induced VEGF secretion.
Future studies that aim to investigate the role of miR-203 further,
may provide novel insights into its mechanisms of action, as well
as identify potential therapeutic strategies for the treatment of
psoriasis.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81673055 and
81402595) and the Program for Liaoning Excellent Talents in
University (grant no. LR2012026).
References
|
1
|
Schön MP and Boehncke WH: Psoriasis. N
Engl J Med. 352:1899–1912. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wei T, Xu N, Meisgen F, Ståhle M, Sonkoly
E and Pivarcsi A: Interleukin-8 is regulated by miR-203 at the
posttranscriptional level in primary human keratinocytes. Eur J
Dermatol. 19:2013.
|
|
3
|
Wolk K, Witte E, Wallace E, Döcke WD, Kunz
S, Asadullah K, Volk HD, Sterry W and Sabat R: IL-22 regulates the
expression of genes responsible for antimicrobial defense, cellular
differentiation, and mobility in keratinocytes: A potential role in
psoriasis. Eur J Immunol. 36:1309–1323. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Heidenreich R, Röcken M and Ghoreschi K:
Angiogenesis: The new potential target for the therapy of
psoriasis? Drug News Perspect. 21:97–105. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hongqin T, Xinyu L, Heng G, Lanfang X,
Yongfang W and Shasha S: Triptolide inhibits IFN-γ signaling via
the Jak/STAT pathway in HaCaT keratinocytes. Phytother Res.
25:1678–1685. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu Y, Xu X, Gao X, Chen H and Geng L:
Shikonin suppresses IL-17-induced VEGF expression via blockage of
JAK2/STAT3 pathway. Int Immunopharmacol. 19:327–333. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Koga C, Kabashima K, Shiraishi N,
Kobayashi M and Tokura Y: Possible pathogenic role of Th17 cells
for atopic dermatitis. J Invest Dermatol. 128:2625–2630. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Primo MN, Bak RO, Schibler B and Mikkelsen
JG: Regulation of pro-inflammatory cytokines TNFα and IL24 by
microRNA-203 in primary keratinocytes. Cytokine. 60:741–748. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cai Y, Fleming C and Yan J: New insights
of T cells in the pathogenesis of psoriasis. Cell Mol Immunol.
9:302–309. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nickoloff BJ and Wrone-Smith T: Animal
models of psoriasis. Nat Med. 3:475–476. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zamore PD and Haley B: Ribo-gnome: The big
world of small RNAs. Science. 309:1519–1524. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lerman G, Avivi C, Mardoukh C, Barzilai A,
Tessone A, Gradus B, Pavlotsky F, Barshack I, Polak-Charcon S,
Orenstein A, et al: MiRNA expression in psoriatic skin: Reciprocal
regulation of hsa-miR-99a and IGF-1R. PLoS One. 6:e209162011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pittelkow MR: Psoriasis: More than skin
deep. Nat Med. 11:17–18. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen CZ, Li L, Lodish HF and Bartel DP:
MicroRNAs modulate hematopoietic lineage differentiation. Science.
303:83–86. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Creamer D, Sullivan D, Bicknell R and
Barker J: Angiogenesis in psoriasis. Angiogenesis. 5:231–236. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huh JE, Baek YH, Lee MH, Choi DY, Park DS
and Lee JD: Bee venom inhibits tumor angiogenesis and metastasis by
inhibiting tyrosine phosphorylation of VEGFR-2 in LLC-tumor-bearing
mice. Cancer Lett. 292:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheng AM, Byrom MW, Shelton J and Ford LP:
Antisense inhibition of human miRNAs and indications for an
involvement of miRNA in cell growth and apoptosis. Nucleic Acids
Res. 33:1290–1297. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sonkoly E, Ståhle M and Pivarcsi A:
MicroRNAs: Novel regulators in skin inflammation. Clin Exp
Dermatol. 33:312–315. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cho A and Seok SH: Ethical guidelines for
use of experimental animals in biomedical research. J Bacteriol
Virol. 43:18–26. 2013. View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xiong H, Du W, Zhang YJ, Hong J, Su WY,
Tang JT, Wang YC, Lu R and Fang JY: Trichostatin A, a histone
deacetylase inhibitor, suppresses JAK2/STAT3 signaling via inducing
the promoter-associated histone acetylation of SOCS1 and SOCS3 in
human colorectal cancer cells. Mol Carcinog. 51:174–184. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hueber W, Patel DD, Dryja T, Wright AM,
Koroleva I, Bruin G, Antoni C, Draelos Z, Gold MH; Psoriasis Study
Group, ; et al: Effects of AIN457, a fully human antibody to
interleukin-17A, on psoriasis, rheumatoid arthritis and uveitis.
Sci Transl Med. 2:52ra722010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zibert JR, Løvendorf MB, Litman T, Olsen
J, Kaczkowski B and Skov L: MicroRNAs and potential target
interactions in psoriasis. J Dermatol Sci. 58:177–185. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Darnell JE Jr: STATs and gene regulation.
Science. 277:1630–1635. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Valencia-Sanchez MA, Liu J, Hannon GJ and
Parker R: Control of translation and mRNA degradation by miRNAs and
siRNAs. Genes Dev. 20:515–524. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Starnes T, Robertson MJ, Sledge G, Kelich
S, Nakshatri H, Broxmeyer HE and Hromas R: Cutting edge: IL-17F, a
novel cytokine selectively expressed in activated T cells and
monocytes, regulates angiogenesis and endothelial cell cytokine
production. J Immunol. 167:4137–4140. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang Y, Zhou B, Zhang F, Wu J, Hu Y, Liu
Y and Zhai Q: Amyloid-β induces hepatic insulin resistance by
activating JAK2/STAT3/SOCS-1 signaling pathway. Diabetes.
61:1434–1443. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nickoloff BJ: Cracking the cytokine code
in psoriasis. Nat Med. 13:242–244. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Song B, Jin H, Yu X, Zhang Z, Yu H, Ye J,
Xu Y, Zhou T, Oudit GY, Ye JY, et al: Angiotensin-converting enzyme
2 attenuates oxidative stress and VSMC proliferation via the
JAK2/STAT3/SOCS3 and profilin-1/MAPK signaling pathways. Regul
Pept. 185:44–51. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zheng Y, Danilenko DM, Valdez P, Kasman I,
Eastham-Anderson J, Wu J and Ouyang W: Interleukin-22, a T(H)17
cytokine, mediates IL-23-induced dermal inflammation and
acanthosis. Nature. 445:648–651. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Krebs DL and Hilton DJ: SOCS:
Physiological suppressors of cytokine signaling. J Cell Sci.
113:2813–2819. 2000.PubMed/NCBI
|
|
33
|
Numasaki M, Fukushi J, Ono M, Narula SK,
Zavodny PJ, Kudo T, Robbins PD, Tahara H and Lotze MT:
Interleukin-17 promotes angiogenesis and tumor growth. Blood.
101:2620–2627. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liew SC, Das-Gupta E, Chakravarthi S, Wong
SF, Lee N, Safdar N and Jamil A: Differential expression of the
angiogenesis growth factors in psoriasis vulgaris. BMC Res Notes.
5:2012012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sankar L, Arumugam D, Boj S and Pradeep P:
Expression of angiogenic factors in psoriasis vulgaris. J Clin
Diagn Res. 11:EC23–EC27. 2017.PubMed/NCBI
|