Introduction
Urotensin II (UII) is a highly potent
vasoconstrictor, with stronger vasoconstrictive effects than those
of endothelin-1 (ET-1). GPR14, also known as the urotensin receptor
(UT), is the specific receptor of UII. Both UII and its receptor
are primarily expressed in cardiovascular tissues, such as
cardiomyocytes, vascular smooth muscle cells (VSMCs) and
endothelial cells (1). In addition
to its vasoconstrictor activity, UII exerts various effects on cell
proliferation, migration, hypertrophy, apoptosis, fibrosis,
immunity and inflammatory responses in a wide variety of cell types
in the cardiovascular system (2–5). UII
also inhibits insulin release, modulates catecholamine release, and
helps regulate food intake and the sleep cycle (2).
UII and UT were upregulated in cardiovascular
diseases and metabolic syndrome (6), and UII receptor antagonism was shown
to significantly attenuate diabetes-associated atherosclerosis in
diabetic apolipoprotein E knockout mice (7). UII plays a pivotal role in the
development of diseases, such as cardiovascular disease and
diabetes mellitus (1).
Furthermore, increased expression of UII was related to
subendothelial inflammation in the pathogenesis of coronary
atherosclerosis (8). In human
umbilical vein endothelial cells and human monocytes, UII induced
IL-1β and IL-6 expression (9).
Vascular inflammation plays critical roles in the
development of various cardiovascular disease, such as
hypertension, heart failure, vascular restenosis and
atherosclerosis (10). Previously,
this inflammation was considered an ‘inside-out’ response involving
monocyte adhesion to the intima of blood vessels, as described in
the oxidative lipid hypothesis (11). However, increasing evidence
supports a new paradigm, which can be described as the ‘outside-in’
hypothesis, in which vascular inflammation initiates in the
adventitia and progresses toward the intima (12).
Following paracrine/autocrine stimulation, AFs are
activated, and acquire a myofibroblast phenotype, characterized by
increased expression of the cytoskeletal protein α-smooth muscle
actin (α-SMA) (13–16). Then, AFs synthesize a panel of
cytokines and other molecules, such as interleukin-6 (IL-6) and
monocyte chemoattractant protein-1 (MCP-1), both of which
contribute to proinflammatory activation and vascular remodeling
(10,12).
Aldosterone (ALD), the key hormone in the
mineralocorticoid pathway, binds to mineralocorticoid receptors
(MRs) in the kidney. By affecting cell adhesion and cytokine
expression, MRs play an important role in salt and water
homeostasis, blood pressure regulation, and cardiovascular
remodeling. Through MR signaling and subsequent genomic events, as
well as through nongenomic pathways, ALD exerts effects in
non-epithelial cells, such as cardiomyocytes, VSMCs, endothelial
cells, mesangial cells and podocytes. Furthermore, MR expression
has recently been discovered in non-epithelial tissues, including
cardiac tissue and VSMCs. ALD causes inflammation and insulin
resistance, which lead to fibrosis and remodeling in the heart,
vasculature and kidney (17,18).
In a previous study, we reported that UII is
abundantly expressed in the adventitia (4) and that UII stimulated phenotypic
conversion, proliferation, collagen synthesis, and production of
transforming growth factor-β1 and LTC4 in AFs and intracellular
signal transduction pathways, including calcineurin, PKC,
Ca2+, MAPK, and Rho kinase, may be involved in these
processes (4,19–21).
However, the role of the vascular adventitia and the mechanisms
driving the inflammatory response during the genesis of UII-induced
cardiovascular diseases are poorly understood. In the present
study, we assessed the effect of UII on ALD and ALD receptor
(ALD-R) mRNA expression in AFs and tunica adventitia of rat vessels
and elucidated the molecular mechanisms underlying these
effects.
Materials and methods
Animals
Male Sprague-Dawley rats weighing 180–200 g were
supplied by the Experimental Animal Center Hubei University of
Medicine. This study was carried out in strict accordance with the
recommendations in the Guide for the Care and Use of Laboratory
Animals of the Hubei University of Medicine. The protocol was
approved by the Committee on the Ethics of Animal Experiments of
the Hubei University of Medicine. All surgery was performed under
sodium pentobarbital anesthesia, and all efforts were made to
minimize suffering.
Materials and reagents
Rat UII was acquired from Phoenix Pharmaceuticals
Inc. (Belmont, CA, USA). A rat ALD enzyme-linked immunosorbent
assay (ELISA) kit was obtained from Sangon Biotech (Shanghai,
China). Fetal bovine serum (FBS) was acquired from HyClone (Logan,
UT, USA). The ALD antibody from Novus Biologicals (Littleton, CO,
USA), the ALD-R antibody from Enzo Life Sciences (Lausen,
Switzerland). The calcineurin inhibitor cyclosporin A (CSA), the
Ca2+ channel blocker nicardipine and Dulbecco's modified
Eagle's medium/F12 (DMEM/F12) were purchased from Sigma-Aldrich;
Merck KGaA (St. Louis, MO, USA). The signal transduction blockers
PD98059 (for mitogen-activated protein kinase), Y-27632 (for
Rho-associated protein kinase) and H-7 (for protein kinase C) were
acquired from Calbiochem (Darmstadt, Germany); High Capacity cDNA
Reverse Transcription kits and SYBR Select Master Mix were
purchased from Applied Biosystems (Foster City, CA, USA); and RIPA
lysis buffer and TRIzol reagent were acquired from Invitrogen
(Carlsbad, CA, USA). PCR primers were designed and synthesized by
Sangon Biotech (Shanghai, China), and their sequences are shown in
Table I. All other chemicals and
reagents were of analytical grade.
 | Table I.Forward and reverse primers for the
rat ALD receptor and β-actin. |
Table I.
Forward and reverse primers for the
rat ALD receptor and β-actin.
| Primer | Sequence | Amplicon size
(bp) | Annealing
temperature (°C) |
|---|
| ALD-R | F:
5′-CGGCAAATCTCAACAACTCAAGG-3′ | 240 bp | 58 |
| ALD-R | R:
5′-CCTCTGTCTTAGGGAAAGGAACG-3′ |
|
|
| β-actin | F:
5′-CACGATGGAGGGGCCGGACTCATC-3′ | 240 bp | 58 |
| β-actin | R:
5′-TAAAGACCTCTATGCCAACACAGT-3′ |
|
|
Hematoxylin and eosin (H&E)
staining and immunohistochemistry
Vessels were performed in 10% formalin-fixed,
paraffin-embedded, cut into 10 µm serial sections and then mounted
on slides. The slides were stained with H&E for histological
examination. For immunohistochemical staining, sections were
deparaffinized with xylene and rehydrated by immersion into
decreasing concentrations of ethanol. Endogenous peroxidase
activity was blocked using 3% hydrogen peroxide solution for 10
min, followed by citric acid buffer (pH 6.0) microwave antigen
retrieval. Nonspecific protein binding was blocked by 30 min
incubation in 5% bovine serum (Wuhan Boster Biological Technology,
Ltd., Wuhan, China). Sections were incubated with primary
antibodies: Antibodies against aldosterone (1:50), aldosterone
receptor (1:100), overnight at 4°C, followed by 5 min wash in PBS
for 3 times. Sections were then incubated for 1 h at RT with
HRP-labelled secondary antibodies. DAB (Boster) for 2 min at RT and
counterstaining was made using hematoxylin, dehydrated and
clarified by a conventional method, and prepared for examination
under a light microscope.
Tissue incubation
Rats were anesthetized and rapidly euthanized by
decapitation. The full length of the thoraco-abdominal aorta was
excised under aseptic conditions and placed into DMEM/F12. The
aorta was sectioned longitudinally after the loose connective
tissue and collateral vessels were removed. The blunt side of
ophthalmic bending forceps was used to removed endothelial cells
and medial layer by gently rubbing. Eye scissors were used to cut
the remaining tissue, which was predominantly composed of the
adventitia, as small as possible. The adventitia fragments were
distributed into 1.5 ml Eppendorf tubes after they were weighed
(~100 mg/tube) and incubated on a shaker under different
intervention conditions at 37°C in a cell incubator (22).
The experimental groups consisted of: i) Control
groups, with tissues incubated in serum-free DMEM/F12; ii) the UII
groups, in which 10−10 to 10−6 mol/l UII was
added to the serum-free medium; and iii) the UII + inhibitor
groups, in which tissues were pretreated with different signal
transduction blockers (concentration was 10−5 mol/l),
including Y-27632, H7, PD98059, CSA and nicardipine in serum-free
medium for 0.5 h prior to addition of UII. After 6 h of UII
incubation, the samples were collected.
Cell culture
AFs from the aorta were prepared as described by Kim
et al (23) and Tsuruda
et al (24) with slight
modifications. Adventitia tissues mentioned above were placed in a
25 cm2 tissue culture flask containing DMEM/F12
supplemented with 20% FBS. After 5–8 days, the fibroblasts were
harvested with trypsin prior to forming a confluent monolayer and
seeded onto new dishes containing DMEM/F12 medium supplemented with
10% FBS. The growth characteristics and morphology of the cells
were typical of fibroblasts and were distinguished from VSMCs by
immunohistochemical staining positive for ‘Vimentin’ and negative
for ‘α-SMA’. AFs used for experiments in this study were at
passages two and three.
RNA isolation
Total RNA was isolated directly from AFs using
TRIzol reagent according to the manufacturer's instructions
(Invitrogen). Then, the RNA was treated with DNase I to remove
residual traces of DNA. The RNA concentration was quantified using
a spectrophotometer (NanoDrop 2000; Thermo Fischer Scientific,
Inc., Waltham, MA, USA) measuring the OD260/280 ratio (1.8–2.0).
Agarose gel electrophoresis (2%) and GoldView nucleic acid staining
were used to examine RNA integrity.
RT-PCR
Reverse transcription was performed with High
Capacity cDNA Reverse Transcription kits (Applied Biosystems),
which included buffer, dNTP mix, random primer and reverse
transcriptase, according to the manufacturer's instructions. The
PCR was conducted on a PCR instrument (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), initial denaturing was performed at 95°C for 5
min; then, 35 cycles at 95°C for 45 sec (denaturing), at 58°C for
ALD-R and β-actin for 45 sec (annealing), at 72°C for 60 sec
(extension) and a further extension at 72°C for 3 min were carried
out. After amplification, the RT-PCR products were electrophoresed
on 1% agarose gel containing with ethidium bromide, viewed by UV
light.
Quantitative real-time PCR
ALD-R and β-actin complementary DNA (cDNA) was
synthesized as previously described. For quantitative real-time
RT-PCR, gene expression was quantified using SYBR select master mix
(Applied Biosystems). The reagent concentrations used were based on
the manufacturer's instructions. Primers targeting rat ALD-R and
β-actin are listed in Table I. PCR
conditions were 95°C for 10 min, followed by 40 cycles of 94°C for
15 sec, 60°C for 1 min. The relative mRNA levels of ALD-R in AFs
were determined using the comparative threshold cycle (CT) method
using the 2−ΔΔCT equation with β-actin as an internal
control. Each experimental condition was conducted in
triplicate.
ELISA
The ELISA was performed with a rat ALD ELISA kit to
assess the release of ALD into the culture medium. Briefly, after
treatment with the respective stimuli, the culture medium was
collected and then centrifuged to obtain the supernatant. The ELISA
was conducted according to the manufacturer's directions. The
absorbance was read at 450 nm by a microplate reader.
Statistical analysis
Data are expressed as mean ± SEM. Statistical
differences among multiple groups were analyzed by one-way analysis
of variance (ANOVA) followed by Dunnett's test for multiple
comparisons, and a Student t-test was used for the statistical
analysis of differences between two groups. All data were analyzed
with the statistical software GraphPad Prism 5.0 software (GraphPad
Software, San Diego, CA, USA). P-values <0.05 were considered to
indicate a statistically significant difference.
Results
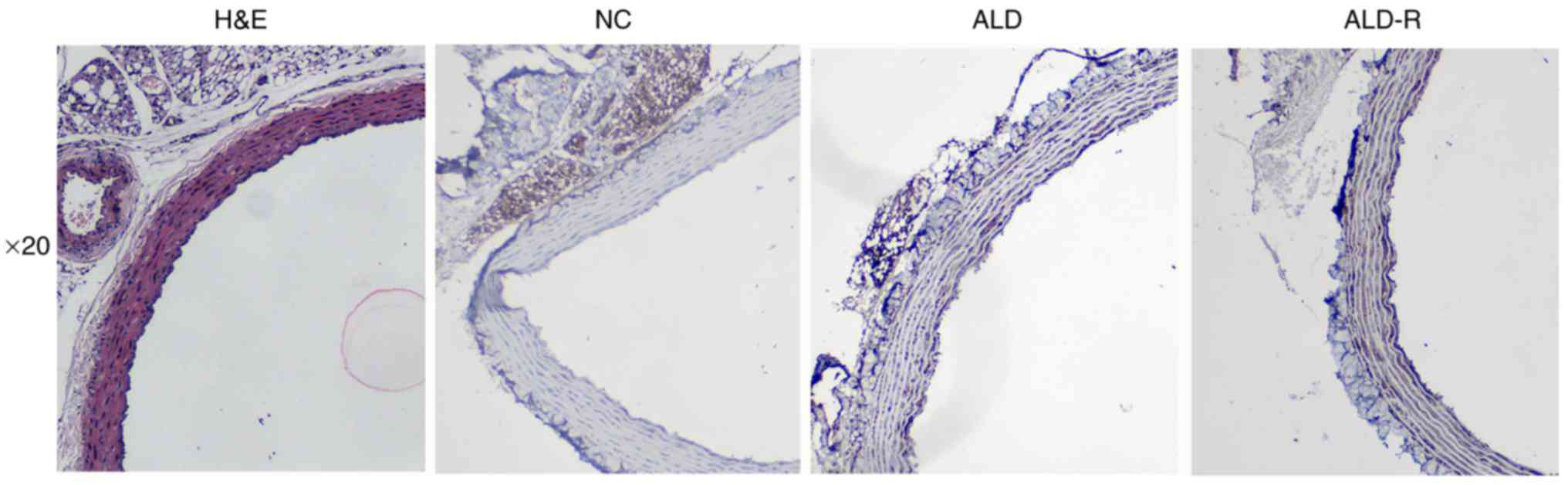
ALD and its receptors were expressed
on adventitia
H&E staining was used to observe the morphology
of the blood vessels, and Immunohistochemistry was used to confirm
whether aldosterone and its receptors were expressed on adventitia.
As shown in Fig. 1, both ALD and
its receptors were expressed on tunica adventitia of vessels.
Effect of UII on ALD synthesis in
AFs
To determine the role of UII in the synthesis of
vasoactive substances in AFs and the signaling pathways associated
with this process, we tested the effects of UII on ALD secretion,
which is an important indicator of vascular remodeling. After UII
stimulation, AFs underwent the phenotypic transformation from
fibroblasts to myofibroblasts, demonstrating proliferation,
migration, intense ALD secretion and α-SMA expression.
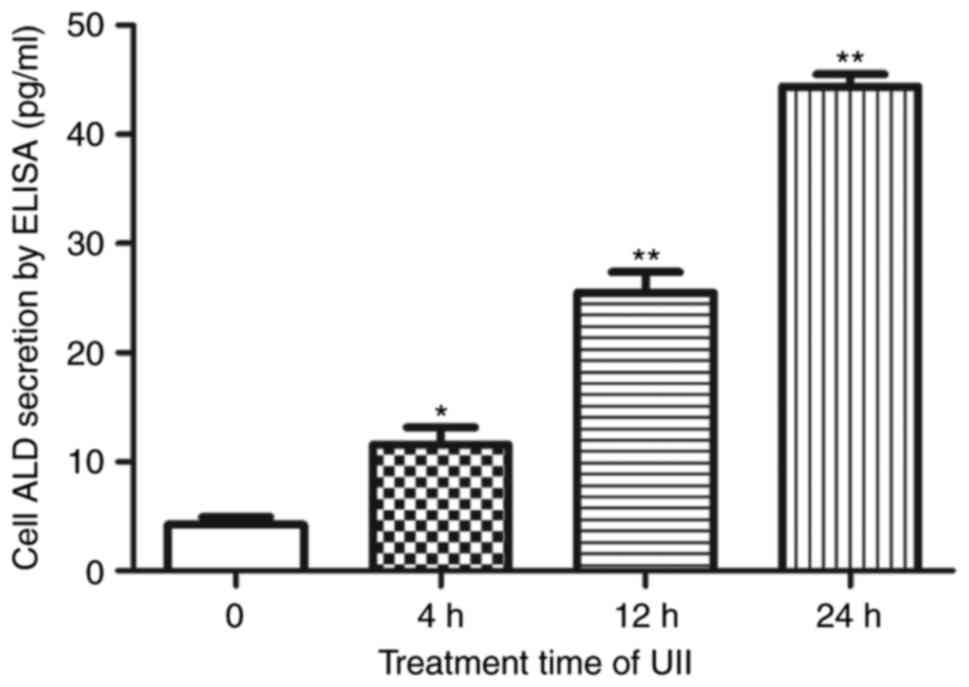
As shown in Fig. 2,
the ELISA results indicated that ALD secretion increased after 4 h
of UII (10−8 mol/l) stimulation (P<0.05) and reached
a maximum at 24 h (P<0.01) (Fig.
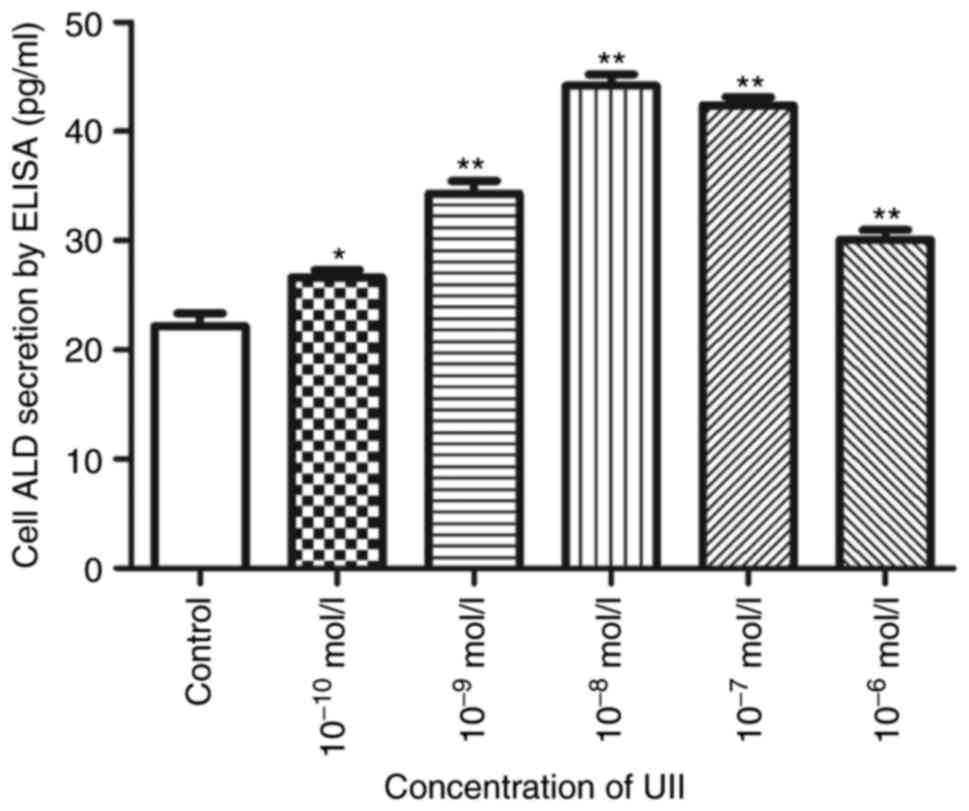
2). UII also upregulated ALD secretion in a
concentration-dependent manner (Fig.
3). Maximal effectiveness was achieved at 10−8 mol/l
(P<0.01), as ALD secretion increased by 20.1% (10−10
mol/l, P<0.05), 54.7% (10−9 mol/l, P<0.05), 99.2%
(10−8 mol/l, P<0.01), 91% (10−7 mol/l,
P<0.01) and 35.7% (10−6 mol/l; P<0.01).
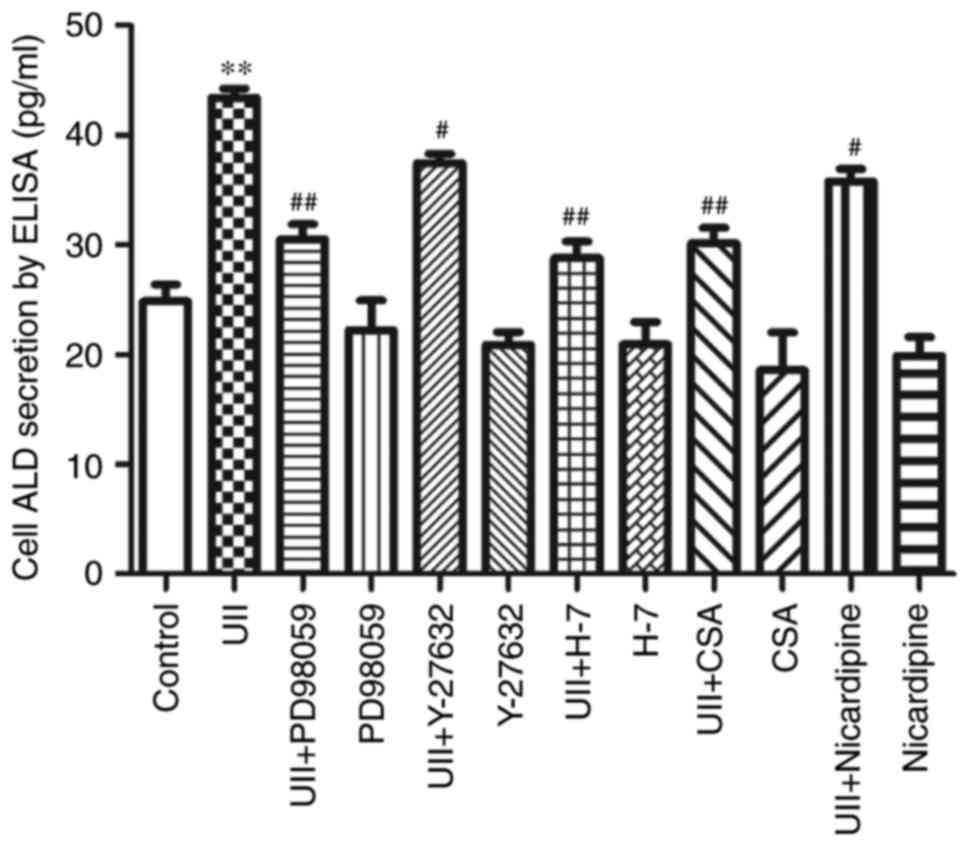
After treatment with UII and different inhibitors
(Fig. 4), including PD98059,
Y-27632, H7, CSA and nicardipine, the ELISA results (Fig. 3) showed that ALD secretion was
inhibited to varying degrees under all conditions, suggesting that
MAPK, Rho, PKC, calcineurin and Ca2+ signaling,
respectively, may be involved in UII-induced ALD synthesis.
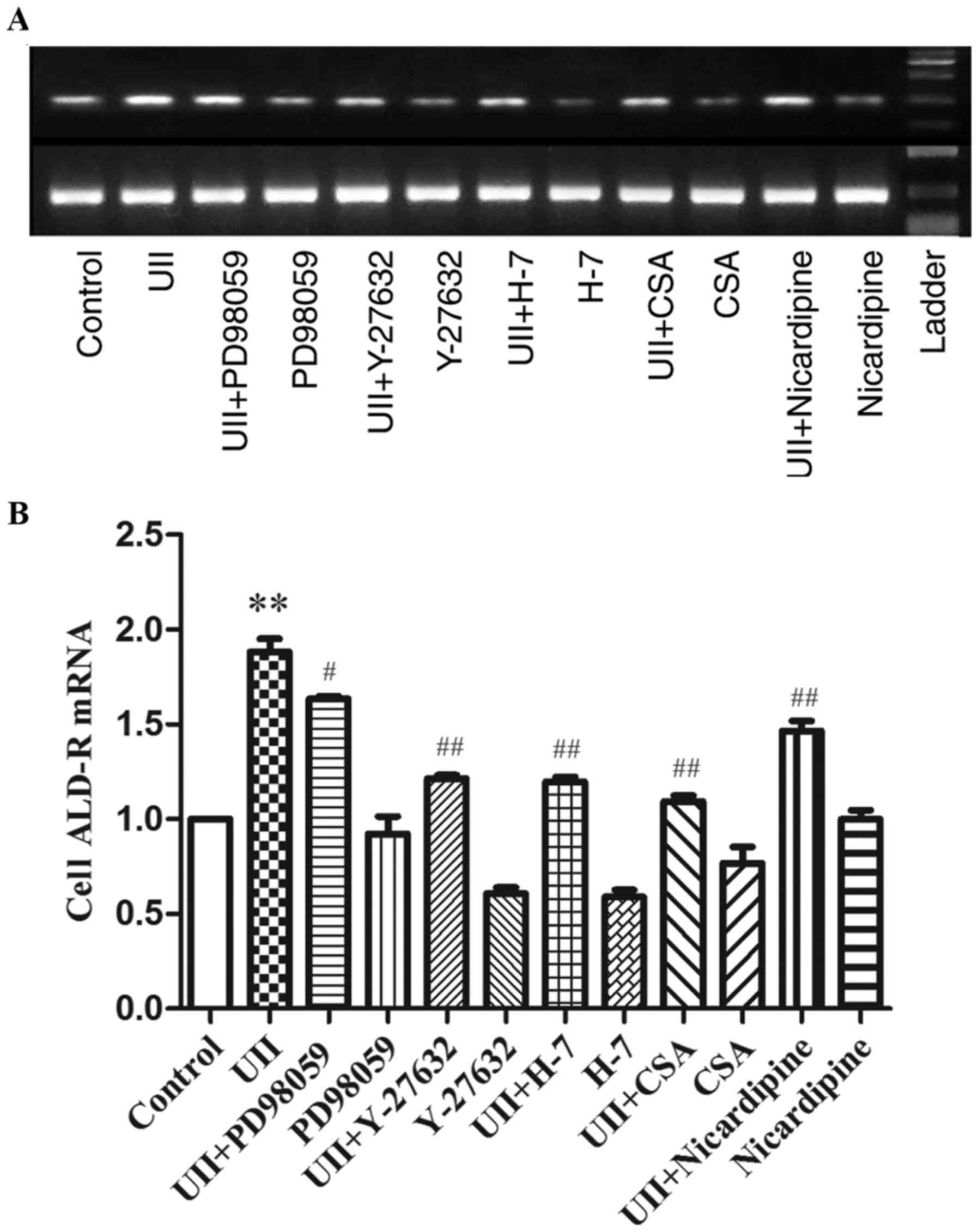
Effect of UII on ALD-R mRNA expression
in AFs
UII induced dose- and time-dependent increases in
ALD expression, with the maximal effect observed at a dose of
10−8 mol/l for 24 h (P<0.01, Figs. 2 and 3). We used this fixed condition to
investigate the effect of UII on ALD-R expression in AFs. As shown
in Fig. 5 RT-PCR results indicated
that ALD-R expression increased after 24 h of UII stimulation
compared with that of the control group without UII stimulation
(P<0.01). Furthermore, this UII-induced effect was attenuated
following pretreatment with kinase inhibitors, including PD98059,
Y-27632, H7, CSA or nicardipine, suggesting that MAPK, Rho, PKC,
calcineurin and Ca2+ signaling, respectively, may be
involved in UII-induced ALD-R expression (Fig. 5).
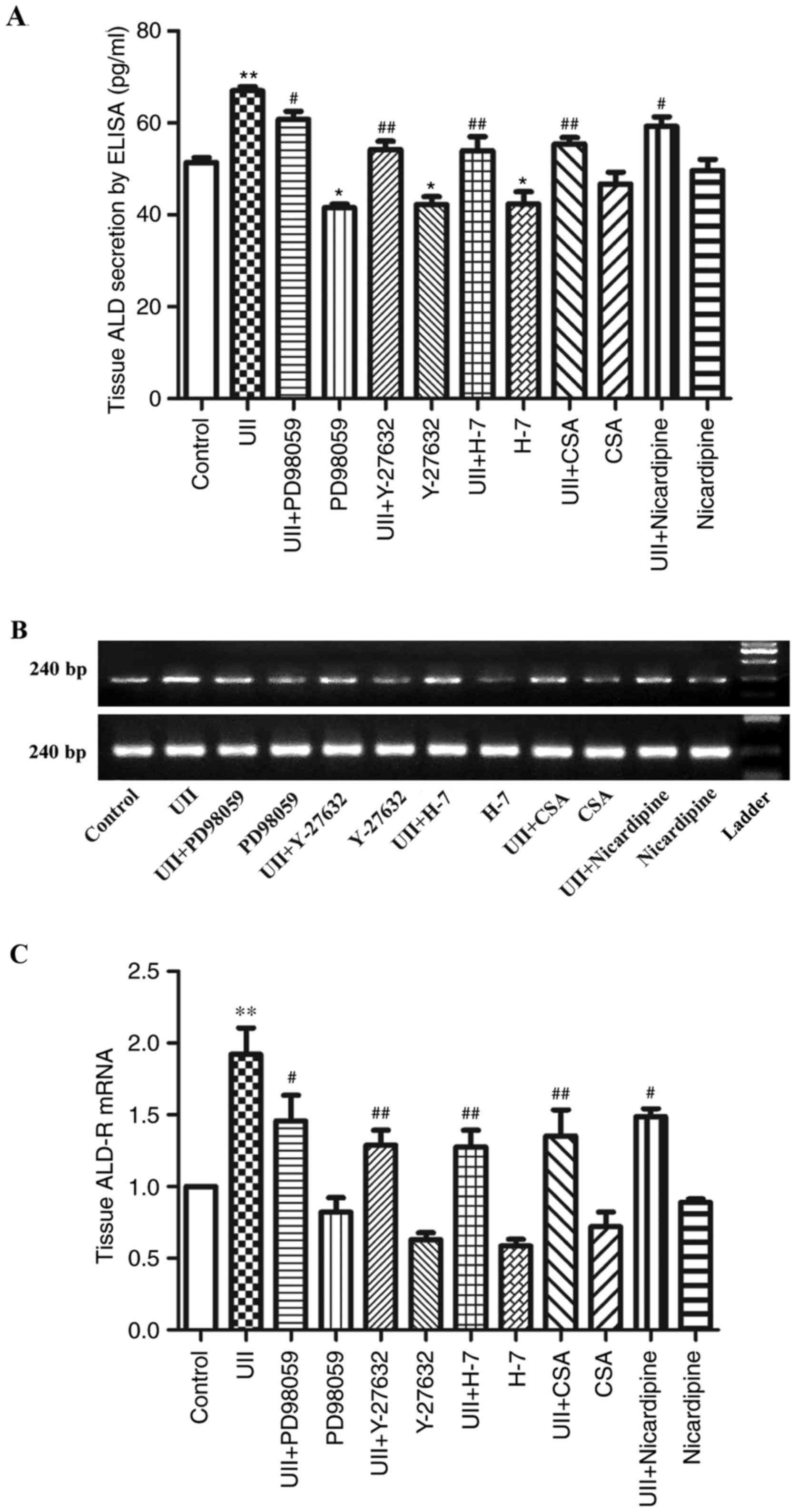
Effect of UII on ALD synthesis and
ALD-R expression in adventitia
We used the above-described fixed UII treatment
conditions to examined the effect of UII on the expression and
secretion of ALD and ALD-R in the adventitia and on the synthesis
of vasoactive substances in the adventitia, as well as on the
signaling pathways associated with these processes. After UII
treatment, the adventitia showed intense ALD and ALD-R
secretion.
After cotreatment with UII and the various
inhibitors for 6 h, the ELISA results showed that ALD secretion was
inhibited to varying degrees (Fig.
6), suggesting that MAPK, Rho, PKC, calcineurin and
Ca2+ signaling may be involved in UII-induced ALD and
ALD-R expression.
Discussion
ALD is an important hormone in the
renin-angiotensin-ALD system (RAAS) and acts in the distal renal
tubule. Angiotensin II and potassium primarily stimulate ALD
release. ALD binds to the MR and, in conjunction with various
transcription factors, induces the production of proteins involved
in sodium reabsorption and potassium excretion. The serum levels of
ALD the levels of glucocorticoids are particularly important in
this process.
Increasing evidence has shown that ALD plays a
pivotal role in the vascular inflammatory response by increasing
the expression of adhesion molecules, cytokines and chemokines as
well as growth factors. These molecules in turn promote the
recruitment and adhesion of inflammatory cells, inducing the
initiation and progression of many cardiovascular diseases,
including hypertension, atherosclerosis and restenosis. Blockade of
the RAAS is the most effective strategy to prevent renal disease
progression, heart failure, and cardiovascular death. ALD
administration causes perivascular inflammation and fibrosis, and
an MR antagonist reduced atherosclerotic lesions in different
models of atherosclerosis, supporting this strategy (25–27).
Additionally, this reduction in atherosclerosis progression was
accompanied by a reduction in inflammatory markers (26,27).
Some of these beneficial effects can be attributed to either ALD
reduction or MR antagonism, both of which provide advantages beyond
controlling hypertension, such as improvements in glucose
metabolism, fibrosis, and inflammation. Effector molecules whose
downstream activities can be affected by ALD include angiotensin
II, endothelin, growth factors, plasminogen activator inhibitor-1
(PAI-1), and oxidative stress (28). MR is also expressed in VSMCs and
endothelium in the vasculature and contributes to vascular function
and remodeling (29).
The major pathogenesis of cardiovascular disease is
vascular inflammation, which is observed throughout the whole
process of initiation, progression and complications of
cardiovascular disease. Atherosclerosis is a cardiovascular disease
characterized by a chronic low-grade inflammatory process (30). Numerous studies have demonstrated
that ALD disorder is associated with a proinflammatory vascular
response. Rocha et al found that in coronary arteries, a
short-term infusion of ALD in uninephrectomized rats fed a
high-salt diet elicited infiltration of inflammatory cells in these
arteries, which was associated with necrotic lesions and ischemia
of the adjacent myocardium. This cell infiltration was preceded by
an increase in proinflammatory mediators, such as MCP-1, ICAM-1,
COX-2 and cytokines (31,32).
Vascular remodeling is critical in cardiovascular
physiology and has the greatest potential for useful translation
from basic research to clinical applications. The adventitia plays
a key role in vascular remodeling, as recent studies have
demonstrated that the adventitia is the initiation site for this
remodeling. Following injury and stimulation, AFs are activated and
differentiate into myofibroblasts, which show contractile
properties as well as significant proliferative and synthetic
activities. These myofibroblasts migrate to the intima and
contribute to neointima formation. AF proliferation, migration,
differentiation and collagen synthesis are involved in vascular
remodeling (19); however, the
mechanisms underlying vascular remodeling in the adventitia are
still poorly understood.
A limited number of studies have examined the
effects of UII on ALD expression in AFs and its role in vascular
remodeling. Although ALD was shown to be critical in inducing
vascular remodeling in vivo, the role of ALD in UII-induced
vascular remodeling remains unclear, and little is known about
whether AFs are a novel source of ALD. As vascular remodeling is a
complex process in vivo, we performed an in vitro
experiment to elucidate the role of ALD in UII-induced vascular
remodeling.
Previously, little was known regarding ALD
expression in AFs. In the present study, we showed for the first
time that both ALD and its receptors were expressed on tunica
adventitia of vessels, and UII promotes ALD secretion and mRNA
expression of its receptor in AFs in a time- and dose-dependent
manner, suggesting that UII may stimulate ALD expression.
In the past, we have confirmed that UII induces AFs
phenotypic transformation (Fig. 7)
and Secretion of inflammatory factors via the UT receptor followed
by various intracellular signal transduction mechanisms, such as Gq
protein, protein tyrosine kinases of ERK and PKC, and the
RhoA/ROCK-related pathways (19,21).
In the present study, we found that the effects of UII upregulared
ALD expression could be blocked by inhibitors targeting Rho kinase,
PKC, MAPK, calcineurin, and Ca2+ channels. Therefore,
activation of these signaling pathways may be involved in
UII-induced ALD expression.
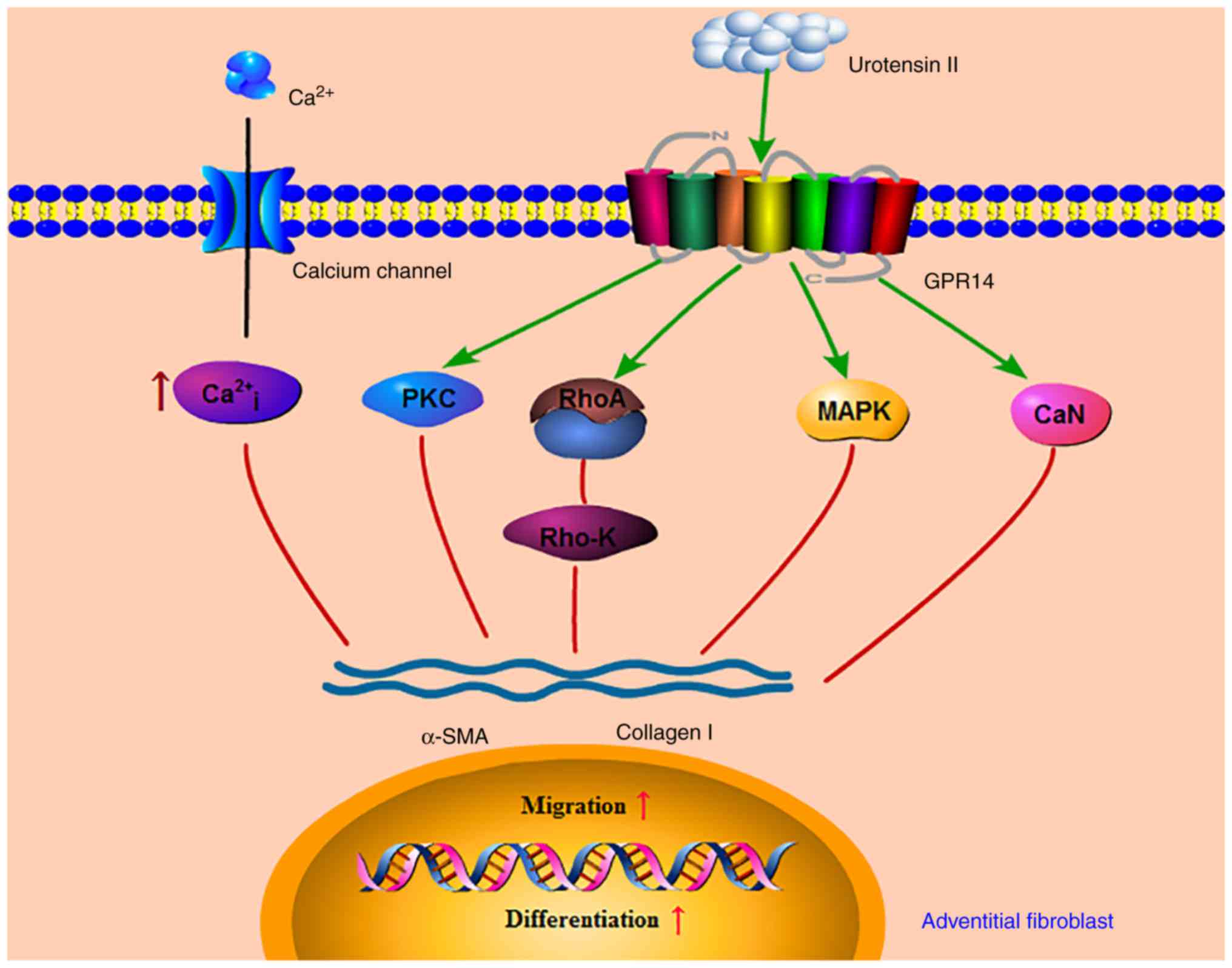 | Figure 7.Mechanism of UII promoting phenotypic
transformation of AFs. UII may stimulate AFs phenotypic conversion,
migration, and collagen I synthesis via the PKC, MAPK, calcineurin,
Rho kinase and Ca2+ signal transduction pathways,
contributing to the development of vascular remodeling through AFs
activation. UII, urotensin II; PKC, protein kinase C; CSA,
cyclosporine A; MAPK, mitogen-activated protein kinase; CaN,
calcineurin; Rho-K, Rho protein kinase; α-SMA, α-smooth muscle
actin. |
We previously showed that UII can activate AFs in an
autocrine/paracrine manner (4,20).
Others researchers have demonstrated that UII stimulates monocyte
chemotaxis (33); promotes foam
cell formation in human monocyte-derived macrophages (5); induces inflammatory activation of
endothelial cells by promoting expression of the proinflammatory
cytokines IL-1β and IL-6, the adhesion molecule VCAM-1, and tissue
factors in endothelial cells; and promotes the adhesion of
monocytes to endothelial cells (9). Here, we found that UII may directly
induce ALD expression in AFs, possibly in an autocrine/paracrine
manner. ALD may mediate the effects of UII in adventitial
inflammatory activation, which is a new mechanism through which UII
accelerates vascular remodeling, such as that observed in
atherosclerosis.
In conclusion, the results of this study indicated
for the first time that UII can stimulate the secretion of ALD in
cultured AFs in a time- and dose-dependent manner. In addition,
UII-induced secretion of ALD was significantly blocked by the MAPK
kinase inhibitor PD98059, the Rho protein kinase inhibitor Y-27632,
the PKC inhibitor H-7, the calcineurin inhibitor CSA and the
Ca2+ channel blocker nicardipine. These findings
elucidate the mechanisms responsible for the progression of
cardiovascular disease, such as atherosclerosis, vascular
restenosis, and hypertension associated with UII. Moreover, these
results may contribute to broaden our understanding of the novel
effects of UII and may provide new insights into the mechanism
underlying adventitial inflammation. Finally, inhibiting these
pathways may be a novel therapeutic approach for the treatment of
vascular inflammation in cardiovascular disease.
Acknowledgements
This study was supported by grants from the Natural
Science Foundation of Hubei Province (no. 2011CDC049) and the Taihe
Hospital Foster Fund for the National Natural Science Foundation of
China (no. 2014PY03). The experiments were further supported by the
Institute of Basic Medical Research, Hubei University of Medicine,
Clinical Laboratory of Shiyan Taihe Hospital and Institute of Life
Sciences of Shiyan Taihe Hospital. We are grateful to Ms. Han-Qin
Wang (Institute of Basic Medical Research, Hubei University of
Medicine), Mr. Zong-Tao Yu and Ji-Cai Zhang (Clinical Laboratory of
Shiyan TaiHe Hospital) for their helpful suggestions.
Glossary
Abbreviations
Abbreviations:
|
ALD
|
aldosterone
|
|
ALD-R
|
aldosterone receptor
|
|
AFs
|
adventitial fibroblasts
|
|
ET-1
|
endothelin-1
|
|
FBS
|
fetal bovine serum
|
|
IL-6
|
interleukin-6
|
|
MRs
|
mineralocorticoid receptors
|
|
MCP-1
|
monocyte chemoattractant protein 1
|
|
α-SMA
|
α-smooth muscle actin
|
|
UII
|
urotensin II
|
|
VSMCs
|
vascular smooth muscle cells
|
References
|
1
|
Ames RS, Sarau HM, Chambers JK, Willette
RN, Aiyar NV, Romanic AM, Louden CS, Foley JJ, Sauermelch CF,
Coatney RW, et al: Human urotensin-II is a potent vasoconstrictor
and agonist for the orphan receptor GPR14. Nature. 401:282–286.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ross B, McKendy K and Giaid A: Role of
urotensin II in health and disease. Am J Physiol Regul Integr Comp
Physiol. 298:R1156–R1172. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Iglewski M and Grant SR: Urotensin
II-induced signaling involved in proliferation of vascular smooth
muscle cells. Vasc Health Risk Manag. 6:723–734. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang Y, Li Y, Wei R, Wang Z, Bu D, Zhao
J, Pang Y and Tang C: Urotensin II is an autocrine/paracrine growth
factor for aortic adventitia of rat. Regul Pept. 151:88–94. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Watanabe T, Suguro T, Kanome T, Sakamoto
Y, Kodate S, Hagiwara T, Hongo S, Hirano T, Adachi M and Miyazaki
A: Human urotensin II accelerates foam cell formation in human
monocyte-derived macrophages. Hypertension. 46:738–744. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsoukas P, Kane E and Giaid A: Potential
clinical implications of the urotensin II receptor antagonists.
Front Pharmacol. 2:382011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Watson AM, Olukman M, Koulis C, Tu Y,
Samijono D, Yuen D, Lee C, Behm DJ, Cooper ME, Jandeleit-Dahm KA,
et al: Urotensin II receptor antagonism confers vasoprotective
effects in diabetes associated atherosclerosis: Studies in humans
and in a mouse model of diabetes. Diabetologia. 56:1155–1165. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hassan GS, Douglas SA, Ohlstein EH and
Giaid A: Expression of urotensin-II in human coronary
atherosclerosis. Peptides. 26:2464–2472. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Park SL, Lee BK, Kim YA, Lee BH and Jung
YS: Inhibitory effect of an urotensin II receptor antagonist on
proinflammatory activation induced by urotensin II in human
vascular endothelial cells. Biomol Ther (Seoul). 21:277–283. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Enzerink A and Vaheri A: Fibroblast
activation in vascular inflammation. J Thromb Haemost. 9:619–626.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li AC and Glass CK: The macrophage foam
cell as a target for therapeutic intervention. Nat Med.
8:1235–1242. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maiellaro K and Taylor WR: The role of the
adventitia in vascular inflammation. Cardiovasc Res. 75:640–648.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang W, Yang JH, Pan CS, Qi YF, Pang YZ
and Tang CS: Effects of adrenomedullin on cell proliferation in rat
adventitia induced by aldosterone. J Hypertens. 22:1953–1961. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu F, Ji J, Li L, Chen R and Hu W:
Activation of adventitial fibroblasts contributes to the early
development of atherosclerosis: A novel hypothesis that complements
the ‘Response-to-Injury Hypothesis’ and the ‘Inflammation
Hypothesis’. Med Hypotheses. 69:908–912. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stenmark KR, Davie N, Frid M,
Gerasimovskaya E and Das M: Role of the adventitia in pulmonary
vascular remodeling. Physiology (Bethesda). 21:134–145. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sartore S, Chiavegato A, Faggin E, Franch
R, Puato M, Ausoni S and Pauletto P: Contribution of adventitial
fibroblasts to neointima formation and vascular remodeling: From
innocent bystander to active participant. Circ Res. 89:1111–1121.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gilbert KC and Brown NJ: Aldosterone and
inflammation. Curr Opin Endocrinol Diabetes Obes. 17:199–204. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xanthakis V and Vasan RS: Aldosterone and
the risk of hypertension. Curr Hypertens Rep. 15:102–107. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang YG, Li J, Li YG and Wei RH:
Urotensin II induces phenotypic differentiation, migration, and
collagen synthesis of adventitial fibroblasts from rat aorta. J
Hypertens. 26:1119–1126. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang YG, Hu YC, Mao YY, Wei RH, Bao SL,
Wu LB and Kuang ZJ: Transforming growth factor-β1 involved in
urotensin II-induced phenotypic differentiation of adventitial
fibroblasts from rat aorta. Chin Med J (Engl). 123:3634–3639.
2010.PubMed/NCBI
|
|
21
|
Dong X, Ye X, Song N, Zhao J, Di B, Peng
F, Tang C and Ding W: Urotensin II promotes the production of LTC4
in rat aortic adventitial fibroblasts through NF-κB-5-LO pathway by
p38 MAPK and ERK activations. Heart Vessels. 28:514–523. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kelm M: Nitric oxide metabolism and
breakdown. Biochim Biophys Acta. 1411:273–289. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim DK, Huh JE, Lee SH, Hong KP, Park JE,
Seo JD and Lee WR: Angiotensin II stimulates proliferation of
adventitial fibroblasts cultured from rat aortic explants. J Korean
Med Sci. 14:487–496. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tsuruda T, Kato J, Cao YN, Hatakeyama K,
Masuyama H, Imamura T, Kitamura K, Asada Y and Eto T:
Adrenomedullin induces matrix metalloproteinase-2 activity in rat
aortic adventitial fibroblasts. Biochem Biophys Res Commun.
325:80–84. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rajagopalan S, Duquaine D, King S, Pitt B
and Patel P: Mineralocorticoid receptor antagonism in experimental
atherosclerosis. Circulation. 105:2212–2216. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Takai S, Jin D, Muramatsu M, Kirimura K,
Sakonjo H and Miyazaki M: Eplerenone inhibits atherosclerosis in
nonhuman primates. Hypertension. 46:1135–1139. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Suzuki J, Iwai M, Mogi M, Oshita A, Yoshii
T, Higaki J and Horiuchi M: Eplerenone with valsartan effectively
reduces atherosclerotic lesion by attenuation of oxidative stress
and inflammation. Arterioscler Thromb Vasc Biol. 26:917–921. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Epstein M: Aldosterone and the
hypertensive kidney: Its emerging role as a mediator of progressive
renal dysfunction: A paradigm shift. J Hypertens. 19:829–842. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ehrhart-Bornstein M, Lamounier-Zepter V,
Schraven A, Langenbach J, Willenberg HS, Barthel A, Hauner H,
McCann SM, Scherbaum WA and Bornstein SR: Human adipocytes secrete
mineralocorticoid-releasing factors. Proc Natl Acad Sci USA.
100:pp. 14211–14216. 2003; View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Libby P, Ridker PM and Maseri A:
Inflammation and atherosclerosis. Circulation. 105:1135–1143. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Joffe HV and Adler GK: Effect of
aldosterone and mineralocorticoid receptor blockade on vascular
inflammation. Heart Fail Rev. 10:31–37. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rocha R, Martin-Berger CL, Yang P,
Scherrer R, Delyani J and McMahon E: Selective aldosterone blockade
prevents angiotensin II/salt-induced vascular inflammation in the
rat heart. Endocrinology. 143:4828–4836. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Segain JP, Rolli-Derkinderen M, Gervois N,
Raingeard de la Blétière D, Loirand G and Pacaud P: Urotensin II is
a new chemotactic factor for UT receptor-expressing monocytes. J
Immunol. 179:901–909. 2007. View Article : Google Scholar : PubMed/NCBI
|