Introduction
Ischemia-reperfusion (IR) injury occurs when blood
flow to an organ is disrupted and subsequently re-introduced, and
it is associated with a variety of diseases, including myocardial
infarction, shock liver, and acute ischemic renal failure (1–3).
Ischemic and reperfusion conditions lead to ATP depletion,
accumulation of toxic metabolites, such as reactive oxygen species
(ROS), and initiation of apoptotic and necrotic cascades in the
affected organs (4,5). The molecular mechanism by which cells
and tissues adapt to IR involves the upregulation of cytoprotective
genes that maintain cellular homeostasis. For instance, expression
of the anti-apoptotic protein Bax inhibitor-1 (BI-1) is induced
during IR injury, and bi-1−/− mice exhibit increased sensitivity to
renal IR injury, suggesting that bi-1 gene induction is needed to
protect tissues (5).
Cyclin dependent kinase inhibitor 1A (termed p21) is
important in cell cycle regulation (6). Previously, Megyesi et al
(7) have demonstrated that
expression of p21 is significantly upregulated following renal
ischemia. In p21−/− mice, renal function is more impaired and
overall mortality is significantly increased compared with
wild-type mice following renal ischemia, suggesting a protective
role of p21 in ischemic acute renal failure. p21 has also been
demonstrated to be involved in recovery from hepatic IR injury
(8). However, the molecular
mechanisms by which p21 exerts its protective effect on IR injury
are not fully understood.
Oxidative stress is a major causes of cell death
during IR injury (1). p21 protects
cells against oxidative stress by upregulating the ER-resident
chaperone heat shock protein family A (HSPA5, also known as BIP and
GRP78) and suppressing endoplasmic reticulum stress (9). Chen et al (10) reported that the antioxidant
function of p21 is mediated through stabilizing NF-E2-related
factor-2 (Nrf2), a transcription factor that regulates expression
of various detoxification and antioxidant enzymes (11). Given the importance of oxidative
stress in IR injury, the present study aimed to investigate whether
p21 protects cardiomyocytes against IR injury by suppressing
oxidative stress.
Materials and methods
Cell culture
Rat H9c2 cardiomyocytes (provided by Dr Yuanli Chen,
Faculty of Basic Medicine, Kunming Medical University, Kumming,
China) were cultured in DMEM (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) with 10% fetal bovine serum (Invitrogen;
Thermo Fisher Scientific, Inc.) and maintained in a humidified, 5%
CO2/95% air incubator at 37°C.
Oxygen-glucose
deprivation/reoxygenation (OGD/R)
Cells were subjected to OGD/R as previously
described (12), with minor
modifications. When cells reached 60–80% confluency, they were
washed with PBS and cultured in glucose-free, serum-free DMEM
medium in an anoxia chamber (94% N2/5% CO2/1%
O2) for 4 h, then cultured in high glucose (4,500 mg/l)
DMEM medium containing 10% FBS in a normal cell incubator (95%
air/5% CO2) for an additional 12 h. To test the role of
p53 in p21 expression, cells were pre-incubated with 30 µM p53
inhibitor pifithrin-α (Sigma-Aldrich; Merck Millipore, Darmstadt,
Germany) for 30 min.
Cell viability assay
Cell survival was measured using the
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
inner salt (MTS) assay (Promega Corporation, Madison, WI, USA).
Briefly, 1×105 H9c2 cells/well were seeded into 96-well
plates at 37°C overnight. Following OGD/R, 20 µl MTS solution was
added in each well and cells were incubated for a further 2 h at
37°C. Absorbance at 490 nm was measured using a microplate reader
(Molecular Devices, LLC, Sunnyvale, CA, USA).
Measurement of ROS
ROS levels in cells were measured using
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA;
Thermo Fisher Scientific, Inc.), as per the manufacturer's
instructions. Briefly, H9c2 cells were seeded at a density of
5×104 into a 96-well plate. Following OGD/R treatment,
10 µM H2DCFDA were added and cells were incubated for a
further 1 h at 37°C. Dichlorofluorescein fluorescence intensity was
measured by using a Spectra Max M5 fluorescent microplate reader
(Molecular Devices, LLC).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated using TRIzol reagent (Thermo
Fisher Scientific, Inc.). Random-primed cDNAs were generated by
reverse transcription of total RNA samples with SuperScript II, as
per the manufacturer's instructions (Thermo Fisher Scientific,
Inc.). qPCR was performed using the SYBR Premix Ex Taq kit (Takara
Biotechnology, Co., Ltd., Dalian, China) on a LightCycler 480
system (Roche Applied Science, Penzberg, Germany), as per the
manufacturer's instructions. The cycle number at which the reaction
crossed an arbitrarily placed threshold (CT) was determined for
each gene, and the relative amount of each mRNA to mRNA of β-actin
was described using the 2−∆∆Cq method (13). The primers used for qPCR were as
follows: p21 forward, 5′-TCTGCTGCTCTCCCTTCCT-3′ and reverse,
5′-CACCACCACCACATACCAC-3′; tumor protein p53 (p53) forward,
5′-CCCATCCTTACCATCATCACG-3′ and reverse, 5′-CAGGCACAAACACGAACCT-3′;
Nrf2 forward, 5′-CCCAGCACATCCAGACAGAC-3′ and reverse
5′-TCCAGGGCAAGCGACTCAT-3′; NAD (P)H quinone oxidoreductase 1 (NQO1)
forward, 5′-GAAGAAAGGATGGGAGGTGGT-3′ and reverse,
5′-GGTGGTGATGGAAAGCAAGG-3′; heme oxygenase-1 (HO-1) forward,
5′-GCGAAACAAGCAGAACCCA-3′ and reverse, 5′-GGCTGGTGTGTAAGGGATGG-3′;
β-actin forward, 5′-GATGGTCTTGGTTTCTGTGCC-3′ and reverse,
5′-TGCTGTTTCCGCCTTCTGG-3′.
Western blotting
Cells were lysed on ice for 30 min in lysis buffer
(BioTeke Corporation, Beijing, China). The lysates were centrifuged
at 14,000 × g for 15 min at 4°C, and the supernatants were
collected. The protein concentration was determined by a Bradford
protein assay kit (BioTeke Corporation). Total cell lysates (30 µg)
were separated on a 10% SDS-polyacrylamide gel. Proteins were then
transferred to polyvinylidene fluoride membranes. Membranes were
blocked for non-specific binding with 5% bovine serum albumin in
Tris phosphate buffered saline (TPBS) at room temperature for 30
min, following which they were incubated overnight at 4°C with
primary antibodies. Primary antibodies were as follows:
Anti-β-actin (cat. no. A2066; 1:1,000 dilution; Sigma-Aldrich;
Merck Millipore), anti-p21 (cat. no. sc-817; 1:500 dilution; Santa
Cruz Biotechnology, Inc. Dallas, TX, USA), anti-p53 (cat. no.
sc-126; 1:1,000 dilution; Santa Cruz Biotechnology, Inc.) and
anti-Nrf2 (cat. no. sc-722; 1:500 dilution; Santa Cruz
Biotechnology, Inc.). Membranes were subsequently washed with TPBS
three times, and incubated with horseradish peroxidase-conjugated
anti-rabbit or -mouse immunoglobin G secondary antibodies [cat.
nos. M21002 and H20704 respectively; 1:5,000 dilution; Abmart
(Shanghai) Co., Ltd., Shanghai, China] for 1 h at room temperature.
Following three washes with TPBS, membranes were exposed to
Amersham Hyperfilm Enhanced Chemiluminescence (GE Healthcare
Bio-Sciences, Pittsburgh, PA, USA). Following this, membranes were
exposed to Kodak X-OMAT film (Kodak, Rochester, NY, USA) and the
films developed.
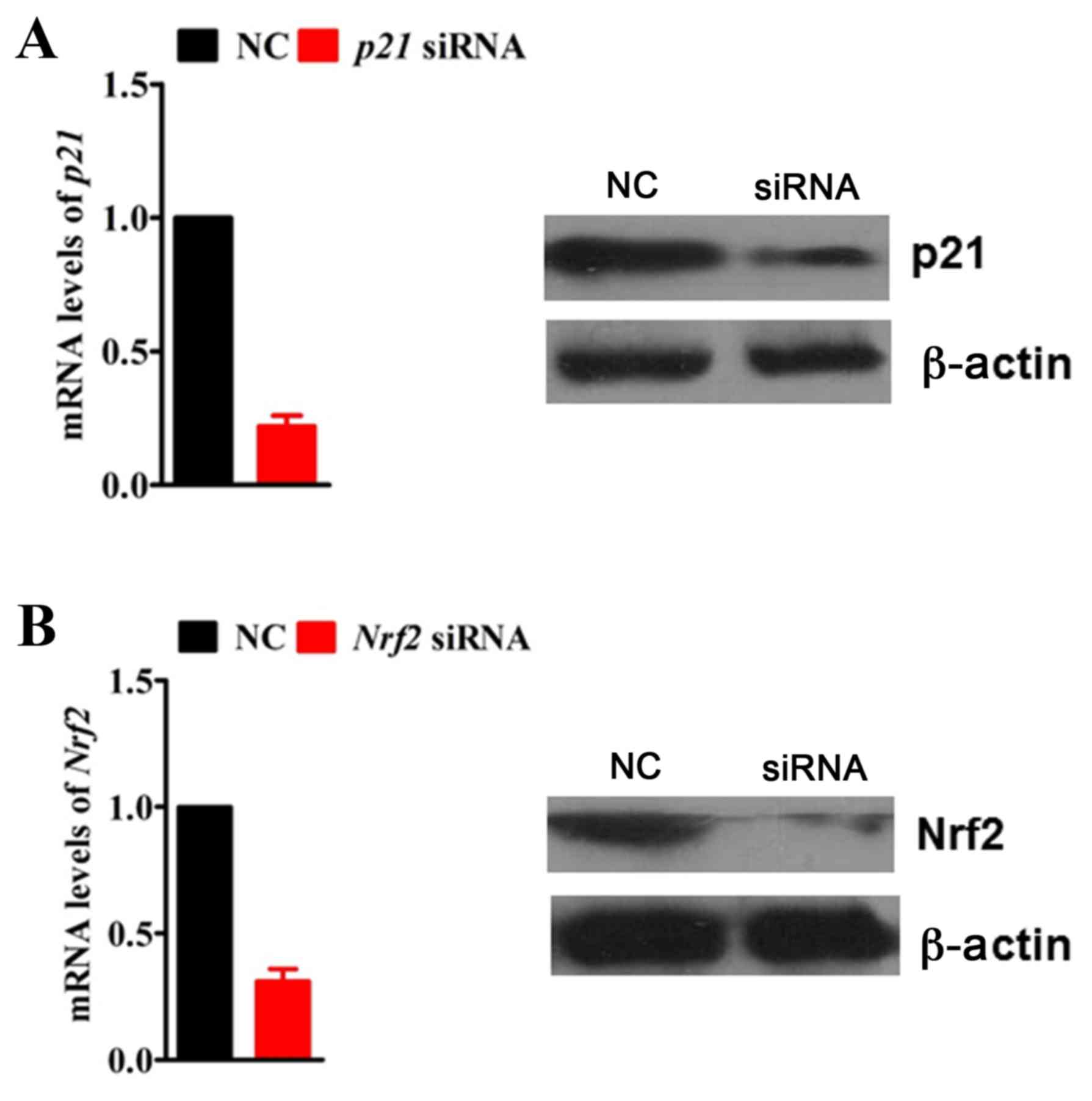
RNA interference
To knockdown the expression of p21 and Nrf2 by RNA
interference, H9c2 cells were transfected at 50% confluence with
100 nM of p21 or Nrf2-specific small interfering RNA (siRNA;
GenePharma Co., Ltd., Shanghai, China) in Opti-MEM medium (Thermo
Fisher Scientific, Inc.) using Lipofectamine 2000 transfection
agent (Thermo Fisher Scientific, Inc.), as per the manufacturer's
instructions. Gene silencing efficiency was determined by RT-qPCR
and western blotting 72 h post-transfection. A non-specific siRNA
sequence was used as a negative control. Endogenous p21 and Nrf2
mRNA and protein expression levels were significantly ablated by
siRNA in H9c2 cells compared with the negative control siRNA
(Fig. 1). siRNA sequences were as
follows (sense strand): p21, 5′-UCCAAUUCCCCUUAACUCGGG-3′; Nrf2,
5′-UUGUUUUCCGUAUUAAGACA-3′, and negative control
5′-UUCUCCGAACGUGUCACGUTT-3′.
Statistical analysis
Data were expressed as the mean ± standard
deviation. Statistical differences between groups were analyzed
using one-way analysis of variance, followed by a
Student-Newman-Keuls test. P<0.05 was considered to indicate a
statistically significant difference.
Results
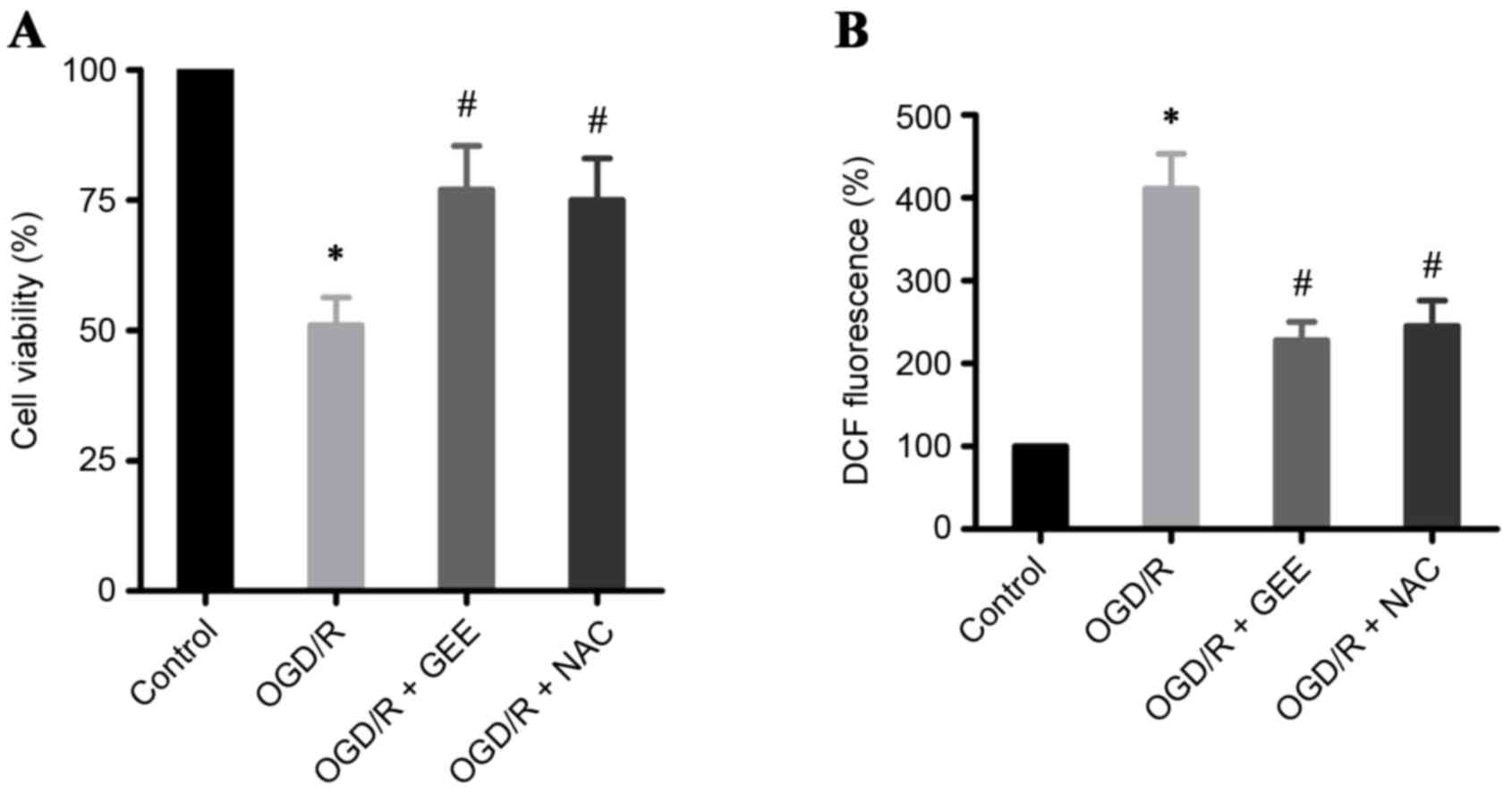
OGD/R induces cell death through
ROS
In the present study, OGD/R in H9c2 heart-derived
myocytes was used as an in vitro model to study IR. A
significant increase in cell death was detected by MTS assay in
OGD/R-treated H9c2 cells compared with untreated cells (Fig. 2A). Oxidative stress has been
demonstrated to be important in acute myocardial infarction
(1,14). Therefore, ROS levels were assessed
in OGD/R-treated H9c2 cells using an H2DCF-DA probe. The
results demonstrated that OGD/R induced a 4-fold increase in ROS
levels in H9c2 cells, compared with the untreated cells (Fig. 2B). To investigate whether cell
death in OGD/R-treated cells was mediated by ROS, the experiment
was repeated in the presence of two antioxidants, glutathione ethyl
ester (GEE) and N-acetylcysteine (NAC). Both GEE and NAC
significantly reduced cell death ROS production in OGD/R-treated
H9c2 cells, compared with cells that received no antioxidant
treatment (Fig. 2A and B,
respectively). The present results further confirm that ROS is
important in OGD/R-induced cell death in cardiomyocytes.
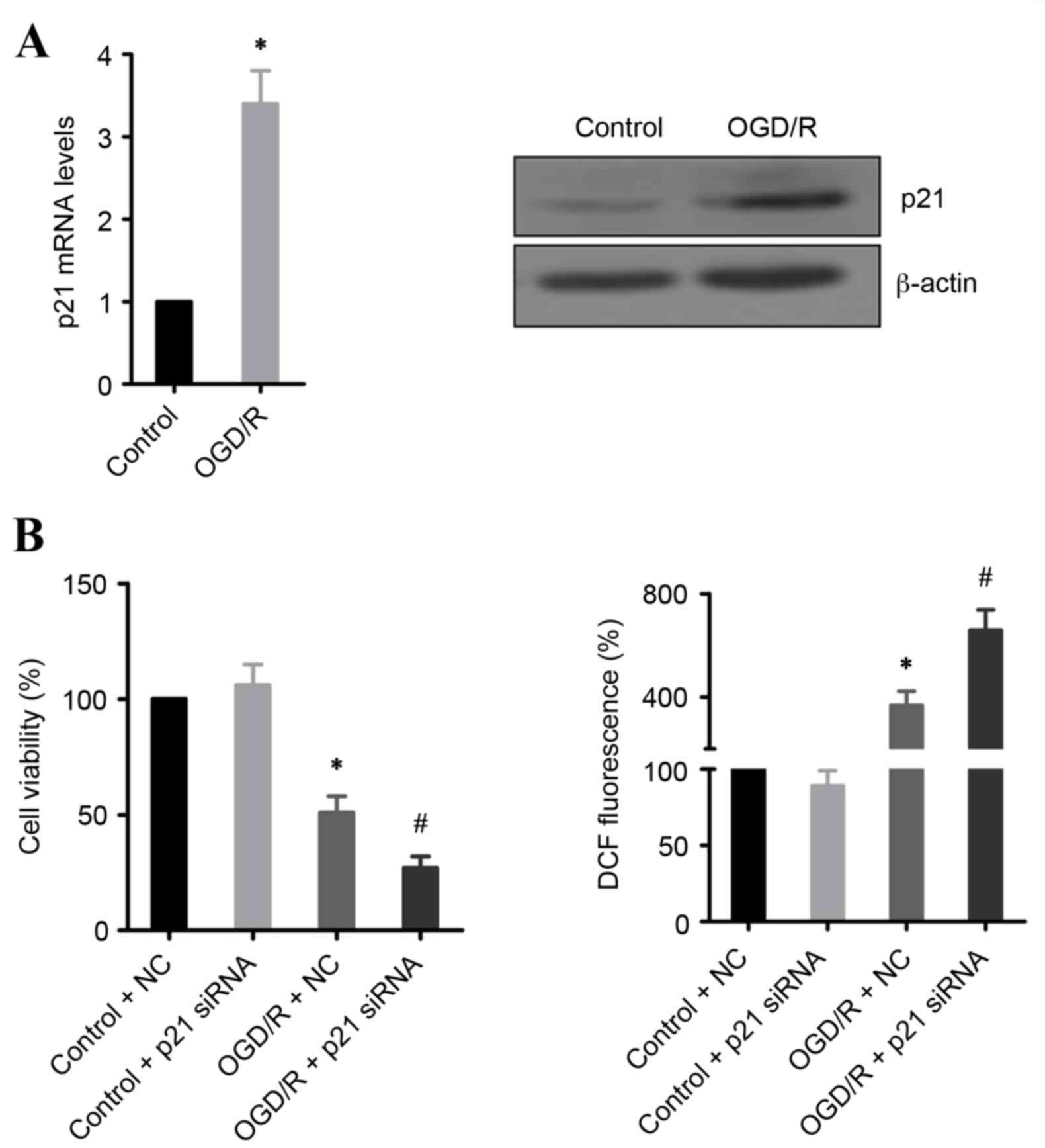
p21 prevents OGD/R-induced cell
death
p21 expression is often upregulated during IR injury
(8,15). In the present study, mRNA and
protein expression levels of p21 were demonstrated to be
significantly increased in OGD/R-treated H9c2 cells, compared with
untreated cells (Fig. 3A). To
address whether this induction of p21 expression is required for
cell survival, p21 expression was silenced using siRNA. The results
demonstrated that knockdown of p21 significantly promoted cell
death and increased ROS production in OGD/R-treated H9c2 cells,
compared with OGD/R-treated cells transfected with control siRNA
(Fig. 3B).
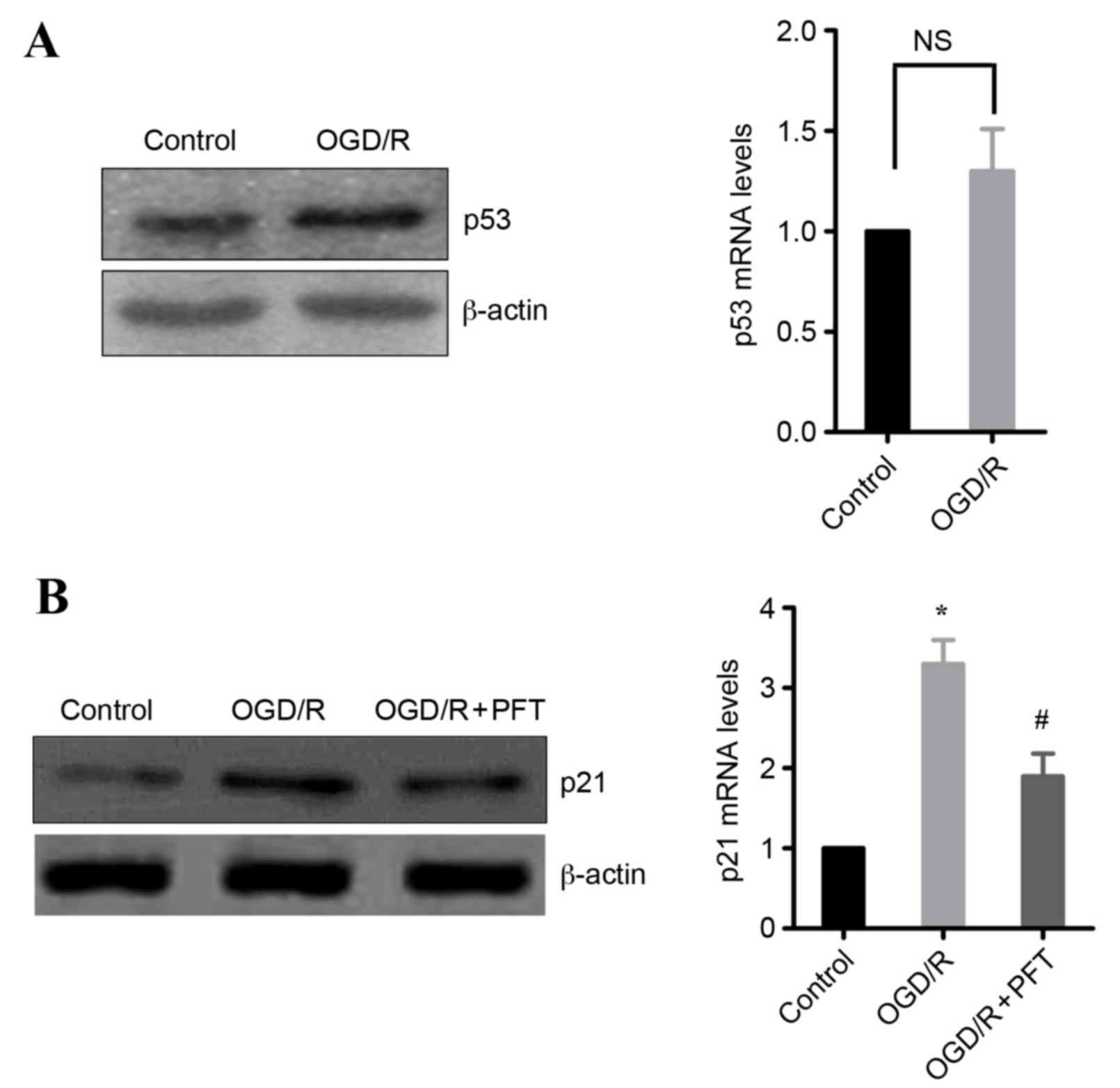
Upregulation of p21 is mediated by
p53
As p21 is a target gene of p53, whether induction of
p21 in OGD/R-treated cells may be dependent on p53 was
investigated. Initially, the effect of OGD/R treatment on p53
expression was examined. The results demonstrated that OGD/R
treatment in H9c2 cells markedly increased the protein expression
levels of p53, whereas the mRNA expression levels were not
significantly different from untreated cells (Fig. 4A). Subsequently, the effect of the
p53 inhibitor pifithrin-α (16) on
p21 expression was examined. The results demonstrated that p53
inhibition significantly decreased the OGD/R-induced expression of
p21 in H9c2 cells, at the protein and the mRNA level (Fig. 4B). These data indicate that p53 is
involved in the upregulation of p21 following OGD/R.
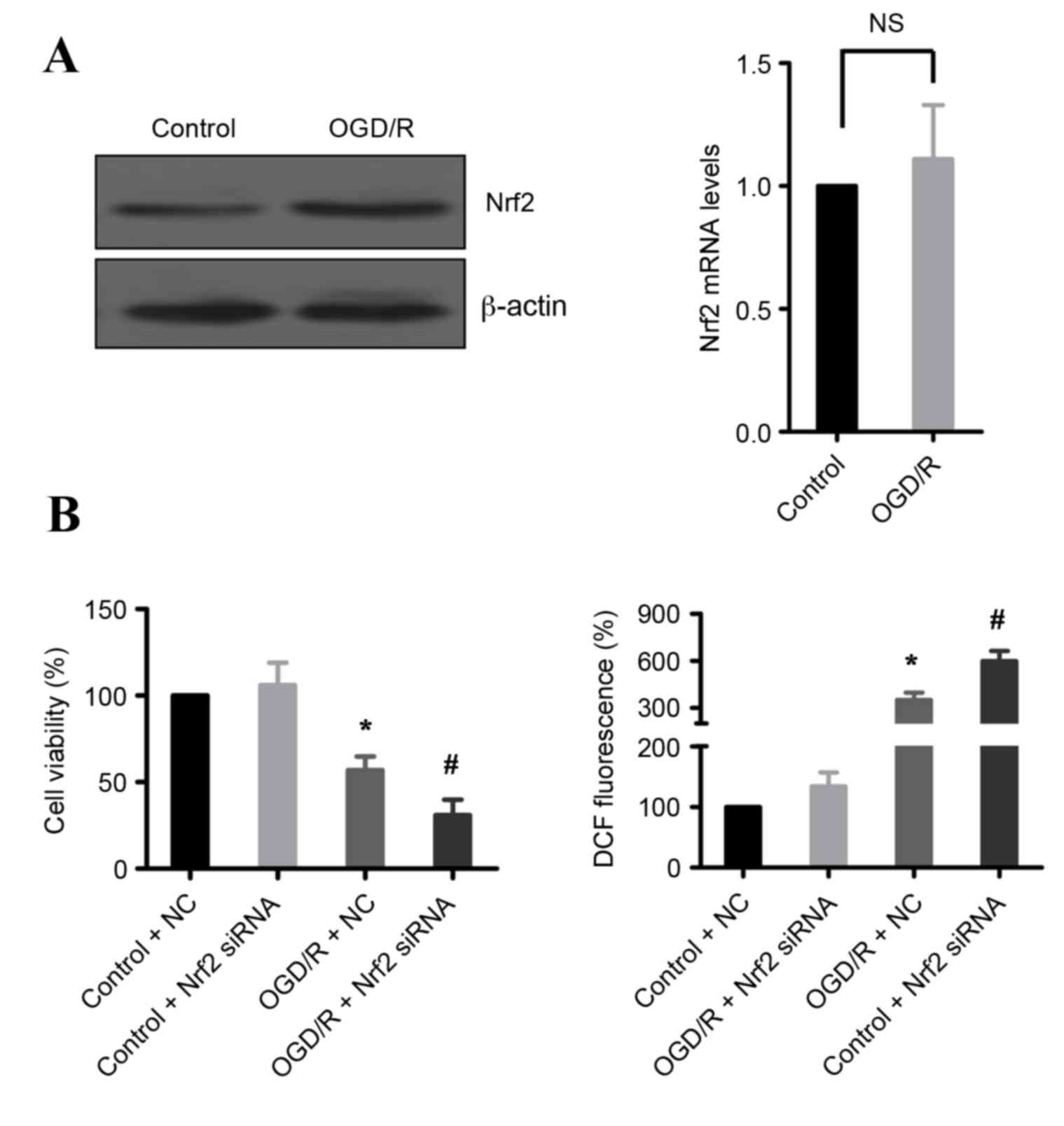
Nrf2 is required for survival
following OGD/R
Transcription factor Nrf2 is a regulator of cell
antioxidant responses, and it is elevated in IR injury (4). Therefore, expression of Nrf2 was
examined following OGD/R treatment in H9c2 cells. As demonstrated
in Fig. 5A, OGD/R treatment
increased Nrf2 protein expression levels, but the mRNA levels were
not changed, compared with untreated cells. These data suggest that
Nrf2 may be important in the cellular defense against OGD/R-induced
cell death. Indeed, knockdown of Nrf2 by siRNA significantly
reduced cell death and increased ROS production in OGD/R-treated
H9c2 cells compared with OGD/R-treated cells transfected with
control siRNA (Fig. 5B).
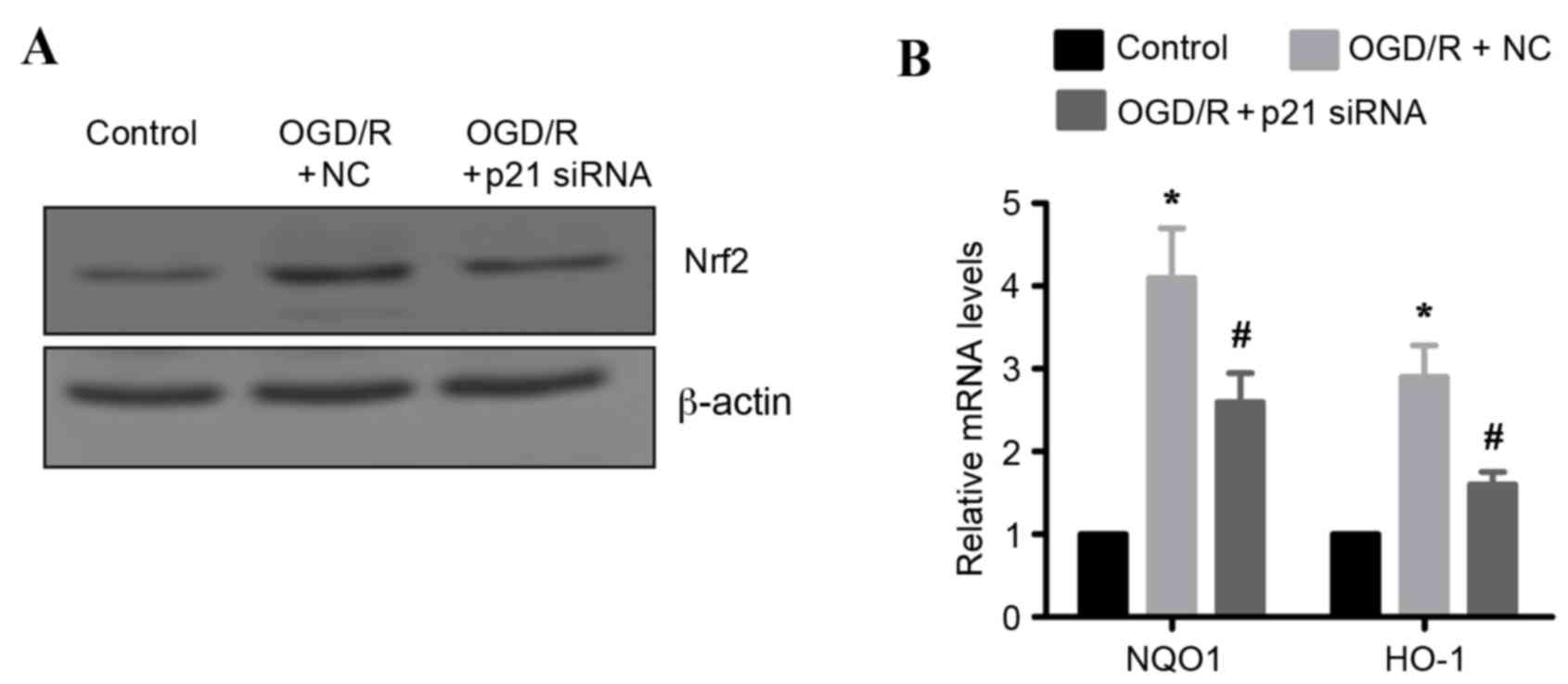
OGD/R induced Nrf2-regulated
antioxidant gene expression via p21
Previous studies have demonstrated that p21 may
stabilize Nrf2 by inhibiting its degradation (10), raising the hypothesis that
OGD/R-induced Nrf2 protein upregulation may be mediated by p21. To
test this hypothesis, endogenous p21 expression was silenced by
siRNA in H9c2 cells. Knockdown of p21 resulted in markedly reduced
protein expression levels of Nrf2 in OGD/R-treated H9c2 cells
compared with OGD/R-treated cells transfected with control siRNA
(Fig. 6A). In addition, the mRNA
expression levels of two known oxidative stress markers, and gene
targets of Nrf2 (NQO1 and HO-1) were examined. As expected, the
mRNA expression levels of NQO1 and HO-1 were significantly
upregulated following OFD/R treatment in H9c2 cells compared with
untreated cells (Fig. 6B).
However, this effect was significantly attenuated in p21 silenced
OFD/R-treated cells, compared to OFD/R-treated cells transfected
with a control siRNA (Fig. 6B).
Taken together, these results indicate that p21 is important in
activating the Nrf2-mediated antioxidant response following
OGD/R.
Discussion
Myocardial apoptosis is critical in the pathogenesis
of acute myocardial infarction and heart failure (17). Oxidative stress is one of the main
causes of myocardial apoptosis following heart IR damage (14). The present study demonstrated that
the p53/p21/Nrf2 pathway protects against OGD/R-induced H9c2 cell
death by suppressing ROS production. Thus, induction of the
p53/p21/Nrf2 pathway may represent an important adaptive cell
response to limit oxidative injury during IR.
The mechanism underlying the upregulation of p21
during IR injury remains controversial. p21 expression is
significantly elevated in the liver of mice that are subjected to
hepatic IR injury, through activation of p53, a known regulator of
p21 (8). However, Yoshida et
al (15) demonstrated that the
expression of activating transcription factor 3 (ATF3) is
significantly increased in the kidney of a mouse model of IR
injury. Overexpression of ATF3 leads to upregulation of p21 and
downregulation of p53 in human kidney-2 cells subjected to
oxidative stress (15), suggesting
that p21 may be a downstream target of ATF3, rather than p53,
during IR injury. In the present study, it was demonstrated that
p53 inhibition suppressed the p21 expression, indicating that
OGD/R-induced p21 upregulation is p53-dependent. Notably, p53
protein levels, rather than mRNA levels, were upregulated in H9c2
cells following OGD/R. Thus, post-transcriptional regulation may be
a key process in the control of p53 expression. A recent study has
revealed that OGD/R significantly inhibits the phosphorylation of
AKT serine/threonine kinase 1 (Akt) in H9c2 cells (18). It is well established that Akt
phosphorylates the E3 ubiquitin ligase MDM2 proto-oncogene,
enhancing MDM2-mediated degradation of p53 by ubiquitination
(19). It is very likely therefore
that inactivation of Akt may lead to accumulation of p53 protein
following OGD/R, due to decreased MDM2-mediated degradation.
Further studies will be required to clarify this hypothesis.
The transcription factor Nrf2A is a master regulator
of phase-II detoxification enzymes and antioxidant genes through
binding with antioxidant response elements within the promoters of
these genes (20,21). Previous studies have demonstrated
that IR induces Nrf2 expression or upregulates Nrf2-dependent gene
expression in liver and kidney (4,22).
Furthermore, serum urea nitrogen levels in Nrf2-null mice are
higher than those in wild-type mice following IR (22). These data suggest that Nrf2 is a
crucial molecule for the coordinated cytoprotective response to
oxidative stress during IR. In the present study, it was
demonstrated that the upregulation of Nrf2 expression by OGD/R was
mediated by p21 in cardiomyocytes. Under basal conditions, an E3
ubiquitin ligase, kelch like ECH associated protein 1 (Keap1),
targets Nrf2 for ubiquitination-mediated degradation (23). Notably, a previous study has
demonstrated that p21 can directly interact with Nrf2 and thus
competes with Keap1 for Nrf2 binding, suppressing ubiquitination of
Nrf2 (10). Thus, the
OGD/R-induced upregulation of Nrf2 via p21 observed in the present
study, may be explained by this previously reported p21 capacity to
stabilize Nrf2 and suppress its ubiquitination and degradation.
In summary, the present study demonstrated that
upregulation of p21 following OGD/R was p53-dependent. p21
protected cardiomyocytes from IR injury by suppressing oxidative
stress, in part through Nrf2, a master regulator of antioxidant
responses. These findings provided valuable insights into the
p53/p21/Nrf2 signaling in ischemic heart disease. Further in
vivo investigations will be required in the future to fully
comprehend the role of this pathway in IR-damaged
cardiomyocytes.
Acknowledgements
We thank Dr Yuanli Chen for kindly providing the
H9c2 cells. The present study was supported in part by a grant
(grant no. 81170171) from the National Natural Science Foundation
of China (to JFY), and a grant (grant no. 2012FB034) from the Joint
fund for Yunnan Department of Science and Technology-Kunming
Medical University.
References
|
1
|
Sen CK, Khanna S and Roy S: Perceived
hyperoxia: Oxygen-induced remodeling of the reoxygenated heart.
Cardiovasc Res. 71:280–288. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schriewer JM, Peek CB, Bass J and
Schumacker PT: ROS-mediated PARP activity undermines mitochondrial
function after permeability transition pore opening during
myocardial ischemia-reperfusion. J Am Heart Assoc. 2:e0001592013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechanism to translation. Nat Med. 17:1391–1401.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leonard MO, Kieran NE, Howell K, Burne MJ,
Varadarajan R, Dhakshinamoorthy S, Porter AG, O'Farrelly C, Rabb H
and Taylor CT: Reoxygenation-specific activation of the antioxidant
transcription factor Nrf2 mediates cytoprotective gene expression
in ischemia-reperfusion injury. FASEB J. 20:2624–2626. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bailly-Maitre B, Fondevila C, Kaldas F,
Droin N, Luciano F, Ricci JE, Croxton R, Krajewska M, Zapata JM,
Kupiec-Weglinski JW, et al: Cytoprotective gene bi-1 is required
for intrinsic protection from endoplasmic reticulum stress and
ischemia-reperfusion injury. Proc Natl Acad Sci USA. 103:pp.
2809–2814. 2006; View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weinberg WC and Denning MF: P21Waf1
control of epithelial cell cycle and cell fate. Crit Rev Oral Biol
Med. 13:453–464. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Megyesi J, Andrade L, Vieira JM Jr,
Safirstein RL and Price PM: Positive effect of the induction of
p21WAF1/CIP1 on the course of ischemic acute renal failure. Kidney
Int. 60:2164–2172. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barone S, Okaya T, Rudich S, Petrovic S,
Tenrani K, Wang Z, Zahedi K, Casero RA, Lentsch AB and Soleimani M:
Distinct and sequential upregulation of genes regulating cell
growth and cell cycle progression during hepatic
ischemia-reperfusion injury. Am J Physiol Cell Physiol.
289:C826–C835. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vitiello PF, Wu YC, Staversky RJ and
O'Reilly MA: p21 (Cip1) protects against oxidative stress by
suppressing ER-dependent activation of mitochondrial death
pathways. Free Radic Biol Med. 46:33–41. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen W, Sun Z, Wang XJ, Jiang T, Huang Z,
Fang D and Zhang DD: Direct interaction between Nrf2 and p21
(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol
Cell. 34:663–673. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lau A, Villeneuve NF, Sun Z, Wong PK and
Zhang DD: Dual roles of Nrf2 in cancer. Pharmacol Res. 58:262–270.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu X, He L, Chen F, He X, Cai Y, Zhang G,
Yi Q, He M and Luo J: Impaired autophagy contributes to adverse
cardiac remodeling in acute myocardial infarction. PLoS One.
9:e1128912014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C (T))Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ide T, Tsutsui H, Kinugawa S, Utsumi H,
Kang D, Hattori N, Uchida K, Arimura Ki, Egashira K and Takeshita
A: Mitochondrial electron transport complex I is a potential source
of oxygen free radicals in the failing myocardium. Circ Res.
85:357–363. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yoshida T, Sugiura H, Mitobe M, Tsuchiya
K, Shirota S, Nishimura S, Shiohira S, Ito H, Nobori K, Gullans SR,
et al: ATF3 protects against renal ischemia-reperfusion injury. J
Am Soc Nephrol. 19:217–224. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Komarov PG, Komarova EA, Kondratov RV,
Christov-Tselkov K, Coon JS, Chernov MV and Gudkov AV: A chemical
inhibitor of p53 that protects mice from the side effects of cancer
therapy. Science. 285:1733–1737. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Abbate A, Bussani R, Amin MS, Vetrovec GW
and Baldi A: Acute myocardial infarction and heart failure: Role of
apoptosis. Int J Biochem Cell Biol. 38:1834–1840. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu F, Yu H, Liu J and Cheng L:
Pyrroloquinoline quinone inhibits oxygen/glucose
deprivation-induced apoptosis by activating the PI3K/AKT pathway in
cardiomyocytes. Mol Cell Biochem. 386:107–115. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ogawara Y, Kishishita S, Obata T, Isazawa
Y, Suzuki T, Tanaka K, Masuyama N and Gotoh Y: Akt enhances
Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem.
277:21843–21850. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jaiswal AK: Nrf2 signaling in coordinated
activation of antioxidant gene expression. Free Radic Biol Med.
36:1199–1207. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kobayashi M and Yamamoto M: Molecular
mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene
regulation. Antioxid Redox Signal. 7:385–394. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tanaka Y, Maher JM, Chen C and Klaassen
CD: Hepatic ischemia-reperfusion induces renal heme oxygenase-1 via
NF-E2-related factor 2 in rats and mice. Mol Pharmacol. 71:817–825.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kobayashi A, Kang MI, Watai Y, Tong KI,
Shibata T, Uchida K and Yamamoto M: Oxidative and electrophilic
stresses activate Nrf2 through inhibition of ubiquitination
activity of Keap1. Mol Cell Biol. 26:221–229. 2006. View Article : Google Scholar : PubMed/NCBI
|