Introduction
Ischemic stroke is an important disease worldwide
and a leading cause of mortality and long-term disability (1). Ischemic stroke often results in focal
motor weakness, sensory loss, visual damage, impaired speech
comprehension and memory disturbances (2). Despite the high incidence of the
disease, there remain only a few treatments available, namely
thrombolysis and neurosurgery. These are beneficial to only a small
proportion of patients with acute stage stroke and thus, more
widely applicable therapies are required (1–3).
In the brain, energy is stored mainly in the form of
the high energy phosphate compound, adenosine triphosphate (ATP).
The normal brain requires complete oxidation of glucose to fulfill
its energy requirements (4).
However, under ischemic conditions, oxygen depletion forces the
brain to switch to anaerobic glycolysis. As a result, the majority
of ATP is produced by the glycolytic pathway, converting pyruvate
to lactate and hydrogen ions, leading to a gradual decrease in
overall brain pH. A stable intracellular pH [(pH)(i)] is
critical for normal cellular function, and the majority of
biological processes are markedly sensitive to pH. Even small
changes in (pH)(i) affect the properties of various ion
channels, plasma membrane excitability, and cellular metabolism
(5–7). It has been proposed that this drop in
pH results in acidotoxicity, which contributes to neuronal injury
following cerebral ischemia (8).
Under ischemic conditions, plasma membrane
depolarization leads to an increased synaptic release of glutamate.
This released glutamate can cause an excessive influx of
Na+ through glutamate-gated ion channels, leading to the
subsequent influx of CI− and H2O, which
results in plasma membrane rupture and further release of
cytoplasmic glutamate into the extracellular space (9). Excessive glutamate overactivates
glutamate receptors, specifically N-methyl-D-aspartate receptor
(NMDAR), causing high levels of calcium ions (Ca2+) to
influx into the postsynaptic cell and increase intracellular
calcium [(Ca2+)(i)] (10). NMDAR overactivation disrupts
antioxidant defenses and critical survival pathways, which
increases the susceptibility of neurons and glia to ischemic
damage, and also triggers numerous ischemic cascades, leading to
further neuronal degeneration, or in some cases mortality.
Therefore, improving neuronal defenses against glutamate-induced
excitotoxicity and/or decreasing the
(Ca2+)(i) influx into the ischemic neurons is
crucial following ischemic stroke (9,11).
3-(5′-Hydroxymethyl-2′-furyl)-1-benzylindazole
(YC-1) is a nitric oxide (NO)-independent and direct activator of
soluble guanylyl cyclase, and a hypoxia-inducible factor-1
inhibitor. YC-1 has been demonstrated to downregulate vascular
endothelial growth factor, erythropoietin, endothelin-1 and
inducible NO synthase signaling and protein levels (12,13).
YC-1 can downregulate matrix metalloproteinase-9 protein levels in
the human retina and protect blood-brain-barrier permeability
following ischemia/reperfusion-induced injury in rats (14).
The present study assessed YC-1-mediated
neuroprotection and its therapeutic window in an in vitro
model of a cultured neurons exposed to glutamate-induced
excitotoxicity, and further determined the crucial therapeutic
window using intraperitoneal administration of YC-1 in a mouse
model of transient focal cerebral ischemia.
Materials and methods
Animal preparation
A total of 74 2-day-old Sprague-Dawley rats and 43
mice (20–26 g) (all female) were supplied by the National Cheng
Kung University Laboratory Animal Center and were allowed free
access to food and water. Unless otherwise indicated, mice were
housed individually in static microisolation caging (45.5×23×20.5
cm). Cages were maintained in temperature-controlled chambers
(29±1°C) on a 12-h light/dark cycle. For in vitro
experiments of cell viability, cell swelling,
(Ca2+)(i) and (pH)(i), 74 rats
underwent cortical isolation for primary cultured neurons. For
in vivo experiments of brain infraction, 43 mice were used
for transient middle cerebral artery (MCA) occlusion.
The present study received affidavits of ethical
approval for the use of animals in the present study; ethical
approval was provided by National Cheng Kung University (Tainan,
Taiwan, R.O.C.). An affidavit of approval for animal use protocols
was obtained for in vivo experiments (approval no. 97175)
and in vitro primary cortical neuron culture experiments
(approval no. 103177).
Primary cortical neuronal culture
According the method described previously (11,15),
cultured neurons were obtained from the cerebral cortices of
1-day-old Sprague-Dawley rats. Following animal sacrifice, the
cortices were excised in ice-cold Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Tissue samples were minced and then incubated with Hank's
Balanced Salt Solution (HBSS; Gibco; Thermo Fisher Scientific,
Inc.) containing papain (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) and DNase I (Bionovas Biotechnology Co., Ltd., Toronto,
Canada) at 37°C to dissociate the cells. Following 30 min
incubation, heat-inactivated fetal bovine serum (FBS; Gibco; Thermo
Fisher Scientific, Inc.) was added to the cell suspensions to
terminate the reaction and the suspensions were then centrifuged at
800 × g for 10 min at 4°C. The pellets were collected and
resuspended in DMEM with 10% FBS and then filtrated with a 70 µm
cell strainer (BD Biosciences, Franklin Lakes, NJ, USA).
Dissociated cells were plated onto poly-D-lysine-coated petri
dishes and incubated at 37°C in a humidified incubator with 5%
CO2. Then, 4 h following plating, the culture medium was
replaced by neurobasal medium containing 25 µM glutamate, 0.5 mM
L-glutamine and 2% B27 supplement (Invitrogen; Thermo Fisher
Scientific, Inc.). Culture medium was changed every 3 days and the
cultured neurons were allowed to grow for ~7–14 days.
Neurotoxicity of YC-1
Neurons were incubated with YC-1 (3–300 µM) or
vehicle (0.1% DMSO) at 37°C overnight. Neuronal damage was assessed
by densitometric measurements of the cellular propidium iodide (PI;
Thermo Fisher Scientific, Inc.) uptake following 24 h. PI was
solubilized in water to a final concentration of 50 µg/ml and
stored in aliquots at −20°C for a maximum of 3 months. Cells were
washed 3 times in PBS and then stained by the addition of 1 ml PI
(50 µg/ml) to each dish for 10 min in the dark at room temperature.
PI positive cell and total cell numbers were measured on an Olympus
IX71 inverted phase/fluorescence microscope (Olympus Corporation,
Tokyo, Japan), equipped with a cooled charge-couple device camera
(300T-RC; Dage-MTI, Inc., Michigan City, IN, USA). PI %=[(PI
positive cell numbers)/(total cell numbers)] × 100.
Glutamate-induced cell
cytotoxicity
Cells were administered the following experimental
treatments: i) Pre-treatment group were administered YC-1 (1–100
µM) or vehicle (0.1% DMSO) for 30 min and then exposed to glutamate
(300 mM) for 24 h; ii) post-treatment group in which YC-1 (30 µM)
or vehicle (0.1% DMSO) was added to the culture medium at 0, 2, 4
or 6 h following glutamate exposure. The neuronal cytotoxicity was
determined at 24 h following treatment using a PI assay kit (Thermo
Fisher Scientific, Inc.). PI positive cell and total cell numbers
were measured on an Olympus IX71 inverted phase/fluorescence
microscope, equipped with a cooled charge-couple device camera as
above. PI %=[(PI positive cell numbers)/(total cell numbers)] ×
100.
Cell swelling measurements
The experimental treatments were divided into four
groups: i) Vehicle (0.1% DMSO); ii) glutamate-treated group; iii)
delayed treatment, YC-1 at 2 h following glutamate exposure; and,
iv) delayed treatment, YC-1 at 4 h following glutamate exposure.
The glutamate (300 mM)-induced neuronal morphological changes were
measured by time-lapse imaging techniques on an Olympus IX71
inverted microscope equipped with a thermo-controllable heating
stage, differential interference contrast (DIC) and an image
analyzer (MCID Elite 6.0 Rev. 1.4, Imaging Research Inc., St.
Catharines, ON, Canada) by the method described previously
(11,16). In each microscopic field ~6 to 12
neurons per culture were measured and compared over time. Data are
expressed as a percentage relative to the baseline values.
(Ca2+)(i)
measurement
The level of (Ca2+)(i) was
measured on a single cell fluorimeter, as previously described
(11). Briefly, neuronal cultures
were incubated with 3 µM Fura 2-Acetoxymethylester (Fura-2 AM;
Invitrogen; Thermo Fisher Scientific, Inc.) and 10 µM ionomycin in
a standard buffer (composition in mM: NaCl, 140; KCl, 3.5;
KH2PO4, 0.4; Na2HPO4,
1.25; CaCl2, 2.2; MgSO4, 2; glucose, 10;
HEPES, 10, pH 7.3) for 30 min, followed by incubation in dye-free
standard buffer for 30 min and then the addition of vehicle or YC-1
(30 µM) for 20 min, and exposure to glutamate (300 µM) all at 37°C.
The glass coverslip was placed into the stage chamber of an Olympus
IX71 inverted microscope, equipped with a 75 W xenon illumination
system, a cooled charge-couple device (CCD) camera (300T-RC;
Dage-MTI, Inc., Michigan City, IN, USA) coupled to an image
intensifier (Gen II S-25 image intensifier; Dage-MTI, Inc.), a
Lambda 10-2 filter-wheel and shutter (Sutter Instruments, Novato,
CA, USA) and a computerized image analyzer (MCID Elite 6.0 Rev.
1.4; Imaging Research Inc.). The cells were alternatively
illuminated with light at 340 and 380 nm wavelengths, and the
emitted light was passed through a 510-nm barrier filter. The 340
and 380 nm images were captured at 6 sec intervals, and the ratio
signals (340 nm excited image/380 nm excited image) were processed
and examined for changes in (Ca2+)(i). A
total of ~10 neurons in the microscope field were individually
measured.
(pH)(i) measurement
According to the method described previously
(17), the fluorescent pH
indicator dye,
2′,7′-Bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF;
Invitrogen; Thermo Fisher Scientific, Inc.) was loaded into cells.
Following 30 min incubation in 5 µM BCECF-AM (acetoxymethyl), the
glass coverslip was placed into the stage chamber of an Olympus
IX71 inverted microscope, equipped with a 75 W xenon illumination
system, a cooled CCD camera (300T-RC; Dage-MTI, Inc.) coupled to an
image intensifier (Gen II S-25 image intensifier; Dage-MTI, Inc.),
a Lambda 10-2 filter-wheel and shutter (Sutter Instruments) and a
computerized image analyzer (MCID Elite 6.0 Rev. 1.4, Imaging
Research Inc.). The cells were alternatively illuminated with light
at 440 and 490 nm wavelengths, and the emitted light was passed
through a 535-nm barrier filter. The 440 and 490 nm images were
captured at 6 sec intervals and the ratio signals (490 nm excited
image/440 nm excited image) were processed and examined for changes
in (pH)(i). A total of ~10 neurons in the microscopic
field were individually measured. pH calibrations using BCECF
measurement were performed by incubating cells with nigericin, a
K+/H+ ionophore. Cells were washed with HBSS
buffer (nigericin 1 µM, KCl 140 mM and MgCl2 1 mM) 3
times and cells were then incubated with HBSS buffer at pH 6, 6.5,
7, 7.5 and 8. The (pH)(i) level was calculated using the
following equation: (pH)(i)=pKa + log
[(R-Rmin)/(Rmax-R)] × [F440
min/F440 max)], where R is the ratio of
fluorescence intensity recorded at 490 and 440 nm, and
Rmin and Rmax are the ratios of 490/440 nm
fluorescence intensity recorded at pH 6 and pH 8. pKa was converted
by known pH concentrations of HBSS buffer.
MCA model and drug administration
Adult male C57 black (C57BL/B6) mice, (n=43, weight
20–26 g, 6–8 weeks old) were housed at 24±1°C, 60% humidity with a
12-h light/dark cycle and free access to food and water prior to
and following surgery. For the surgical procedures performed on
mice, 1–2% halothane in N2O:O2 (70:30%) was
used for anesthesia (3–4% used for induction and 1–2% for
maintenance). A precision vaporizer, adequate ventilation and
scavenging were employed.
Focal cerebral ischemia was employed by
intra-arterial suture occlusion of the proximal right MCA according
to the method described previously (18). Briefly, the external carotid artery
(ECA), internal carotid artery (ICA) and pterygopalatine artery of
the ICA were exposed under an operating microscope. A silicone
rubber coated nylon suture was inserted into the ICA via a slit in
the ECA. The suture was advanced 7.5–8.5 mm along the ICA until the
tip occluded the origin of the MCA. Reperfusion was produced by
gently removed the suture and the incision closed following 1 h of
MCA occlusion. Following the surgical procedures, the animals were
kept in a cage with a heating lamp, monitored for 2 h and then
transferred back into their original cages (ambient temperature,
~24±1°C).
YC-1 was dissolved in a mixture of polyethylene
glycol (PEG) 400 (Sigma-Aldrich; Merck KGaA) and saline (PEG
400:saline=3:7). Animals were administered an intraperitoneal
injection of either YC-1 (25 mg/kg) or vehicle (PEG 400:saline=3:7)
at 1, 2, 3, or 4 h following MCA occlusion to test the window of
opportunity. Brain infarctions were examined 48 h following
ischemia reperfusion.
Animal sacrifice and quantification of
ischemic brain damage
All assessments were based on the method described
previously (18). At 48 h,
sacrifice was performed on the mice under anesthesia via
transcardial perfusion with 4% formaldehyde in 0.1 M PBS following
the ischemia-reperfusion insult. Following fixation via
transcardial perfusion with ice-cold 4% formaldehyde in 0.1 M PBS
and dehydration, the brains were embedded in Optimal Cutting
Temperature compound (OCT; Miles Inc., Elkhart, IN, USA) and frozen
in liquid nitrogen. The brains were sectioned (40 µm) at 8
preselected coronal levels on a cryostat (HM-500O; Microm
International GmbH, Walldorf, Germany). The sections were mounted
on poly-l-lysine-coated slides (Sigma-Aldrich; Merck KGaA), dried
at 37°C overnight, and were then stored at −20°C.
The 40-µm brain sections were stained with 0.5%
Cresyl violet for 4 h at room temperature. Under light microscopy,
the areas with neuronal perikarya displaying typical morphological
features of ischemic damage were delineated using a computerized
image analyzer (MCID Elite 6.0 Rev. 1.4; Imaging Research Inc.).
The number of fields of view analyzed were: Control group, 10; YC-1
treated after 1 h of ischemia, 10; YC-1 treated after 2 h of
ischemia, 7; YC-1 treated after 3 h of ischemia, 9; and, YC-1
treated after 4 h of ischemia 7. Infarct volumes were measured and
expressed as a percentage of the contralateral hemisphere volume
using the following equation: (Contralateral hemisphere
area-ipsilateral non-ischemic hemisphere area)/contralateral
hemisphere area. The ipsilateral brain edema was expressed as a
percentage index relative to the volume of the left hemisphere
[(ischemic hemisphere area/contralateral hemisphere area)-infarct
volume].
Statistical analysis
All data were expressed as the mean ± standard
deviation of 3 repeated experiments. Unpaired Student's t-test or
one-way analysis of variance with a least significant difference
post hoc test were used to evaluate differences between groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
YC-1 reduces glutamate-induced cell
death
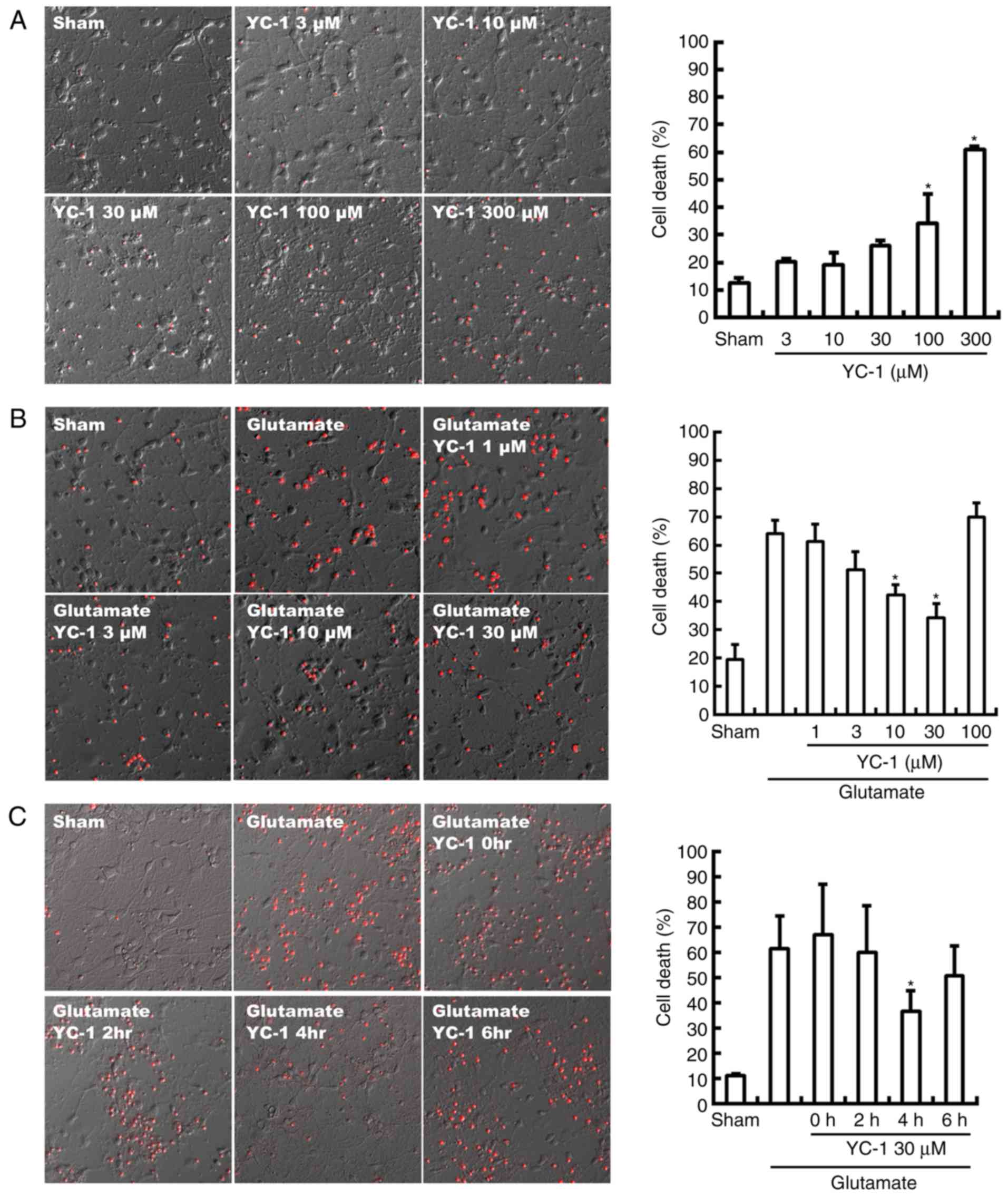
Neurotoxicity of YC-1 was observed with a
concentration beyond 100 µM, compared with sham group (Fig. 1A; P<0.05). Conversely, the
glutamate-induced neurotoxicity was significantly attenuated by
YC-1 at 10–30 µM, compared with the glutamate only group (Fig. 1B; P<0.05). Delayed treatment
with YC-1 (30 µM) at 4 h, though not at 2 or 6 h, significantly
reduced glutamate-induced cell death, compared with the glutamate
group (Fig. 1C; P<0.05).
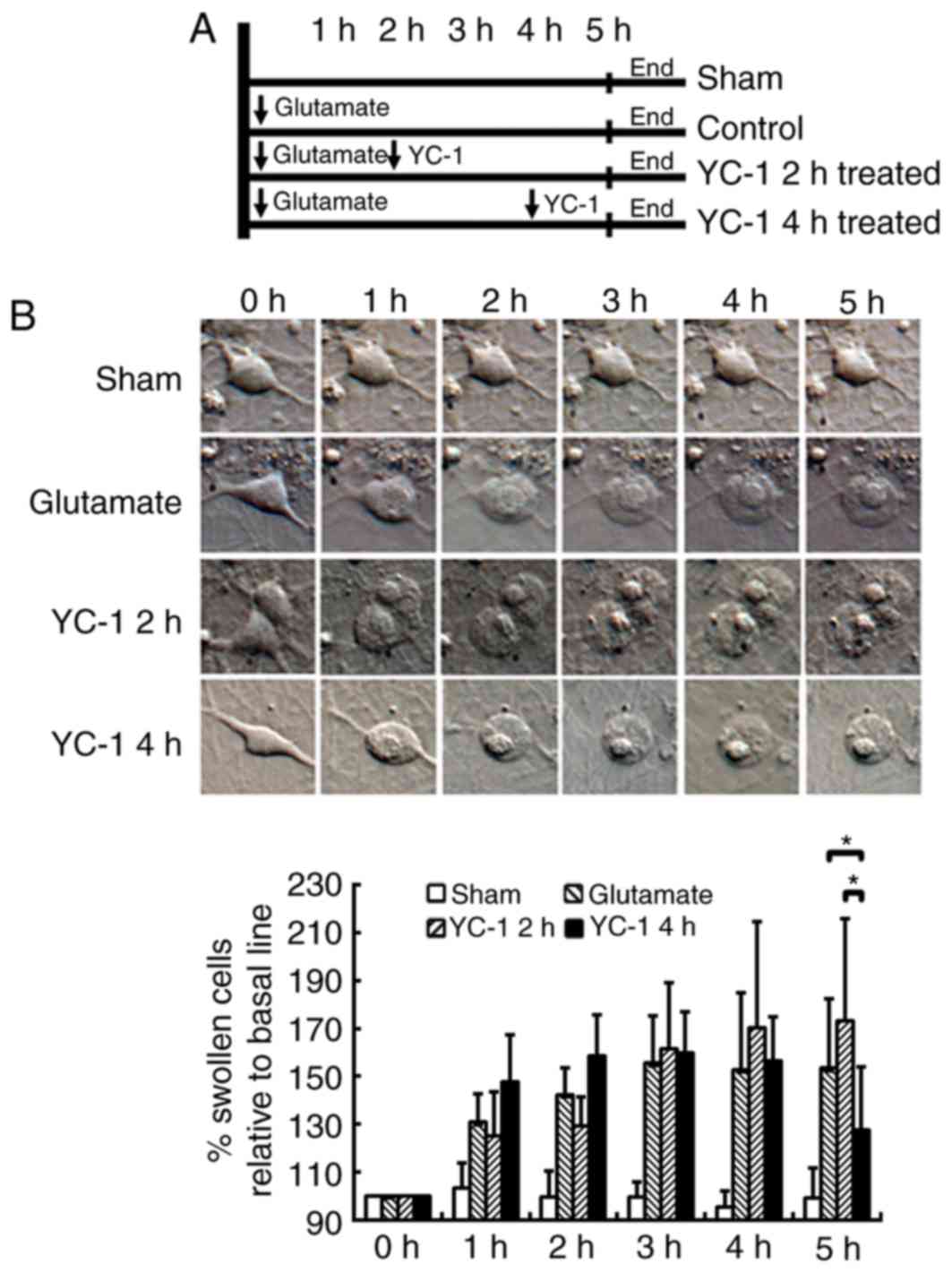
Delayed YC-1 treatment at 4 h
attenuates glutamate-induced cell swelling in cultured neurons
The cultured neurons were exposed to 300 µM
glutamate and then delayed treatment with 30 µM YC-1 at 2 or 4 h
(Fig. 2A). Time-course DIC
photomicrographs of cultured neurons demonstrated that only delayed
treatment with 30 µM YC-1 at 4 h significantly attenuated the
glutamate-induced cell swelling over time (Fig. 2B; P<0.05).
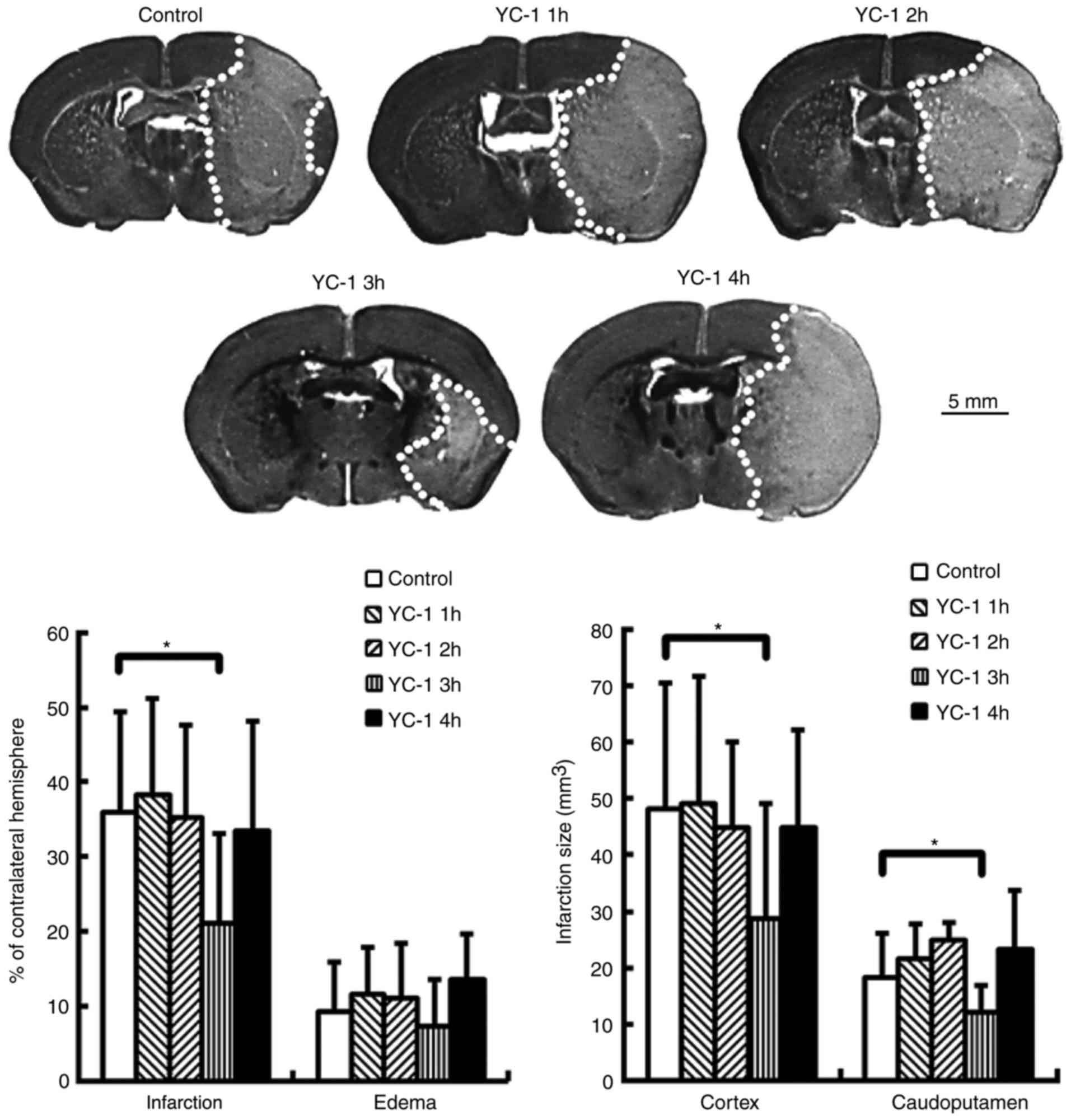
YC-1 reduces brain infarction in mice
subjected to transient MCA occlusion
Adult mice were subjected to transient MCA
occlusion. Compared with controls, delayed treatment with YC-1 (25
mg/kg) at 3 h, though not at 1, 2 or 4 h, significantly reduced
brain infarction at 24 h post-insult (Fig. 3; P<0.05). The infarct volumes
are expressed as a percentage of the contralateral hemisphere.
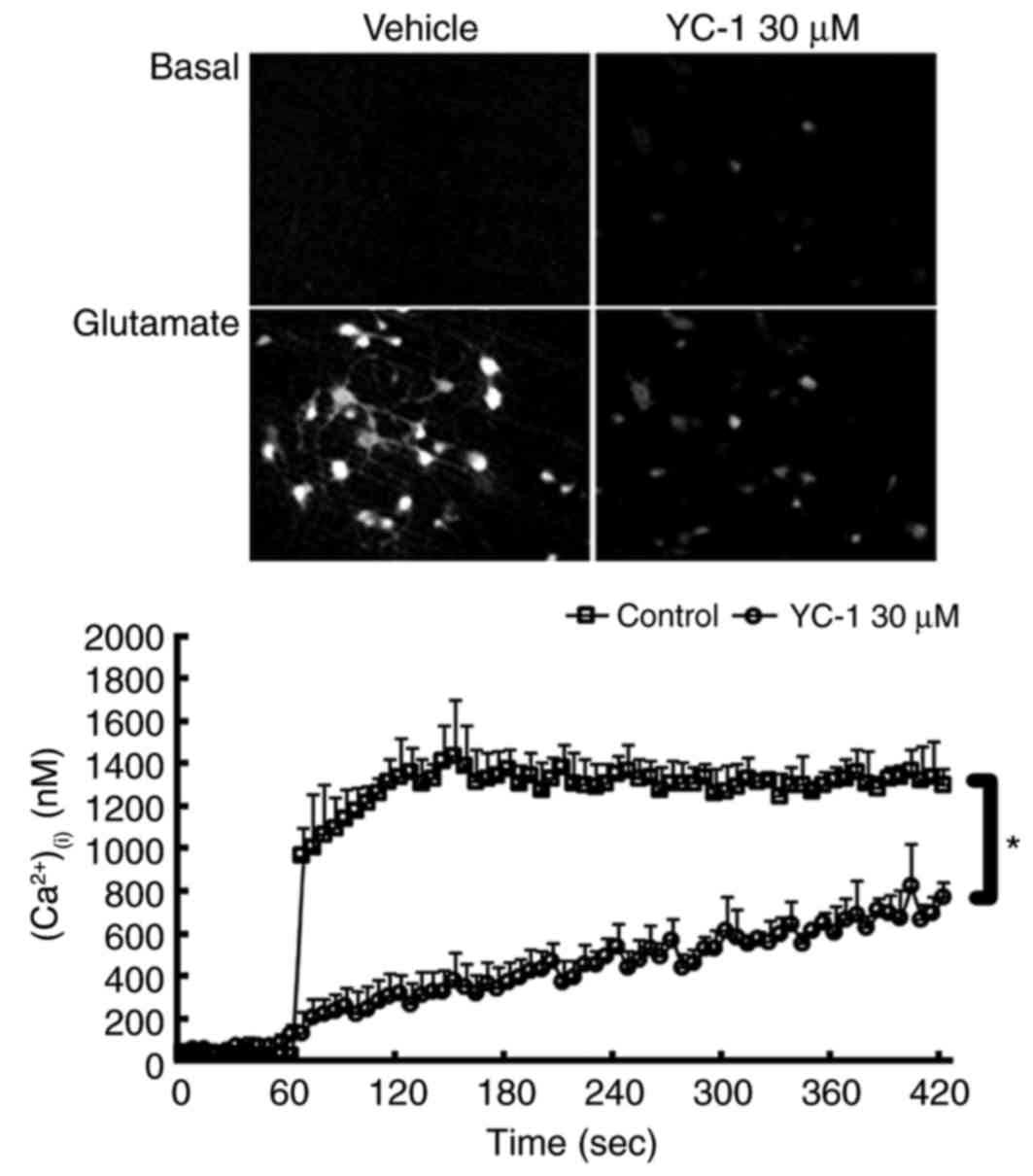
YC-1 attenuates the glutamate-induced
rise in (Ca2+)(i) in cultured neurons
Ratio image detection for
(Ca2+)(i) concentrations demonstrated that
adding 300 µM glutamate induced an abrupt rise in
(Ca2+)(i) levels to ≥1,000 nM in the control
group. Compared with controls, treatment with 30 µM YC-1
effectively inhibited this glutamate-induced rise in
(Ca2+)(i) (Fig.
4; P<0.05).
Immediate treatment with YC-1
following glutamate exposure causes a marked change in
(pH)(i)
BCECF loaded single cortical neurons exposed to 300
µM glutamate then immediately treated with YC-1 (30 µM),
demonstrated a marked reduction in (pH)(i), when
compared with glutamate only, YC-1 only and delayed treatment with
YC-1 at 2 and 4 h (Fig. 5;
P<0.05).
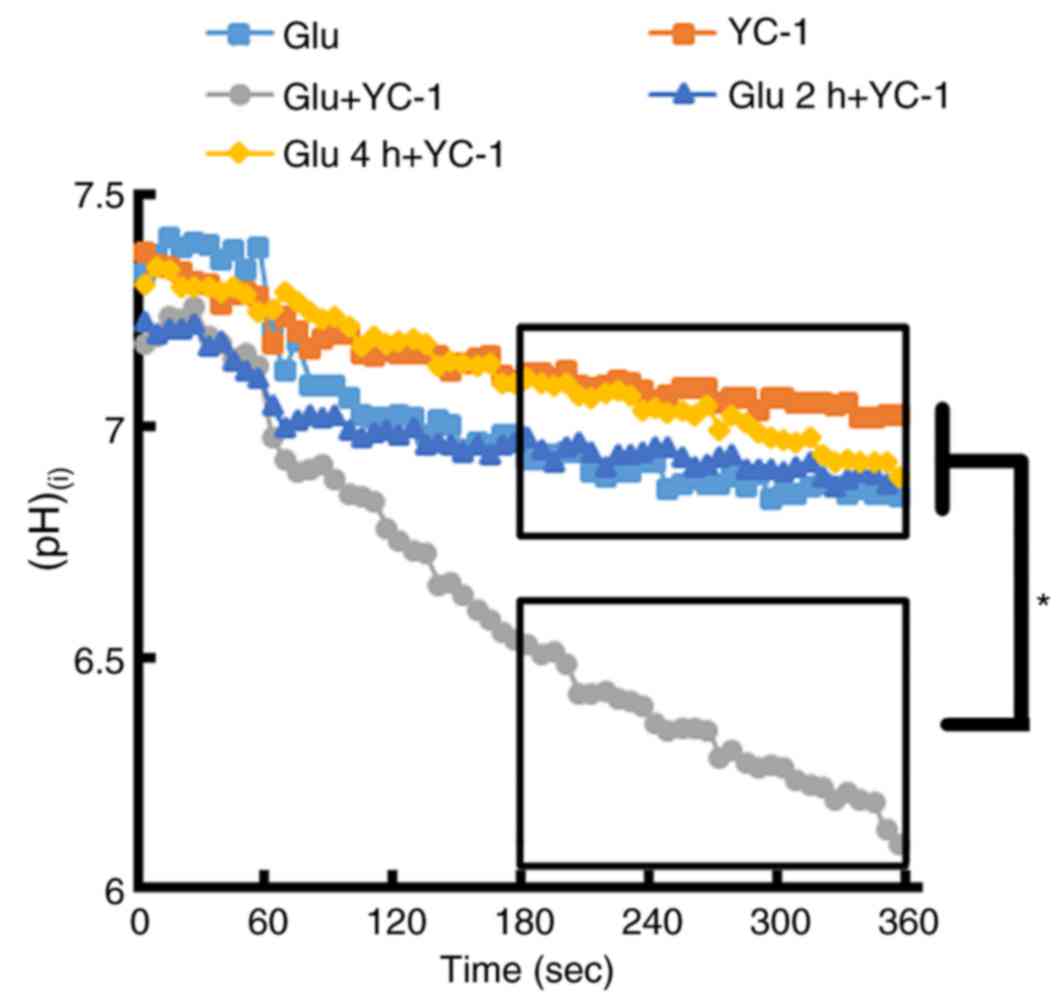 | Figure 5.BCECF-loaded single cortical neurons
were exposed to 300 µM Glu. Following the exposure of cortical
neurons to 300 µM Glu, immediate treatment with YC-1 (30 µM)
demonstrated a marked reduction in (pH)(i) when compared
with Glu only, YC-1 only, and delayed treatment with YC-1 at 2 and
4 h. (Glu n=11; YC-1 n=10; Glu+YC-1 n=9; Glu 2 h+YC-1 n=9; Glu 4
h+YC-1 n=10). *P<0.05, as indicated. BCECF,
2′,7′-Bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein; YC-1,
3-(5′-Hydroxymethyl-2′-furyl)-1-benzylindazole; Glu, glutamate;
(pH)(i), intracellular pH; n, number of rats. |
Discussion
The results of the present study indicated that YC-1
demonstrated neurotoxicity at a concentration >100, and 10–30 µM
YC-1 achieved potent cytoprotection against glutamate-induced
neuronal damage. Additionally, 30 µM YC-1 effectively attenuated
the rise in (Ca2+)(i) levels in cultured
neurons exposed to glutamate. However, only the cultured neurons
administered YC-1 (30 µM) at 4 h following exposure to glutamate
demonstrated protection against glutamate-induced neuronal damage
and cell swelling. Immediate treatment of YC-1 (30 µM) following
the exposure of cortical neurons to glutamate induced a marked
reduction in the (pH)(i). In this animal stroke model,
only delayed treatment of YC-1 at 3 h post-insult had
neuroprotective effects in mice subjected to transient focal
cerebral ischemia.
Hypoxia-ischemia increases extracellular glutamate
resulting in the cytotoxicity of neurons. Excessive glutamate will
stimulate NMDARs and increase the influx of
(Ca2+)(i) (11,19).
Excessive Ca2+ influx overwhelms the compensatory
mechanisms of neurons, causing osmotic volume expansion (swelling).
If the impact is limited and small, it may be reversible if
appropriate therapeutic actions are taken (20). A previous study demonstrated that
pretreatment with 3–30 µM YC-1 could decrease the damaging effect
of glutamate on PC12 cells, a cell line derived from a rat with
adrenal medulla pheochromocytoma (21). In addition, treatment with 10–50 µM
YC-1 to neutrophils stimulated with cyclopiazonic acid produced a
concentration-dependent reduction of
(Ca2+)(i) (21). In agreement with this previous
report (21), the present study
demonstrated that YC-1 (10–30 µM) also effectively reduced
glutamate-induced neuronal damage in primary cortical neurons. YC-1
(30 µM) also effectively attenuated the rise in
(Ca2+)(i) levels in cultured neurons exposed
to glutamate. These results indicated that the neuroprotection
produced by YC-1 may be mediated via its ability to limit
glutamate-induced excitotoxicity.
Pretreatment with YC-1 at 10–30 µM could protect
neurons against glutamate-induced neurotoxicity. The results of the
present study identified that pretreatment with YC-1 at 30 µM
attenuated the glutamate-induced rise in
(Ca2+)(i). Pretreatment with YC-1 produced a
neuroprotective effect, potentially due to the neurons remaining
intact at the time of treatment, and mediated the decrease in
(Ca2+)(i) escalation following ischemia.
However, post-treatment with YC-1 at 30 µM was protective only when
added to the medium at 4 h, though not at 2 h. Similar results were
observed in the cell swelling experiment. Post-treatment with YC-1
at 30 µM reduced glutamate-induced cell swelling when added to the
medium at 4 h, though not at 2 h. Thus, these data indicated that
YC-1 was neurotoxic, potentially due to its effect on aggravating
post-ischemic cell swelling and pH within 4 h of ischemia.
Conversely, these data clearly indicated that this mechanistic
effect diminished following 4 h of ischemia. Therefore, YC-1
regained its neuroprotective effects following 4 h of ischemia.
Immediate treatment with YC-1 at 30 µM demonstrated
a marked reduction of (pH)(i) following stroke. The
(pH)(i) data from the present study also indicated that
YC-1 was neurotoxic to intraischemic cells immediately post-insult,
this may be due to its effect in further decreasing
(pH)(i). In addition, ischemia induced cellular damage
with decreased (pH)(i) up to 4 h. Therefore,
post-treatment with YC-1 may aggravate neuronal damage within 4 h
of ischemia. Thus, acidosis may serve a critical role in the
pathogenesis of brain ischemia. This may also explain how
post-treatment with YC-1 at 30 µM protected neurons against
glutamate-induced neurotoxicity and cellular swelling when added to
the medium at 4 h, though not at 2 h. Although the optimal time
point was slightly different between the in vitro (4 h) and
the in vivo (3 h) data, the possibility that the in
vitro data may not be fully translated to in vivo data
cannot be excluded.
Ischemia increased extracellular glutamate resulting
in excitotoxic damage and an increased influx of
(Ca2+)(i). Although YC-1 could reduce
glutamate-induced neuronal damage, cell swelling and increased
influx of (Ca2+)(i), the protective effects
were not observed in delayed YC-1 treatment at 1–3 h in
vitro and 1–2 h in vivo following stroke induction. This
also indicated that other factors may interfere with or counteract
the therapeutic effect of YC-1. A significant change in
(pH)(i) during the early phase of stroke was identified,
which may account for the phenomena observed above.
The mechanism of ischemic stroke is quite complex.
Following ischemia, oxygen depletion forces the brain to switch to
anaerobic glycolysis. The accumulation of lactic acid as a
byproduct of glycolysis and protons produced by ATP hydrolysis
cause the pH levels to fall in the ischemic brain. During severe
ischemia, pH can fall to <6.0 (4). Even small changes in
(pH)(i) can affect a considerable number of important
mechanisms and induce cell death following stroke. The results of
the present study demonstrated that neurons exposed to glutamate or
delayed treatment with YC-1 at 2 h then glutamate exposure produced
similar changes in (pH)(i) (a reduction of ~0.4 in pH);
however, immediate treatment with YC-1 following the exposure of
cortical neurons to glutamate resulted in a marked reduction in
(pH)(i) (a reduction of ~1.0 in pH). In addition, only
neurons treated with YC-1 or delayed treatment with YC-1 at 4 h
following glutamate exposure produced minor changes in
(pH)(i) (reduction of ~0.2 in pH). These changes in
(pH)(i) may be the cause of the protective effect of
YC-1 at 4 h in vitro and 3 h in vivo following brain
injury.
A previous study indicated that a change in
(pH)(i) affects the acid-induced increase of
(Ca2+)(i), membrane depolarization and
acidosis-mediated neuronal injury (22). Acidosis serves a critical role in
brain ischemia. Acid-sensing ion channels (ASICs) are a key
mediator of acidosis-induced neuronal injury (23). A previous study indicated that
ASIC1a is involved in synaptic plasticity, learning/memory and fear
conditioning (4). In addition, a
previous study indicated that the function of ASICs is modulated by
extracellular pH and also (pH)(i) (5). In the results from the present study,
treatment with YC-1 immediately following neuronal exposure to
glutamate resulted in a marked reduction in (pH)(i).
However, whether such a change is associated with ASICs remains
unclear. Thus, further studies on whether YC-1 affects ASICs in
early stroke are required.
In conclusion, YC-1 offers neuroprotection against
glutamate-induced neuronal damage in mice subjected to transient
focal cerebral ischemia. Although YC-1 treatment appears to be a
potential strategy for clinical therapy, the timing of treatment
was crucial in the present study and so further studies are
required to elucidate its potential.
References
|
1
|
O'Bryant Z, Vann KT and Xiong ZG:
Translational strategies for neuroprotection in ischemic
stroke-focusing on acid-sensing ion channel 1a. Transl Stroke Res.
5:59–68. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee EJ, Chen HY, Hung YC, Chen TY, Lee MY,
Yu SC, Chen YH, Chuang IC and Wu TS: Therapeutic window for
cinnamophilin following oxygen-glucose deprivation and transient
focal cerebral ischemia. Exp Neurol. 217:74–83. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Demaerschalk BM and Yip TR: Economic
benefit of increasing utilization of intravenous tissue plasminogen
activator for acute ischemic stroke in the United States. Stroke.
36:2500–2503. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J,
Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ and Simon RP:
Neuroprotection in ischemia: Blocking calcium-permeable
acid-sensing ion channels. Cell. 118:687–698. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li MH, Leng TD, Feng XC, Yang T, Simon RP
and Xiong ZG: Modulation of acid-sensing ion channel 1a by
intracellular pH and its role in ischemic stroke. J Biol Chem.
291:18370–18383. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mutch WA and Hansen AJ: Extracellular pH
changes during spreading depression and cerebral ischemia:
Mechanisms of brain pH regulation. J Cereb Blood Flow Metab.
4:17–27. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
von Hanwehr R, Smith ML and Siesjö BK:
Extra- and intracellular pH during near-complete forebrain ischemia
in the rat. J Neurochem. 46:331–339. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mari Y, Katnik C and Cuevas J: ASIC1a
channels are activated by endogenous protons during ischemia and
contribute to synergistic potentiation of intracellular Ca(2+)
overload during ischemia and acidosis. Cell Calcium. 48:70–82.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Szatkowski M and Attwell D: Triggering and
execution of neuronal death in brain ischaemia: Two phases of
glutamate release by different mechanisms. Trends Neurosci.
17:359–365. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dubinsky JM: Intracellular calcium levels
during the period of delayed excitotoxicity. J Neurosci.
13:623–631. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee WT, Lin MH, Lee EJ, Hung YC, Tai SH,
Chen HY, Chen TY and Wu TS: Magnolol reduces glutamate-induced
neuronal excitotoxicity and protects against permanent focal
cerebral ischemia up to 4 hours. PLoS One. 7:e399522012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hwang TL, Hung HW, Kao SH, Teng CM, Wu CC
and Cheng SJ: Soluble guanylyl cyclase activator YC-1 inhibits
human neutrophil functions through a cGMP-independent but
cAMP-dependent pathway. Mol Pharmacol. 64:1419–1427. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
DeNiro M, Al-Halafi A, Al-Mohanna FH,
Alsmadi O and Al-Mohanna FA: Pleiotropic effects of YC-1
selectively inhibit pathological retinal neovascularization and
promote physiological revascularization in a mouse model of
oxygen-induced retinopathy. Mol Pharmacol. 77:348–367. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yan J, Zhou B, Taheri S and Shi H:
Differential effects of HIF-1 inhibition by YC-1 on the overall
outcome and blood-brain barrier damage in a rat model of ischemic
stroke. PLoS One. 6:e277982011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tai SH, Hung YC, Lee EJ, Lee AC, Chen TY,
Shen CC, Chen HY, Lee MY, Huang SY and Wu TS: Melatonin protects
against transient focal cerebral ischemia in both reproductively
active and estrogen-deficient female rats: The impact of
circulating estrogen on its hormetic dose-response. J Pineal Res.
50:292–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
DeLorenzo RJ, Sun DA, Blair RE and Sombati
S: An in vitro model of stroke-induced epilepsy: Elucidation of the
roles of glutamate and calcium in the induction and maintenance of
stroke-induced epileptogenesis. Int Rev Neurobiol. 81:59–84. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Trenholm S and Baldridge WH: The effect of
aminosulfonate buffers on the light responses and intracellular pH
of goldfish retinal horizontal cells. J Neurochem. 115:102–111.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tai SH, Chen HY, Lee EJ, Chen TY, Lin HW,
Hung YC, Huang SY, Chen YH, Lee WT and Wu TS: Melatonin inhibits
postischemic matrix metalloproteinase-9 (MMP-9) activation via dual
modulation of plasminogen/plasmin system and endogenous MMP
inhibitor in mice subjected to transient focal cerebral ischemia. J
Pineal Res. 49:332–341. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Papadia S, Soriano FX, Léveillé F, Martel
MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska
V, et al: Synaptic NMDA receptor activity boosts intrinsic
antioxidant defenses. Nat Neurosci. 11:476–487. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Alzheimer C: Na+ channels and Ca2+
channels of the cell membrane as targets of neuroprotective
substances. Adv Exp Med Biol. 513:161–181. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang X, Wang Y, Luo J, Liu S and Yang Z:
Protective effects of YC-1 against glutamate induced PC12 cell
apoptosis. Cell Mol Neurobiol. 31:303–311. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang WZ, Chu XP, Li MH, Seeds J, Simon RP
and Xiong ZG: Modulation of acid-sensing ion channel currents,
acid-induced increase of intracellular Ca2+, and acidosis-mediated
neuronal injury by intracellular pH. J Biol Chem. 281:29369–29378.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jiang N, Wu J, Leng T, Yang T, Zhou Y,
Jiang Q, Wang B, Hu Y, Ji YH, Simon RP, et al: Region specific
contribution of ASIC2 to acidosis-and ischemia-induced neuronal
injury. J Cereb Blood Flow Metab. 37:528–540. 2017. View Article : Google Scholar : PubMed/NCBI
|