Introduction
Hepatocellular carcinoma (HCC) is one of the most
prevalent cancers worldwide, causing the third most death of
cancers (1). Patients with HCC
have a high risk in liver cirrhosis and other symptoms such as
pain, fatigue, weight loss, and obstructive syndromes including
ascites and jaundice (2). As not
all of HCC patients qualifying surgical treatments including tumor
resection and liver transplantation, the prognosis of these
patients is poor (3). Although
great improvements in HCC treatments have been achieved, further
investigation of safe and accurate diagnosis methods and effective
therapies for HCC should be taken into consideration.
In the past decades, efforts on molecular mechanism
researches of HCC have thrown light on molecular diagnosis and
molecular targeted therapies of the disease. Numerous genes and
transcription factors (TFs) associated with HCC development have
been revealed. Like all the other cancers, cells of HCC lost
control of cell cycle (4). Genes
such as cyclin-dependent kinases and TFs such as E2F transcription
factors (E2Fs) are involved in cell cycle (5,6).
Another hallmark of cancer cells is metastasis. Genes like matrix
metalloproteinase (MMP)2 and MMP9, and TFs like hypoxia inducible
TFs are found to play important role in the invasion and metastasis
of HCC cells (7–9). What's more, signaling pathways such
as retinoblastoma pathways, Ras/MAPK pathway and Wnt/β-catenin
pathway are altered in HCC cells (4). Based on achieved improvements in the
molecular mechanism underlying HCC, many efforts have been made to
investigate promising biomarkers and therapeutic targets for the
diagnosis and treatment of HCC. Quantitative detection of
methylated GSH-sulphur-transferase P1 in serum can be used for the
early diagnosis of HCC (10).
Besides, methylation status of plasma P16 gene has been indicated
to have the value for the detection of primary HCC (11). In addition, it is revealed that the
expression level of glypican 3 (GPC-3) in liver tissues can be used
as an indicator of HCC (12) and
silencing of GPC-3 can inhibit the proliferation of HCC cells
(13). Furthermore, DNA
methyltransferase 1 (DNMT1) knockdown can inhibit HCC cell
proliferation and increase apoptosis, suggesting that DNMT1 may
serve as a targets for HCC therapy (14). Dysruption of pathways involved in
HCC have been proven to be efficient for HCC treatment. The
candidate pathways include pathways involved in signal transduction
such as growth factor receptors, pathways involved in apoptosis
such as intrinsic pathway and pathways participating in cell cycle
(15). Despite these great
advances, the key mechanism underlying HCC remains to be further
elucidated to screen promising biomarkers and potential targets for
the diagnosis and treatment of HCC.
Bioinformatics approaches are effective for
mechanism research. Makowska et al (16) developed the gene expression file
GSE64041 using HCC samples and matched control samples to establish
a molecular classification of human HCC. Based on differential gene
expression analysis, subclass prediction, and pathway analysis,
they found that three subgroups of HCC patients were identified
based on the gene expression data and different subgroups of
patients with different prognosis (16). In comparison with this study, we
downloaded this gene expression file (GSE64041) to further
investigate the potential mechanism underlying HCC by a
comprehensive bioinformatics analysis. In addition to differential
gene expression and pathway analyses, protein-protein interaction
(PPI) network, sub-network and integrated TF-miRNA-target network
were also constructed and analyzed to further elucidate the key
genes, TFs and pathways associated with HCC. Our results may be
helpful for better understanding of the molecular mechanisms
underlying HCC and provide valuable information for the diagnosis
and treatment of this disease.
Materials and methods
Microarray data
Gene expression profile data GSE64041 which was
deposited by Makowska et al (16), was downloaded from Gene Expression
Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) database (17). The datasets were sequenced on the
platform Affymetrix Human Gene 1.0 ST Array (Affymetrix; Thermo
Fisher Scientific, Inc., Waltham, MA, USA). A total of 60 biopsy
pairs from patients with HCC were obtained, including 60 HCC
samples and 60 matched non-tumor liver samples.
Data preprocessing and differentially
expressed genes (DEGs) analysis
The downloaded data were preprocessed using limma
package (18) (version 3.30.11,
http://www.bioconductor.org/packages/release/bioc/html/limma.html)
in R language. Expression calculation was performed after
background correction and normalization. Then the probes were
corresponded to gene symbols according to the downloaded NCBI gene
data. If different probes were corresponding to the same symbol,
the average expression values were taken. DEGs between HCC and
non-tumor control groups were identified using limma package
(18). P-values were calculated
using bayesian t-test method. The cut-off thresholds were P<0.05
and fold-change ≥1.
Functional enrichment analysis
Gene Ontology (GO) (19) terms including molecular function
(MF), cellular component (CC) and biological process (BP), and
Kyoto Encyclopedia of Genes and Genomes (KEGG) (20) pathways were enriched for the
upregulated and downregulated DEGs using the database for
annotation, visualization and integrated discovery (DAVID) (version
6.8, https://david-d.ncifcrf.gov/),
respectively (21). P<0.05 was
considered to indicate a statistically significant difference.
PPI network analysis
Search Tool for the Retrieval of Interacting
Genes/Proteins (STRING, version 10.0; http://www.string-db.org/) (22) is an online database proving
assessment and integration of PPIs. In this study, PPIs were
identified based on the information of STRING, followed by PPI
network construction using Cytoscape software (23) (version 3.4.0; http://www.cytoscape.org/). The topology analysis for
the PPI network was performed using CytoNCA plugin (24) (version 2.1.6; http://apps.cytoscape.org/apps/cytonca).
The parameter setting was network without weight. Results arranged
in descending order included scores of degree centrality,
betweenness centrality and closeness centrality. Nodes with top 10
highest centralities were regarded as hub genes. Besides, we
performed a sub-network analysis to identify sub-networks with
great importance from PPI network using MCODE plugin (25) (version 1.4.2; http://apps.cytoscape.org/apps/mcode) in
cytoscape software. Additionally, functional enrichment analyses
for genes in the screened sub-networks were carried out.
TF-miRNA-target regulatory network
analysis
HCC relating miRNAs and experimentally validated
target genes were extracted from Mirwalk2 database (26) (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/).
HCC relating miRNA-target pairs were identified through comparing
the DEGs with the downloaded miRNA-target pairs. Then the
miRNA-target regulatory network was constructed using Cytoscape
software. Besides, TF-target pairs in the miRNA-target network were
predicted using iRegulon plugin (27) (version 1.3; http://apps.cytoscape.org/apps/iregulon) in Cytoscape
with normalized enrichment score (NES) >5 as threshold. Finally,
TF-miRNA-target regulatory network construction was performed using
Cytoscape software.
Results
Analysis of DEGs
A total of 32,321 probes and 18,710 genes were
obtained after data preprocessing. Among these obtained genes, a
total of 378 genes were differentially expressed in HCC group
compared with control group, including 101 upregulated and 277
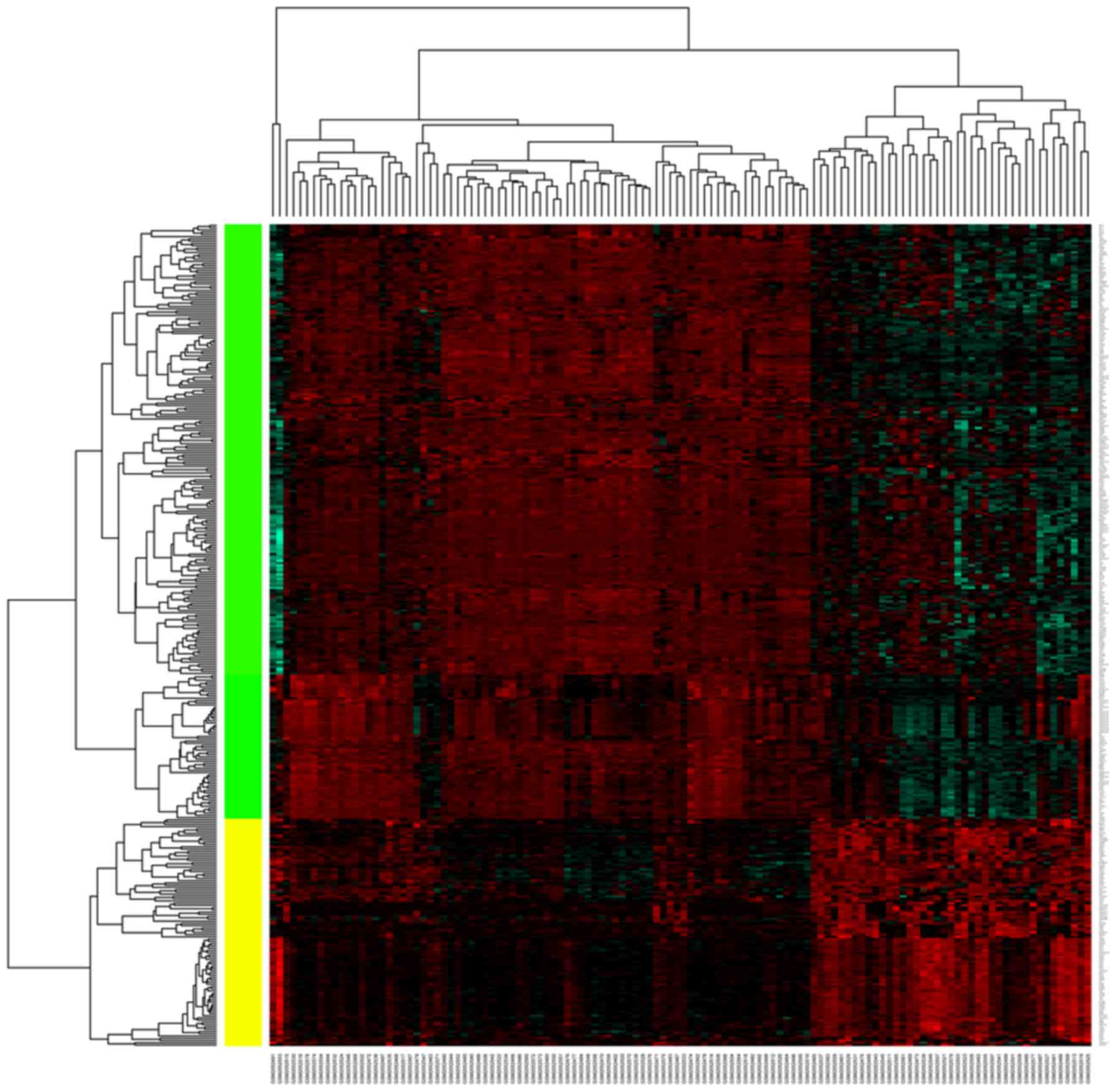
downregulated DEGs. The heatmap of DEGs was shown in Fig. 1.
Functional enrichment analysis
The KEGG pathways of the upregulated and
downregulated DEGs were obtained. The upregulated DEGs were
enriched in nine KEGG pathways such as cell cycle, pathways in
cancer and PI3K-Akt signaling pathway; the downregulated DEGs were
enriched in ten KEGG pathways such as metabolic pathways,
biosynthesis of antibiotics and chemical carcinogenesis. In
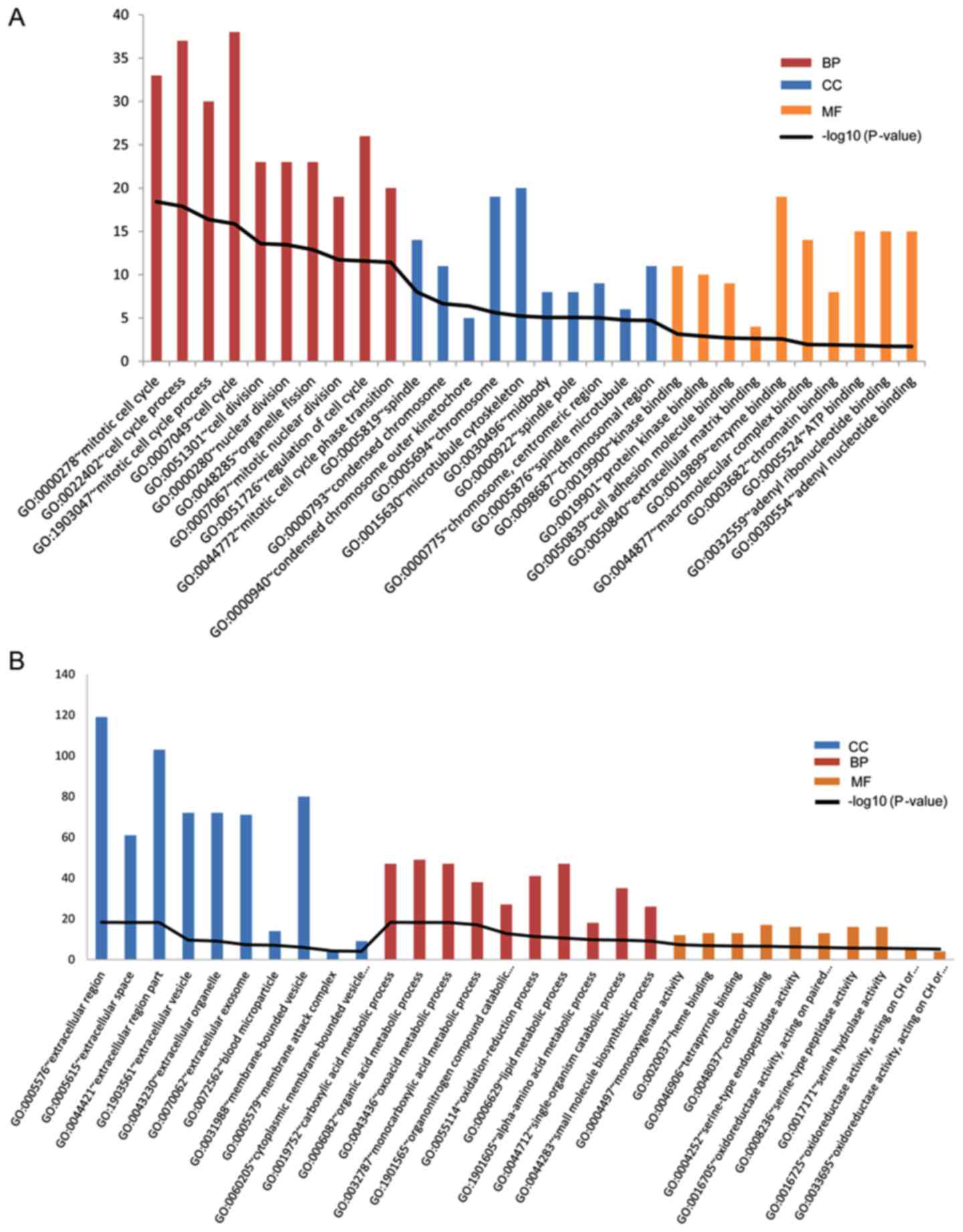
addition, significant GO terms enriched by DEGs were obtained. The
upregulated DEGs were enriched in GO terms such as cell cycle,
microtubule cytoskeleton and enzyme binding (Fig. 2A); and the downregulated DEGs were
enriched in GO terms such as extracellular region, organic acid
metabolic process and cofactor binding (Fig. 2B).
PPI network analysis
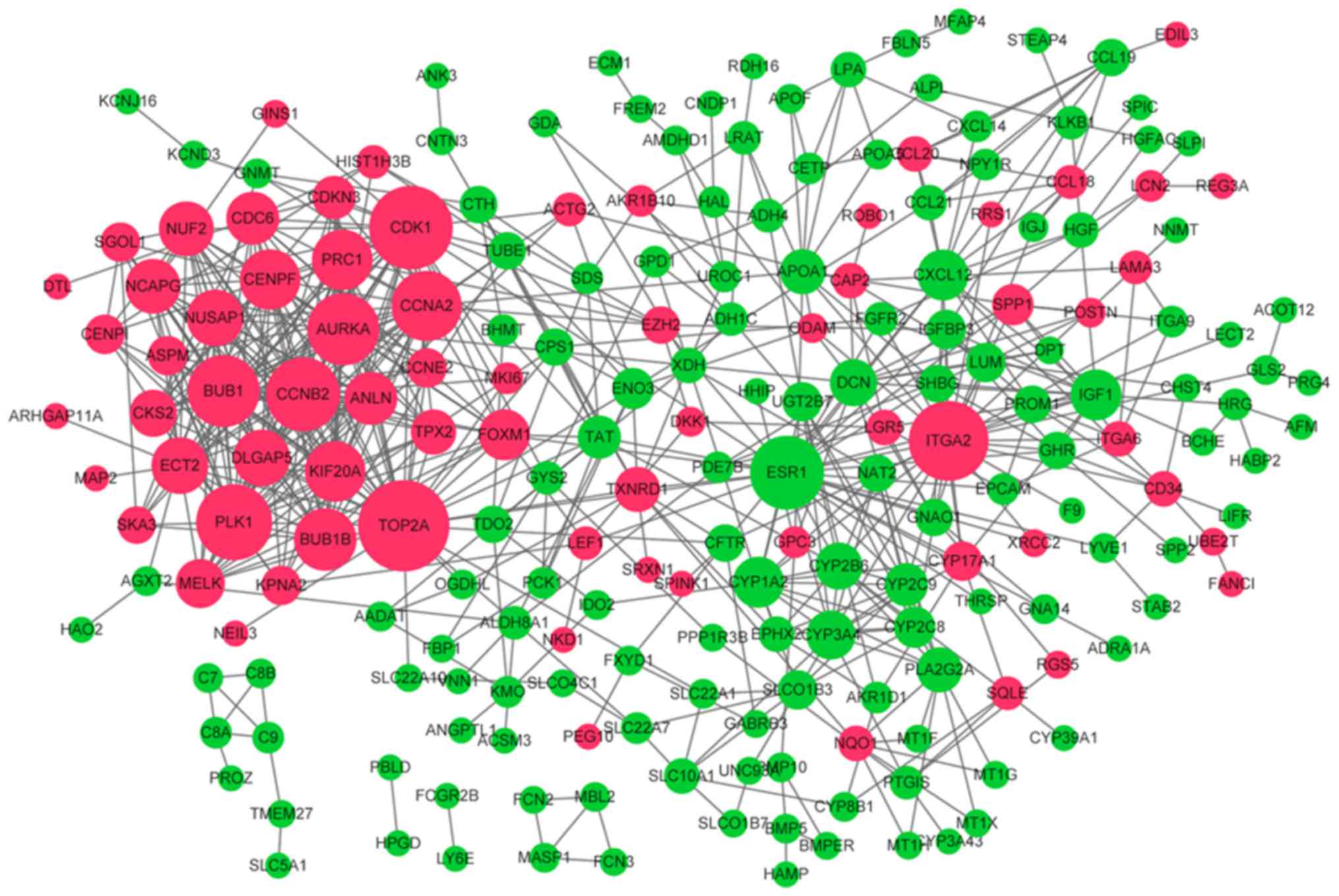
PPI network analysis for the DEGs was performed,
containing 211 nodes and 618 PPI pairs (Fig. 3). Nodes with top ten highest score
in three algorithms were listed in Table I, such as topoisomerase (DNA) IIα
(TOP2A), cyclin dependent kinase 1 (CDK1) and integrin
subunit α2 (ITGA2) (degree centrality); estrogen receptor 1
(ESR1), TOP2A and ITGA2 (betweenness
centrality); and ESR1, TOP2A and ITGA2 (closeness
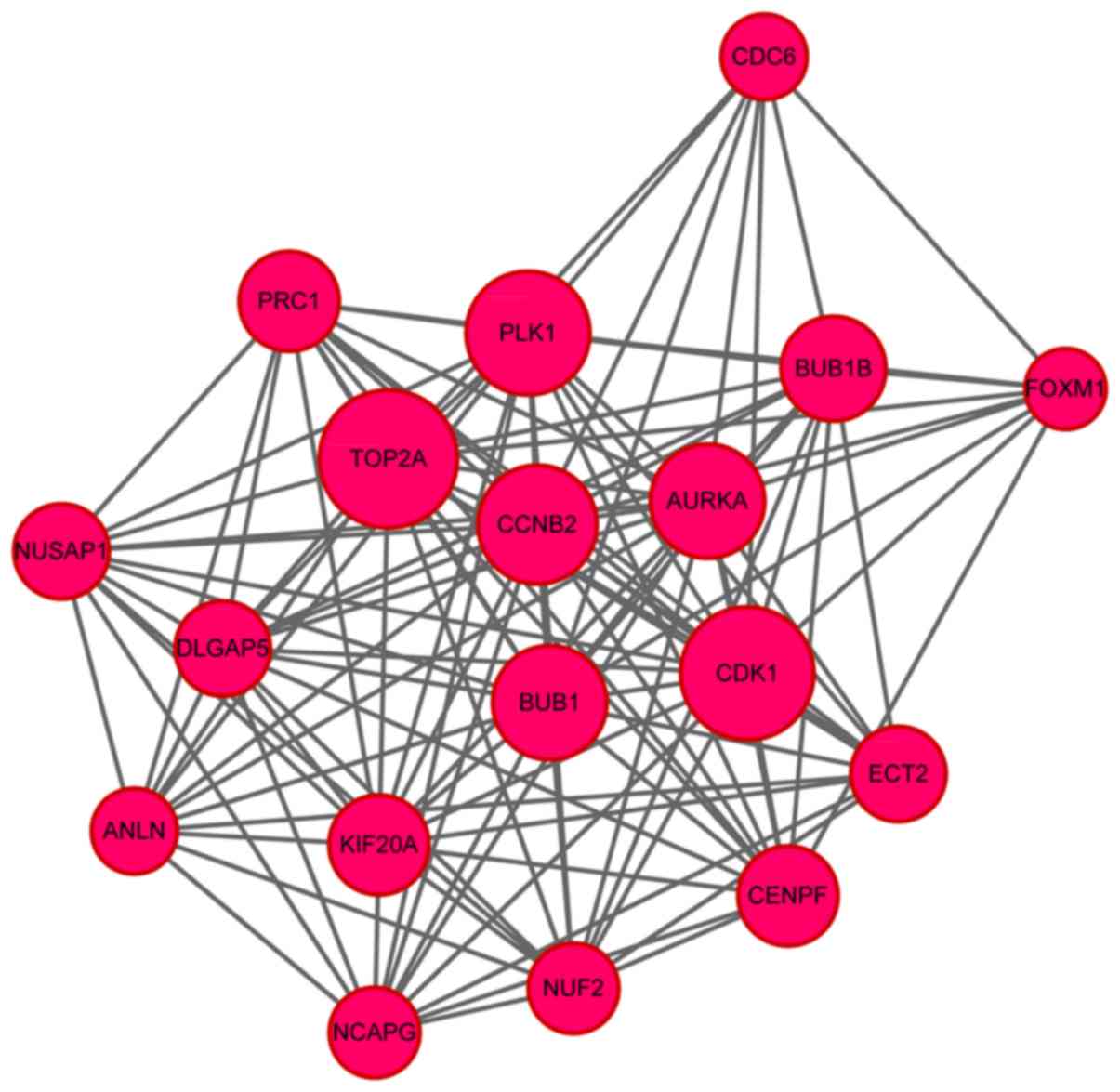
centrality). In addition, a sub-network including the most genes
was identified via sub-network analysis, including 18 nodes and 123
PPI pairs (Fig. 4). In the
identified sub-network, TOP2A (degree=35), CDK1
(degree=31) and polo like kinase 1 (PLK1) (degree=27) were
the nodes with most PPI pairs. Functional enrichment analysis for
the genes in this sub-network revealed that these genes were
significantly enriched in one GO term (cell division) and three
KEGG pathways including cell cycle, progesterone-mediated oocyte
maturation and oocyte meiosis (Table
II). Additionally, CDK1, PLK1 and mitotic checkpoint
serine/threonine kinase (BUB1) were the common genes in the
GO term and KEGG pathways mentioned above (Table II).
 | Table I.List of top 10 highest scoring nodes
in the protein-protein interaction network. |
Table I.
List of top 10 highest scoring nodes
in the protein-protein interaction network.
| Rank | Top 10 genes | Degree | Top 10 genes | Betweenness | Top 10 genes | Closeness |
|---|
| 1 | TOP2A | 35 | ESR1 | 9275.706 | ESR1 | 0.05781939 |
| 2 | CDK1 | 31 | ITGA2 | 7831.359 | ITGA2 | 0.05731441 |
| 3 | ITGA2 | 29 | TOP2A | 6589.938 | TOP2A | 0.05729877 |
| 4 | PLK1 | 27 | APOA1 | 4281.257 | CDK1 | 0.05675676 |
| 5 | ESR1 | 26 | DCN | 4087.898 | IGF1 | 0.05663430 |
| 6 | CCNB2 | 26 | CDK1 | 3732.873 | TXNRD1 | 0.05658852 |
| 7 | AURKA | 25 | TXNRD1 | 3077.681 | CFTR | 0.05657328 |
| 8 | BUB1 | 25 | CXCL12 | 2707.838 | DCN | 0.05652759 |
| 9 | CCNA2 | 24 | IGF1 | 2424.979 | FOXM1 | 0.05651238 |
| 10 | BUB1B | 20 | TAT | 2213.226 | CYP2B6 | 0.05648198 |
| Table II.Functional enrichment analysis of
differentially expressed genes in the module. |
Table II.
Functional enrichment analysis of
differentially expressed genes in the module.
| Category | Term | Count | P-value | Genes |
|---|
| GOTERM_BP_FAT | GO:0051301~‘cell
division’ | 3 | 1.26×10–20 | CDC6, CDK1, PRC1,
NUF2, NUSAP1, CENPF, AURKA, ANLN, ECT2, CCNB2, NCAPG, PLK1, BUB1,
BUB1B, TOP2A, KIF20A |
| KEGG_PATHWAY | hsa04110~‘Cell
cycle’ | 5 | 1.02×10-8 | CDK1, CDC6, CCNB2,
PLK1, BUB1, BUB1B |
|
|
hsa04914~‘Progesterone-mediated oocyte
maturation’ | 11 | 3.75×10-5 | CDK1, CCNB2, PLK1,
BUB1 |
|
| hsa04114~‘Oocyte
meiosis’ | 3 | 7.38×10-5 | CDK1, PLK1, BUB1,
AURKA |
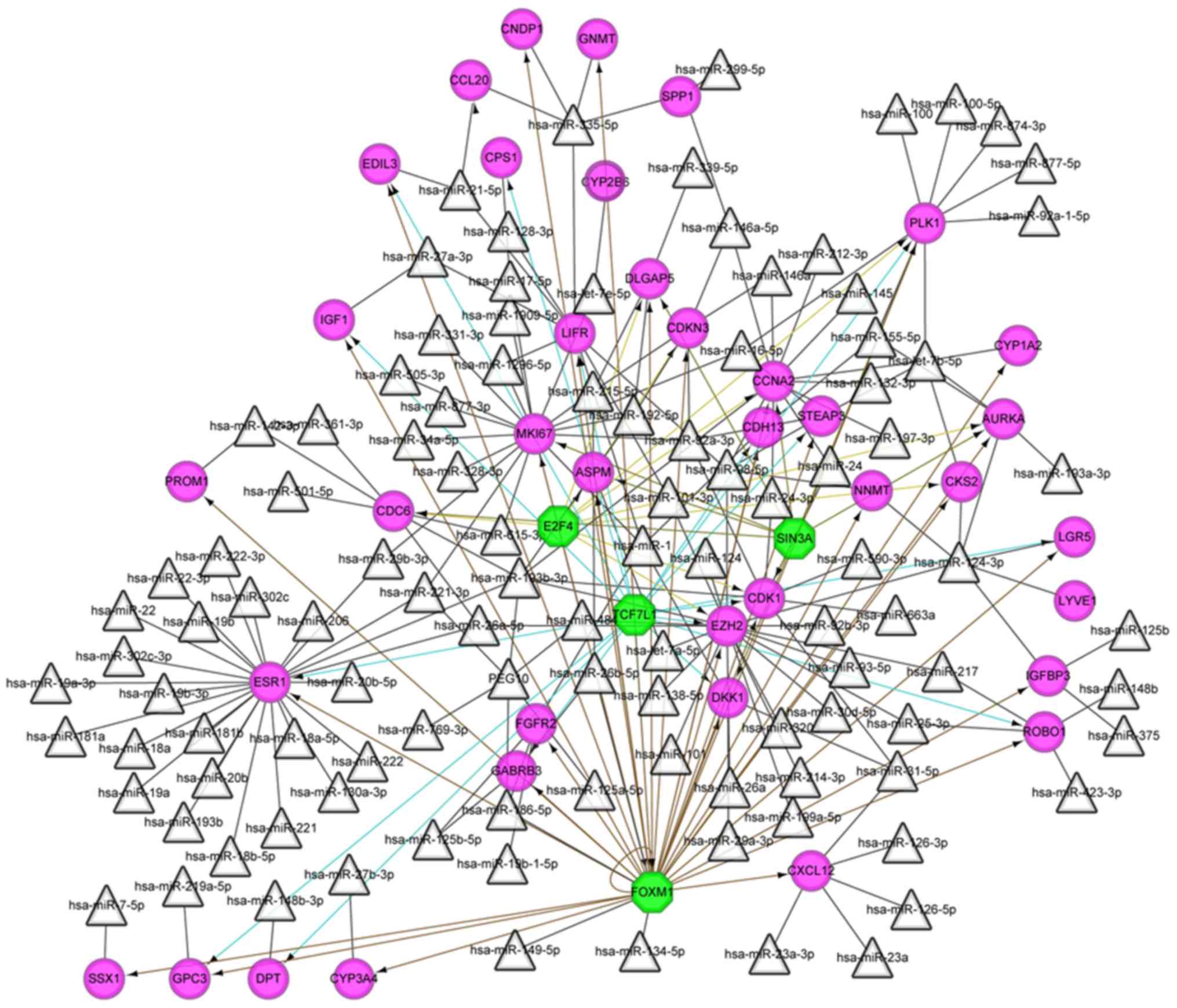
TF-miRNA-target regulatory network
analysis
The miRNA-target network was constructed, including
107 miRNAs and 40 target genes. TF prediction for the DEGs in the
miRNA-target network revealed 4 TFs including forkhead box M1
(FOXM1), E2F4, SIN3 transcription regulator family
member A (SIN3A) and transcription factor 7 like 1
(TCF7L1). As shown in the integrated TF-miRNA-target network
(Fig. 5), FOXM1 (NES=8.642)
targeted 32 genes, followed by TCF7L1 (NES=5.449) targeting
17 genes, E2F4 (NES=5.398) targeting 12 genes and
SIN3A (NES=5.095) targeting nine genes.
Discussion
HCC is among the most deadliest cancers worldwide.
Identification of promising biomarker or targets for the diagnosis
and treatment of HCC is needed. In our study, a comprehensive
microarray analysis of liver tumor and normal samples from patients
with HCC was performed. The results of our analyses revealed that
key upregulated DEGs such as TOP2A, ITGA2, PLK1 and
CDK1 were associated with HCC. Besides, cell division and
cell cycle might play key roles in the development of HCC.
Furthermore, crucial TFs including FOXM1, TCF7L1, E2F4 and
SIN3A were revealed to be key TFs related to HCC.
TOP2A and ITGA2 were identified as the
key nodes in the PPI network. Moreover, TOP2A, PLK1 and
CDK1 were the genes with the highest degrees in the
sub-network. It is reported that TOP2A is overexpressed in
HCC tissues (28), which is in
accordance to our results. Furthermore, it is revealed that the
expression of TOP2A is related to the onset of malignancy
and chemoresistance, shortening the survival time of patients
(29). Silencing of ITGA2
can promote the migration of breast cancer cells (30). What's more, inhibition of
ITGA2 by miR-128 can significantly decrease the metastasis
of HCC cells (31). It is
indicated that the positive expression rate of PLK1 in HCC
is higher than that in healthy controls (32). Additionally, it is reported that
knockdown of PLK1 in HCC tumor-derived endothelial cells can
inhibit the migration of these cells (33). CDK1 is reported to have the
capacity of apoptosis modulation in HCC by co-acting with apoptin
(34). Moreover, it is
demonstrated that miR-582-5p can inhibit the proliferation of HCC
cells through targeting CDK1 and another gene AKT
serine/threonine kinase 3 directly (35). Based on our results, we speculate
that TOP2A, ITGA2, PLK1 and CDK1 may be the key genes
involved in HCC.
Functional enrichment analysis for genes in the
sub-network revealed that PLK1 and CDK1 were the
common genes involved in both cell division and cell cycle. HCC
cells lost control of the cell cycle, which is a common feature
occurring in all tumorigenic cells (4). Inhibition of PLK1 activity can
decrease the polyploid cell population during cell division
(36). Moreover, it is revealed
that the downregulation of PLK1 results in the promotion of
cell cycle arrest and apoptosis (37). It is confirmed that CDK1 is
essential for cell division through knockdown of CDK1 in
mice (38). Besides, CDK1
has been considered to be the only essential cell cycle CDK
(39). Thus, we suspect that
PLK1 and CDK1 may play roles in HCC by participating
in cell cycle and cell division.
The result of TF-miRNA-target network analysis
showed that FOXM1, TCF7L1, E2F4 and SIN3A were TFs
regulating the most DEGs. It is reported that FOXM1
overexpression can promote the metastasis of HCC (40). Meanwhile, inhibition of
FOXM1 is shown to have the capacity of promoting the
senescence of HCC cells (41).
TCF7L1, a member of the T cell factor/lymphoid enhancer
factor family, is reported to modulate colorectal cancer growth via
inhibiting the tumor suppressor EPH Receptor B3 (42). As another member of the family,
TCF7L2 is revealed to be associated with the susceptibility
of HCC in patients with liver cirrhosis (43). As a member of the E2F family,
E2F4 acts as an anti-proliferative TF, playing crucial role
in cell cycle (44). Besides,
comparing with the wild-type E2F4, E2F4 mutants can increase
the growth of colorectal cancer cells (45). It is reported that the upregulation
of miR-210 can inhibit proliferation of HCC cells (46). In addition, miR-210 upregulation is
revealed to inhibit proliferation and induce apoptosis in glioma
cells via targeting SIN3A, a member of the SIN3
transcription regulator family (47). Based on these data, we speculate
that FOXM1, TCF7L1, E2F4 and SIN3A may be key TFs
participating in the development of HCC.
Notably, Maswska et al (16) reported that three subgroups of HCC
patients were identified based on the gene expression data of
GSE64041 and the subgroups of patients have different prognosis,
suggesting that the different expression pattern of genes in
different subgroups may be an important factor to influence HCC
progression and patients' prognosis. However, we did not analyze
the possible mechanism underlying HCC in the different subgroups
due to lack of the related expression data of different subgroups,
let alone the gene expression and the patients' prognosis. Further
analyses should be performed to further elucidate the key mechanism
associated with the patients' prognosis in different subgroups.
Moreover, some experiments, such as expression validation or
knockdown assays, were not performed to determine the role of these
key genes and TFs in HCC development. Taken together, further
investigations remain to be done to confirm the results.
In conclusion, the results of the present study
reveal that TOP2A, ITGA2, PLK1 and CDK1 may act as
key genes associated with HCC. Moreover, cell cycle and cell
division may function as key pathways in HCC through the regulation
of key genes PLK1 and CDK1. Additionally, FOXM1,
TCF7L1, E2F4 and SIN3A are revealed to be key TFs
involved in HCC. Our results might provide valuable data for
selection of biomarker and therapeutic targets of the disease.
Acknowledgements
Not applicable.
Funding
The present study was supported by Innovative
Research Groups of National Natural Science Foundation of China
(grant no. 81421062).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JW contributed to the concept and design of the
study, and drafted and revised the manuscript for important
intellectual content. HL acquired the data. YT analyzed and
interpreted the data. HC performed the statistical analysis. SZ
obtained the data and funding. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sun VC and Sarna L: Symptom management in
hepatocellular carcinoma. Clin J Oncol Nurs. 12:759–766. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maluccio M and Covey A: Recent progress in
understanding, diagnosing, and treating hepatocellular carcinoma.
CA Cancer J Clin. 62:394–399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aravalli RN, Steer CJ and Cressman EN:
Molecular mechanisms of hepatocellular carcinoma. Hepatology.
48:2047–2063. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Henley SA and Dick FA: The retinoblastoma
family of proteins and their regulatory functions in the mammalian
cell division cycle. Cell Div. 7:102012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gopinathan L, Ratnacaram CK and Kaldis P:
Established and novel Cdk/cyclin complexes regulating the cell
cycle and development. Results Probl Cell Differ. 53:365–389. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wilson GK, Tennant DA and Mckeating JA:
Hypoxia inducible factors in liver disease and hepatocellular
carcinoma: Current understanding and future directions. J Hepatol.
61:1397–1406. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hernandez-Gea V, Toffanin S, Friedman SL
and Llovet JM: Role of the microenvironment in the pathogenesis and
treatment of hepatocellular carcinoma. Gastroenterology.
144:512–527. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang JD, Nakamura I and Roberts LR: The
tumor microenvironment in hepatocellular carcinoma: Current status
and therapeutic targets. Semin Cancer Biol. 21:35–43. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ran G, Yang G, Fang W, Yuan Y and Zhang R:
Value of serumGSTP1 gene quantitative methylation analysis for
early diagnosis of hepatocellular carcinoma. Int J Lab Med.
540–542. 2014.
|
|
11
|
Luo S, Zhang FC, Liu CB, Wang X, Zhuang JX
and Zhang J: Methylation of p16 gene in plasma in diagnosis of
primary hepatocellular carcinoma. Acta Scientiarium Naturalium
Universitatis Jilinensis. 55:2004.(In Chinese).
|
|
12
|
Yu JM, Chen Q, Ye YB, et al: Expression of
GPC3 gene and its significance for of diagnosis in hepatocellular
carcinoma. Chin J Clin Oncol Rehabil. 17:2010.(In Chinese).
|
|
13
|
Yao M, Wang L, Dong Z, Qian Q, Shi Y, Yu
D, Wang S, Zheng W and Yao D: Glypican-3 as an emerging molecular
target for hepatocellular carcinoma gene therapy. Tumour Biol.
35:5857–5868. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang C and Gong F: DNA Methyltransferase
1: A potential gene therapy target for hepatocellular carcinoma?
Oncol Res Treat. 39:448–452. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Llovet JM and Bruix J: Molecular targeted
therapies in hepatocellular carcinoma. Hepatology. 48:1312–1327.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Makowska Z, Boldanova T, Adametz D,
Quagliata L, Vogt JE, Dill MT, Matter MS, Roth V, Terracciano L and
Heim MH: Gene expression analysis of biopsy samples reveals
critical limitations of transcriptome-based molecular
classifications of hepatocellular carcinoma. J Pathol Clin Res.
2:80–92. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Barrett T, Suzek TO, Troup DB, Wilhite SE,
Ngau WC, Ledoux P, Rudnev D, Lash AE, Fujibuchi W and Edgar R: NCBI
GEO: Mining millions of expression profiles-database and tools.
Nucleic Acids Res. 33:(Database Issue). D562–D566. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Smyth GK: Limma: linear models for
microarray dataBioinformatics and Computational Biology Solutions
using R and Bioconductor. Gentleman R, Carey V, Dudoit S, Irizarry
R and Huber W: Springer; New York, NY: pp. 397–420. 2005,
View Article : Google Scholar
|
|
19
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
da Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
von Mering C, Huynen M, Jaeggi D, Schmidt
S, Bork P and Snel B: STRING: A database of predicted functional
associations between proteins. Nucleic Acids Res. 31:258–261. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang Y, Li M, Wang J, Pan Y and Wu FX:
CytoNCA: A cytoscape plugin for centrality analysis and evaluation
of biological networks. Biosystems. 127:67–72. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dweep H and Gretz N: miRWalk2.0: A
comprehensive atlas of microRNA-target interactions. Nat Methods.
12:6972015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Janky R, Verfaillie A, Imrichová H, Van de
Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K,
Sanchez Naval M, Potier D, et al: iRegulon: From a gene list to a
gene regulatory network using large motif and track collections.
PLoS Comput Biol. 10:e10037312014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Panvichian R, Tantiwetrueangdet A,
Angkathunyakul N and Leelaudomlipi S: TOP2A amplification and
overexpression in hepatocellular carcinoma tissues. Biomed Res Int
2015: Article ID 381602. 2015. View Article : Google Scholar
|
|
29
|
Wong N, Yeo W, Wong WL, Wong NL, Chan KY,
Mo FK, Koh J, Chan SL, Chan AT, Lai PB, et al: TOP2A overexpression
in hepatocellular carcinoma correlates with early age onset,
shorter patients survival and chemoresistance. Int J Cancer.
124:644–652. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ding W, Fan XL, Xu X, Huang JZ, Xu SH,
Geng Q, Li R, Chen D and Yan GR: Epigenetic silencing of ITGA2 by
MiR-373 promotes cell migration in breast cancer. PLoS One.
10:e01351282015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao X, Wu Y and Lv Z: miR-128 modulates
hepatocellular carcinoma by inhibition of ITGA2 and ITGA5
expression. Am J Transl Res. 7:1564–1573. 2015.PubMed/NCBI
|
|
32
|
He ZL, Zhong DW, Zheng H, Miao XY, Hu JX
and Wen Y: Expression of gene Plk1 and its relationship with
prognosis of hepatocellular carcinoma. World Chin J Digestology.
17:146–150. 2009. View Article : Google Scholar
|
|
33
|
Yu HJ, Wu Y, Li FS, Li W and Jiang QS:
Influence of knocking down Plk1 on migration of human
hepatocellular carcinoma tumor-derived endothelial cells. Chin J
Cancer Prev Treat. 22:584–587. 2015.(In Chinese).
|
|
34
|
Zhao J, Han SX, Ma JL, Ying X, Liu P, Li
J, Wang L, Zhang Y, Ma J, Zhang L and Zhu Q: The role of CDK1 in
apoptin-induced apoptosis in hepatocellular carcinoma cells. Oncol
Rep. 30:253–259. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yi Z, Wei H, Yan R, Xiong Y, Zhong Z, Fan
X, Wang Z and Ye Q: miR-582-5p inhibits proliferation of
hepatocellular carcinoma by targeting CDK1 and AKT3. Tumour Biol.
36:8309–8316. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang Z, Hou SQ, He J, Gu T, Yin Y and
Shen WH: PTEN regulates Plk1 and controls chromosomal stability
during cell division. Cell Cycle. 15:2476–2485. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nihal M, Stutz N, Schmit T, Ahmad N and
Wood GS: Polo-like kinase 1 (Plk1) is expressed by cutaneous T-cell
lymphomas (CTCLs), and its downregulation promotes cell cycle
arrest and apoptosis. Cell Cycle. 10:1303–1311. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Diril MK, Ratnacaram CK, Padmakumar VC, Du
T, Wasser M, Coppola V, Tessarollo L and Kaldis P: Cyclin-dependent
kinase 1 (Cdk1) is essential for cell division and suppression of
DNA re-replication but not for liver regeneration. Proc Natl Acad
Sci USA. 109:3826–3831. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Santamaría D, Barrière C, Cerqueira A,
Hunt S, Tardy C, Newton K, Cáceres JF, Dubus P, Malumbres M and
Barbacid M: Cdk1 is sufficient to drive the mammalian cell cycle.
Nature. 448:811–815. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Meng FD, Wei JC, Qu K, Wang ZX, Wu QF, Tai
MH, Liu HC, Zhang RY and Liu C: FoxM1 overexpression promotes
epithelial-mesenchymal transition and metastasis of hepatocellular
carcinoma. World J Gastroenterol. 21:196–213. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Qu K, Xu X, Liu C, Wu Q, Wei J, Meng F,
Zhou L, Wang Z, Lei L and Liu P: Negative regulation of
transcription factor FoxM1 by p53 enhances oxaliplatin-induced
senescence in hepatocellular carcinoma. Cancer Lett. 331:105–114.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Murphy M, Chatterjee SS, Jain S, Katari M
and Dasgupta R: TCF7L1 modulates colorectal cancer growth by
inhibiting expression of the tumor-suppressor gene EPHB3. Sci Rep.
6:282992016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ling Q, Dong F, Geng L, Liu Z, Xie H, Xu X
and Zheng S: Impacts of TCF7L2 gene polymorphisms on the
susceptibility of hepatogenous diabetes and hepatocellular
carcinoma in cirrhotic patients. Gene. 522:214–218. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Baiz D, Dapas B, Farra R, Scaggiante B,
Pozzato G, Zanconati F, Fiotti N, Consoloni L, Chiaretti S and
Grassi G: Bortezomib effect on E2F and cyclin family members in
human hepatocellular carcinoma cell lines. World J Gastroenterol.
20:795–803. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Paquin MC, Leblanc C, Lemieux E, Bian B
and Rivard N: Functional impact of colorectal cancer-associated
mutations in the transcription factor E2F4. Int J Oncol.
43:2015–2022. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tan W, Lim SG and Tan TM: Up-regulation of
microRNA-210 inhibits proliferation of hepatocellular carcinoma
cells by targeting YES1. World J Gastroenterol. 21:13030–13041.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shang C, Hong Y, Guo Y, Liu YH and Xue YX:
MiR-210 up-regulation inhibits proliferation and induces apoptosis
in glioma cells by targeting SIN3A. Med Sci Monit. 20:2571–2577.
2014. View Article : Google Scholar : PubMed/NCBI
|