Introduction
Gallbladder cancer (GBC) is the most common and
aggressive type of malignant tumor of the biliary tract in humans
(1). The lack of markers for the
timely prediction and diagnosis of GBC, and its frequently silent
and rapid progression confer a poor prognosis. Statistics show
that, due to failure in promptly diagnosing GBC, <10% of
patients are suitable to undergo curative resection (2). Furthermore, the overall mean survival
rate is only 6 months in the majority of patients, even following
surgery, and the five-year survival rate is only 5% (3). To date, the elucidation of several
molecular mechanisms has revealed the roles of genetic and
epigenetic changes involved in the tumorigenicity and progression
of human cancer. However, these factors in GBC remain to be fully
elucidated.
The aberrant methylation of DNA, the most well known
type of epigenetic modification, occurs at CpG islands in promoter
regions and generally leads to methylation-based downregulation or
silencing of gene expression. The downregulation of tumor
suppressors, including P53 (4), phosphatase and tensin homolog
(5), cyclin D2 (6) and Wnt inhibitory factor-1
(WIF-1) (7), caused by
aberrant promoter methylation is integral in the occurrence and
development of several types of cancer. Modifications in
methylation are mediated by DNA methyltransferases (DNMTs),
including DNMT1, DNMT3a, and DNMT3b. DNMT1 maintains its
methyltransferase activity to reestablish methylation patterns
throughout the DNA synthesis process, whereas DNMT3a and DNMT3b act
as de novo methyltransferases (8).
WIF-1, an antagonist of the Wnt/β-catenin
pathway has been demonstrated to be an important tumor suppressor
(7). As a secreted
frizzled-related protein, WIF-1 can directly bind to Wnt molecules
at the cell surface, thereby preventing Wnt from binding to
receptors (9). It has been
demonstrated that the expression of WIF-1 is decreased in
several types of cancer, including breast cancer (10), lung cancer (11) and cervical cancer (12). The low expression of WIF-1
leads to abnormal activation of the Wnt pathway, resulting in the
dysregulation of cell proliferation and differentiation, and
inducing carcinogenesis in humans. There is also increasing
evidence supporting the hypothesis that low expression levels of
WIF-1 can largely be attributed to aberrant hypermethylation
of its promoter region (10–12).
In our previous study, it was demonstrated that WIF-1 was
downregulated in GBC cell lines and tissues, which led to aberrant
activation of the Wnt pathway, altering the processes of
proliferation, invasion, metastasis and apoptosis; WIF-1 was
involved in the tumorigenicity and progression of GBC through these
effects (13). However, the
upstream molecular mechanism regulating WIF-1 remains to be
fully elucidated, particularly in GBC.
c-Jun, the first oncogenic transcription
factor to be identified, is the cellular homolog of the viral
oncoprotein v-Jun, which is transcriptionally activated at Ser63
and Ser73 by Janus kinase (14).
Early experiments revealed that the cooperation between
c-Jun and oncogenic RAS is involved in tumor initiation and
increased invasiveness in humans (15,16).
In addition, c-Jun has been shown to lead to loss of
function of the tumor suppressor p53 (17,18).
c-Jun is widely expressed in different human tumors, is
involved in numerous cell signaling pathways, and contributes to
the pathogenesis, invasion and metastasis of cancer through diverse
mechanisms (19,20).
The present study provided novel evidence
demonstrating that the low expression of WIF-1 in GBC cells
was caused by aberrant hypermethylation of its promoter. The
results revealed a negative correlation between the protein levels
of c-Jun and WIF-1 in 50 GBC specimens. It was subsequently
determined that the knockdown of c-Jun using RNA
interference restored the expression of WIF-1. Based on
these results and previous findings, it was hypothesized that
c-Jun affects the process of methylation, particularly that
performed by major methyltransferases, which leads to
hypermethylation and downregulation of the WIF-1 gene in
GBC.
Materials and methods
Specimens
A total of 50 GBC sample tissues and 20
cholecystitis tissues were obtained from patients at the Department
of Surgery and Pathology of Fujian Medical University Union
Hospital (Fujian, China) between 2006 and 2013. None of the GBC
patients had received any preoperative chemotherapy or
radiotherapy. The tissue samples were fixed in 10% buffered
formalin and embedded in paraffin wax. Written consent was obtained
from each patient to perform experiments on the resected specimens,
and the study was approved by the ethnical committee of the Medical
Faculty of Fujian Medical University in accordance with the 1975
Declaration of Helsinki.
Immunohistochemistry and
evaluation
Serial 4 µm sections were obtained from the
formalin-fixed and paraffin-embedded tissues. Following
deparaffinization in turpentine and rehydration in an alcohol
gradient, the tissue sections were incubated in 3% hydrogen
peroxide for 10 min at room temperature (~25°C) to prevent the
activity of endogenous peroxidases. The tissue sections were
subjected to antigen retrieval through boiling in citrate buffer
(pH 6.0) for 10 min in a microwave, following which they were
cooled at room temperature for 45 min and washed with
phosphate-buffered saline (PBS). Non-specific antigens in the
sections were blocked via incubation for 20 min in 5% normal goat
serum (Beyotime Institute of Biotechnology, Shanghai, China)
diluted in PBS. The tissue sections were subsequently incubated in
a 1:100 dilution of a rabbit polyclonal anti-human c-Jun antibody
(cat. no. ab31419; Abcam, Cambridge, UK) or a 1:100 dilution of a
rabbit polyclonal anti-human WIF-1 antibody (cat. no. ab186845;
Abcam) overnight in humidified boxes at 4°C. Following being washed
in PBS, the sections were incubated according to the instructions
provided with the UltraSensitive S-P kit (Maixin-Bio, Fuzhou,
China). Prior to being dehydrated and mounted, the sections were
stained with 3,3′-diaminobenzidine (DAB) for 3–5 min and
counterstained using hematoxylin for 15 min. PBS was substituted
for the primary antibody as a negative control. Cells showing
deposition of buff-colored granules in the cytoplasm and nucleus
were scored as c-Jun- or WIF-1-positive. The expression levels of
c-Jun or WIF-1 were semiquantitatively analyzed using the mean
optical density (MOD), defined as the integral optical
density/positive area, calculated using Image-Pro Plus 6.0 software
(Media Cybernetics, Inc., Rockville, MD, USA).
Cell culture and treatment
The human GBC cell lines, NOZ (Health Science
Research Resources Bank, Osaka, Japan), GBC-SD (Shanghai Institutes
for Biological Sciences, Shanghai, China) and SGC-996 (Tumor
Cytology Research Unit, Medical College, Tongji University,
Shanghai, China) were cultured in Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) containing 10% fetal bovine serum (FBS; HyClone, GE Healthcare
Life Sciences, Logan, UT, USA) under 5% CO2/95% air, in a
humidified 37°C incubator.
The GBC cell lines were divided into 6-well plates
at a density of 2×105 cells/well (~30% confluence) 12 h
prior to treatment. The cells were treated with
5-aza-2′-deoxycytidine (DAC; Sigma-Aldrich; Merck Millipore,
Darmstadt, Germany) dissolved in dimethyl sulfoxide at a
concentration of 5.0 µM, which was replaced every 24 h for a total
treatment duration of 72 h.
Cell transfection
Appropriate small interfering (si)RNA target
sequences were determined based on the human c-Jun sequence
(GenBank accession no. NM_002228.3). DNA template oligonucleotides
corresponding to three siRNA sequences were designed according to
the guidelines for siRNA design: P-1 sense,
5′-GGACCUUAUGGCUACAGUATT-3′ and antisense,
5′-UACUGUAGCCAUAAGGUCCTT-3′; P-2 sense, 5′-ACGCAAACCUCAGCAACUUTT-3′
and antisense, 5′-AAGUUGCUGAGGUUUGCGUTT-3′; and P-3 sense,
5′-GGAACAGGUGGCACAGCUUTT-3′ and antisense,
5′-AAGCUGUGCCACCUGUUCCTT-3′, which were constructed by GenePharma
Co., Ltd. (Shanghai, China). A non-targeting siRNA was used as a
negative control (NC-siRNA).
Transfection was performed with Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol, when the NOZ cells were at ~90%
confluence. The transfection efficiency was evaluated by counting
the percentage of enhanced green fluorescent protein-positive NOZ
cells using a fluorescence microscope. The cells were harvested 48
h following transfection to extract mRNA for reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis and at 72 h to extract protein for western blot
analysis.
RNA extraction and RT-qPCR
analysis
Total RNA was extracted from the GBC cells with
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and
then quantified using a spectrophotometer. cDNA was synthesized
with the Revert Aid First-Strand cDNA Synthesis kit (Thermo Fisher
Scientific, Inc.) using 2 µg of RNA according to the manufacturer's
protocol. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used
as an internal control. The primers for WIF-1, c-Jun, DNMT1,
DNMT3a, DNMT3b and GAPDH were designed and synthesized according to
standard primer design principles (Table I). PCR reactions were performed
using FastStart Universal SYBR-Green Master Mix (Roche Diagnostics,
Basel, Switzerland) on a 7500 Fast Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The following PCR
conditions were used: Initial denaturation at 95°C for 3 min,
followed by 40 cycles at 95°C for 15 sec and at 60°C for 30 sec.
Each sample was analyzed in triplicate. The relative expression
levels of target genes were calculated based on normalization to
the endogenous mRNA expression of GAPDH, prior to comparative
analysis using the 2−ΔΔCq method (21).
 | Table I.Primers used for polymerase chain
reaction and sequence analyses. |
Table I.
Primers used for polymerase chain
reaction and sequence analyses.
| Gene | Forward primer | Reverse primer |
|---|
| WIF-1 |
5′-CCAGGACTAGAGGGAGAGCA-3′ |
5′-TCGCAGACAGGCTTTGAACA-3′ |
| c-Jun |
5′-AGGAAGCTGGAGAGAATCGC-3′ |
5′-GTTAGCATGAGTTGGCACCC-3′ |
| DNMT1 |
5′-AGAAGTGAAGCCCGTAGAGTG-3′ |
5′-ATGAGATGTGATGGTGGTTTGG-3′ |
| DNMT3a |
5′-TGTAACGAAGTGAAGGAGGAGAA-3′ |
5′-CATCTTGCCGAGGGAGTCT-3′ |
| DNMT3b |
5′-AGAGGAGTGTGAAGCAAGGA-3′ |
5′-TGAGAAATGAGGGTAGCAGACT-3′ |
| GAPDH |
5′-AGGGCTGCTTTTAACTCTGGT-3′ |
5′-TCTCGCTCCTGGAAGATGGTG-3′ |
| USP
(WIF-1) |
5′-GAATTTTATTGGTTGAAAGGGAGAT-3′ |
5′-AAAAATAAAAAAAACAAACAACACT-3′ |
| MSP
(WIF-1) |
5′-AATTTTATTGGTTGAAAGGGAGAC-3′ |
5′-AAAAATAAAAAAAACAAACAACGCT-3′ |
| BSP
(WIF-1) |
5′-GGAATTTTTAAATGTTGGGTGT-3′ |
5′-AAATAATAACTCCTATTCCTCCTCC-3′ |
Protein extraction and western blot
analysis
For protein extraction, the cells were lysed in RIPA
lysis buffer containing a protease inhibitor mixture. The protein
concentration was then measured using a bicinchoninic acid assay
(Beyotime Institute of Biotechnology) according to the
manufacturer's protocol. A total of 20 µg of protein was subjected
to 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis
(SDS-PAGE) and then transferred onto a polyvinylidene fluoride
(PVDF) membrane (GE Healthcare Life Sciences, Chalfont, UK).
Following blocking with 5% nonfat milk for 3 h, the PVDF membranes
were separately incubated with primary antibodies against c-Jun
(1:1,000 dilution; cat. no. ab31419; Abcam), WIF-1 (1:1,000
dilution; cat. no. ab186845; Abcam), DNMT1 (1:1,000 dilution; cat.
no. 5119; Cell Signaling Technology, Inc., Danvers, MA, USA),
DNMT3a (1:1,000 dilution; cat. no. 3598; Cell Signaling Technology,
Inc.), DNMT3b (1:1,000 dilution; cat. no. 67259; Cell Signaling
Technology, Inc.) and β-actin (1:1,000 dilution; cat. no. sc-47778;
Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C.
Following several washes with Tris-buffered saline with Tween
(TBST), the appropriate secondary antibody conjugated to
horseradish peroxidase (1:2,000 dilution; cat. no. ZB-2301 or
ZB-2305; ZSGB-BIO, Beijing, China) was added to the membranes,
followed by incubation for 1 h at room temperature. The quantities
of each protein were visualized using ECL Advance reagent following
use of a chemiluminescence western blot immunodetection kit
(Invitrogen; Thermo Fisher Scientific, Inc.).
DNA extraction, methylation-specific
PCR and bisulfate sequencing PCR (BSP)
Genomic DNA was isolated from the cells and tissues
using the TIANamp Genomic DNA kit (Tiangen Biotech Co., Ltd.)
according to the manufacturer's protocol. Subsequently, 2 µg of
genomic DNA was converted with sodium bisulfite and subsequently
cleaned using a commercial kit (EpiTect Bisulfite kit; Qiagen GmbH,
Hilden, Germany).
EMBOSS CpGplot software (http://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/)
(22) was use to predict a CpG
island spanning nucleotides −562 to −308 following depositing the
2,000 bp prior to the transcriptional start site of the
WIF-1 gene promoter sequence (NC_000012), in which 19 CpG
dinucleotides were present (Fig.
1A). The primers used to amplify bisulfite-treated genomic DNA
(nucleotides −557 to −302) for BSP were designed using MethPrimer
(http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi)
(23) and are listed in Table I. The amplified products were
extracted from a 1.5% agarose gel stained with ethidium bromide,
using a gel extraction kit (E.Z.N.A.TM Gel extraction kit; Omega
Bio-Tek, Inc., Norcross, GA, USA) and then purified for subcloning
into the pMD18-T vector (Takara Bio, Inc., Otsu, Japan).
Subsequently, 10 clones were randomly selected from each sample to
determine the methylation status of the CpG islands of the
WIF-1 promoter. The status of DNA methylation was determined
using BiQ Analyzer software version 2.00 (http://biq-analyzer.bioinf.mpi-inf.mpg.de) (24).
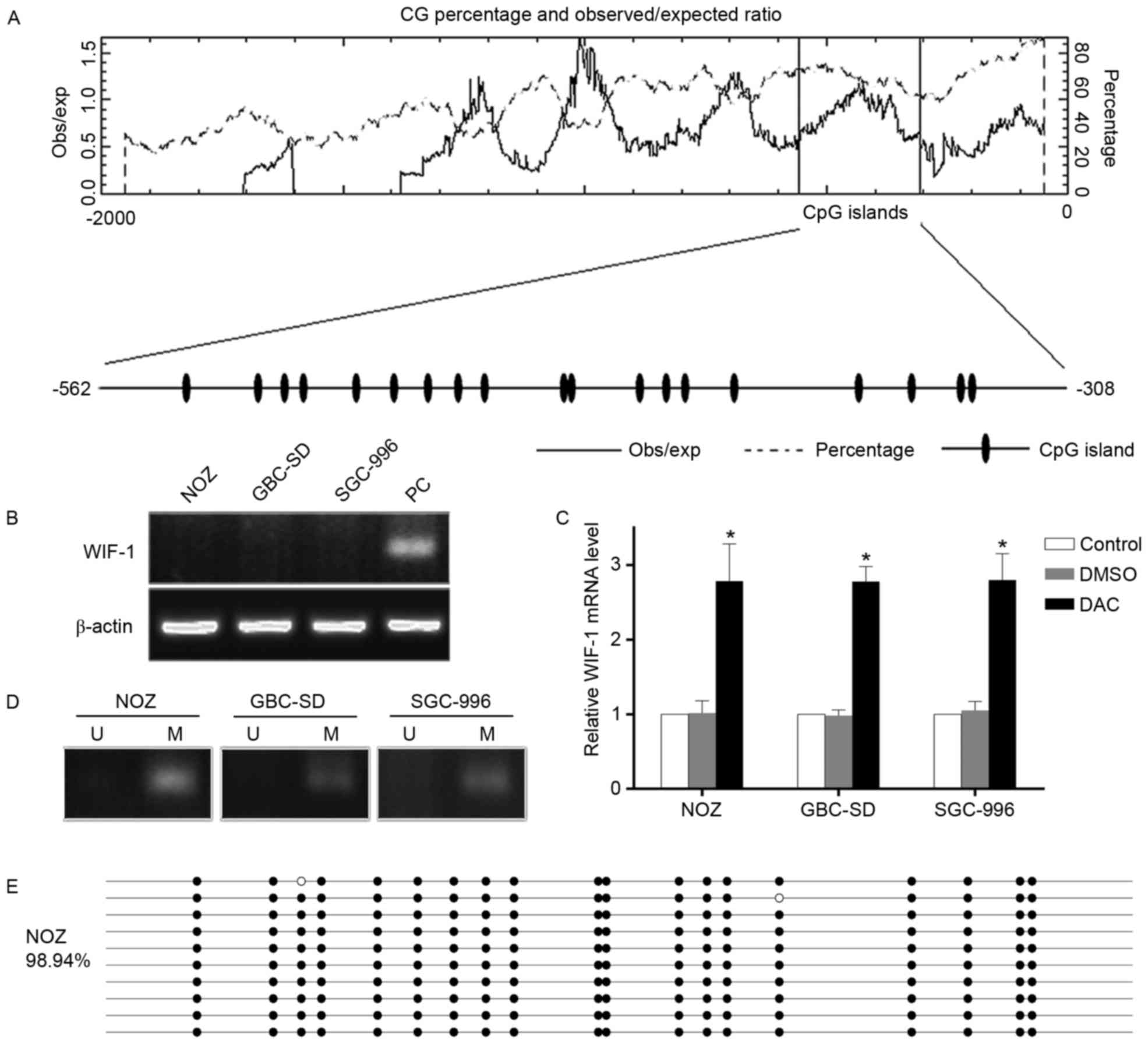 | Figure 1.Loss of the expression of
WIF-1 and aberrant hypermethylation of its promoter region
in GBC cells. (A) A putative CpG island containing 19 CpG
dinucleotides relative to the transcriptional start site meeting
the following criteria: Percent C + percent G >50%; obs/exp
ratio >0.60; length >200 bp. (B) Relative mRNA level of
WIF-1 in GBC cell lines. β-actin served as a loading
control. (C) mRNA expression of WIF-1 following DAC
treatment in GBC cell lines. *P<0.05, vs. control group. (D)
methylation-specific PCR assay of GBC cells. M bands are visible; U
bands are not visible. (E) Bisulfite genomic sequencing of the
WIF-1 promoter region in NOZ cells. Each circle corresponds
to one CpG position; filled (black) circles correspond to
methylated Cs; unfilled (white) circles correspond to unmethylated
Cs. The percentage of methylated Cs was 98.94%. WIF-1, WNT
inhibitory factor 1; GBC, gallbladder cancer; M,
methylation-specific primer; U, unmethylation-specific primer; PC,
positive control normal human lung tissue; obs, observed; exp,
expected; DMSO, dimethyl sulfoxide; DAC,
5-aza-2′-deoxycytidine. |
For the methylation-specific PCR assay,
bisulfite-treated genomic DNA was amplified using either an
unmethylation-specific primer (USP) or a methylation-specific
primer (MSP), as listed in Table
I, targeting the WIF-1 promoter region sequence from
−331/-330 to −164/-164, including five CpG islands. In this assay,
results were defined as positive results when MSP bands were
visible, with negative results defined as those with visible USP
bands with or without MSP bands.
Co-immunoprecipitation assay
Soluble proteins were precleared with 1.0 µg of
normal rabbit IgG and 20 µl of the resuspended volume of protein
A/G plus-agarose (Santa Cruz Biotechnology, Inc.). Subsequently, 1
mg of total cellular protein was mixed with 3 µg of primary
antibodies (c-Jun or DNMT1), following which 20 µl of the
resuspended volume of protein A/G plus-agarose was added, and the
samples were incubated at 4°C on a rotating device overnight. The
immunoprecipitated complexes were washed with lysis buffer and then
analyzed via 10% SDS-PAGE and western blot analysis, using the
respective specific antibodies.
Statistical analysis
The statistical analyses were performed using SPSS
software version 16.0 (SPSS, Inc., Chicago, IL, USA) or GraphPad
Prism software version 6.0 (GraphPad Software, Inc., La Jolla, CA,
USA). Fisher exact test assessed the associations between the
protein expression of WIF-1 and clinical pathological parameters.
Pearson's coefficient was used for the calculation of correlations
of the MOD values of WIF-1 and c-Jun, which represented the protein
levels in GBC tissues. The measurement data are expressed as the
mean ± standard deviation from at least three independent
experiments and were analyzed using independent samples t-tests.
Two-sided P<0.05 (two-sided) was considered to indicate a
statistically significant difference.
Results
Loss of the expression of WIF-1 and
aberrant hypermethylation of its promoter region in GBC cells
The results of the RT-qPCR analysis revealed the
complete loss of WIF-1 in the GBC (NOZ, GBC-SD and SGC-996) cell
lines analyzed (Fig. 1B).
Treatment with the demethylating agent DAC effectively restored the
mRNA levels of WIF-1 (Fig. 1C),
suggesting that genome methylation repressed the expression of
WIF-1 in these cells. To further verify the presence of aberrant
hypermethylation, methylation-specific PCR analysis was performed
on bisulfite-modified genomic DNA harvested from the GBC cells. A
high level of amplification was observed with MSPs, compared with
the absence of amplification with the USPs (Fig. 1D). BSP of the NOZ promoter region
was subsequently performed. As shown in Fig. 1E, the percentage of methylated CpGs
was as high as 98.94%. These findings indicated that the low
expression level of WIF-1 in the GBC cells was caused by aberrant
hypermethylation of the promoter region.
Reduced expression of WIF-1 and
correlation between protein levels of WIF-1 and c-Jun in GBC
specimens
Negative or low expression levels of WIF-1 were
observed in the cytoplasm and nucleus of the GBC mucosal cells
using an immunohistochemistry technique. The expression of WIF-1
was only detected in 10 of the 50 gallbladder cancer samples, with
a positive rate of 20%. By contrast, positive expression was
detected in the majority of the cholecystitis tissue samples
(18/20; 90%), which represented a significant difference
(P<0.05). The associations between the expression of WIF-1 and
clinicopathological characteristics of the GBC cases, including
patient age and sex, pT stage, lymph node metastasis, distant
metastasis and histological grade, are shown in Table II, which revealed that the
expression of WIF-1 was not associated with any of these
factors.
| Table II.Association between
clinicopathological characteristics and the expression of
WIF-1 in gallbladder cancer. |
Table II.
Association between
clinicopathological characteristics and the expression of
WIF-1 in gallbladder cancer.
|
|
| Expression of
WIF-1 |
|
|---|
|
|
|
|
|
|---|
| Characteristic | Cases (n) | − | + | P-value |
|---|
| Age (years) |
|
|
| 0.723 |
|
<60 | 21 | 16 | 5 |
|
|
≥60 | 29 | 24 | 5 |
|
| Sex |
|
|
| 0.736 |
|
Male | 23 | 19 | 4 |
|
|
Female | 27 | 21 | 6 |
|
| pT stage |
|
|
| 0.171 |
|
T1-T2 | 20 | 14 | 6 |
|
|
T3-T4 | 30 | 26 | 4 |
|
| Lymph node
metastasis |
|
|
| 0.171 |
|
Negative | 20 | 14 | 6 |
|
|
Positive | 30 | 26 | 4 |
|
| Distant
metastasis |
|
|
| 0.138 |
|
Negative | 34 | 25 | 9 |
|
|
Positive | 16 | 15 | 1 |
|
| Histological
grade |
|
|
| 0.707 |
|
High | 16 | 12 | 4 |
|
|
Moderate/poor | 34 | 28 | 6 |
|
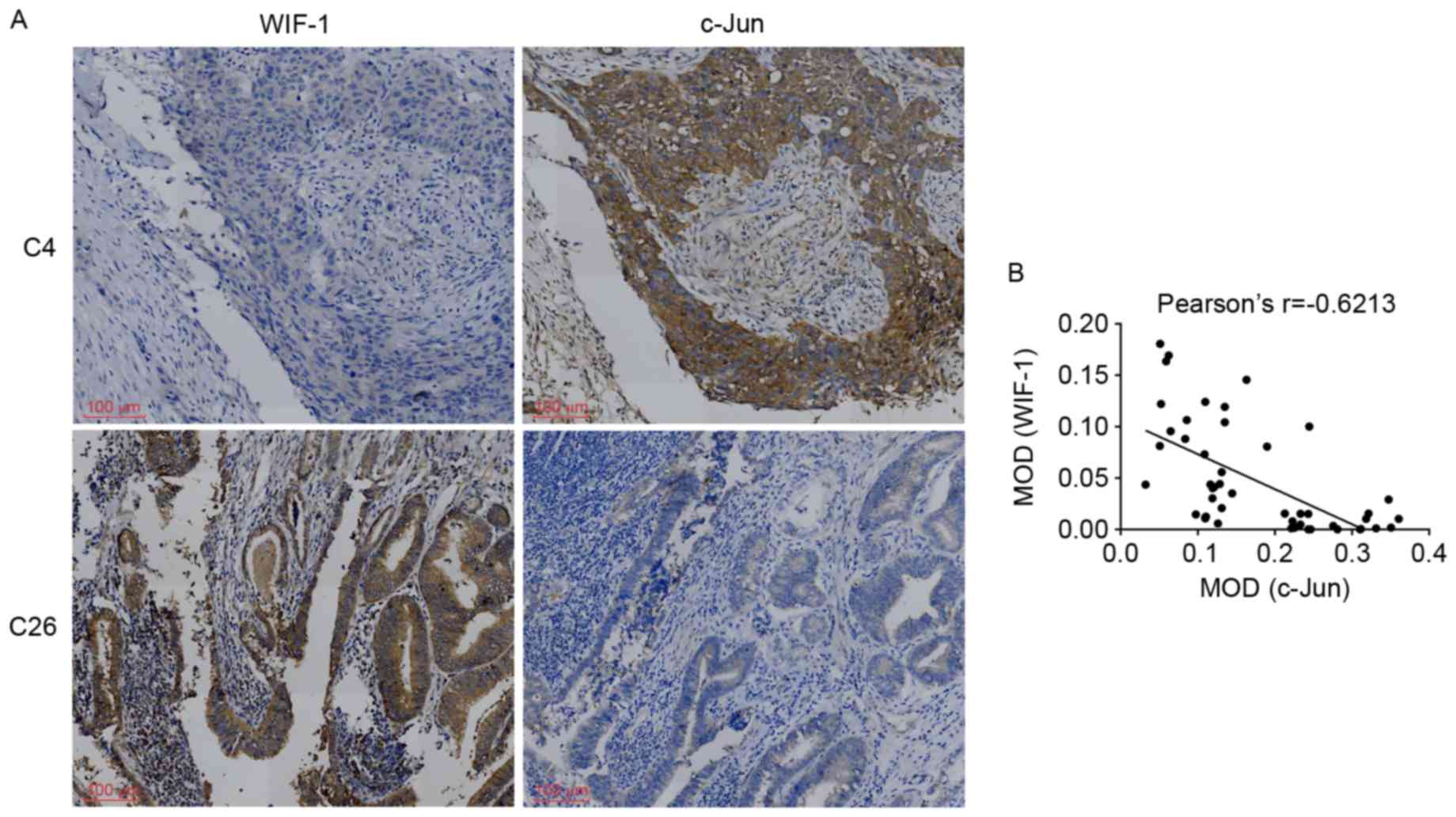
A high level of c-Jun-positivity was detected in
serial sections of the same GBC specimens; a representative sample
(case 4) is shown in Fig. 2A. By
contrast, a high expression of WIF-1 and low expression of c-Jun
was observed in case 26. There are currently no reports on the
association between the expression of WIF-1 and c-Jun
in GBC specimens. To reveal correlations between these proteins,
the present study used the MOD to semiquantitatively analyze the
results. The data showed a negative correlation between the protein
levels of WIF-1 and c-Jun in all GBC specimens (Pearson's
r=−0.6213; P<0.05), as shown in Fig. 2B.
Knockdown of c-Jun increases the mRNA
and protein expression of WIF-1
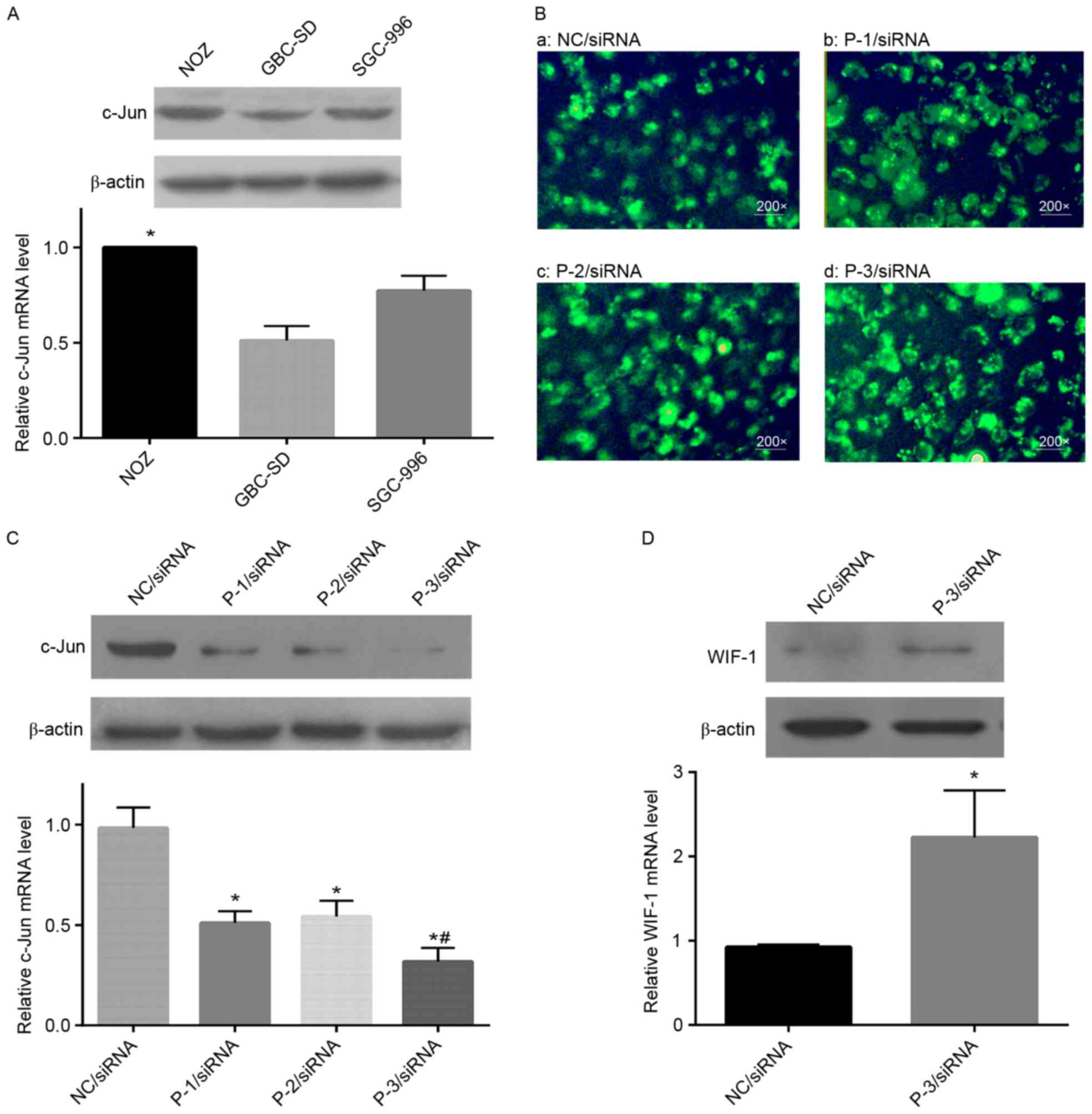
Among the cell lines examined (NOZ, GBC-SD and
SGC-996 cells), the highest mRNA and protein expression levels of
c-Jun, detected using RT-qPCR and western blot analyses, were found
in the NOZ cells (Fig. 3A).
Therefore, NOZ cells were used to further examine the role of c-Jun
in GBC. To identify an effective siRNA for silencing the expression
of c-Jun, three siRNA sequences (P-1, P-2 and P-3) were designed,
chemically synthesized and transiently transfected into NOZ cells.
The transfection efficiency was observed using fluorescence
microscopy (blue light), which was high in all the groups listed
above (Fig. 3B). The P-3/siRNA
vector resulted in the most marked suppression of mRNA and protein
expression of c-Jun, compared with the P-1/siRNA and P-2/siRNA
vectors, whereas the NC/siRNA group had no effect (Fig. 3C). Therefore, for further
experiments, P-3/siRNA was selected to induce knockdown, following
which the mRNA and protein expression levels of WIF-1 were
examined. As shown in Fig. 3D, the
expression of WIF-1 was markedly restored in the P-3/siRNA group,
which also supported the association between c-Jun and WIF-1 in
vitro.
c-Jun represses the expression of
WIF-1 by inducing hypermethylation of the WIF-1 promoter
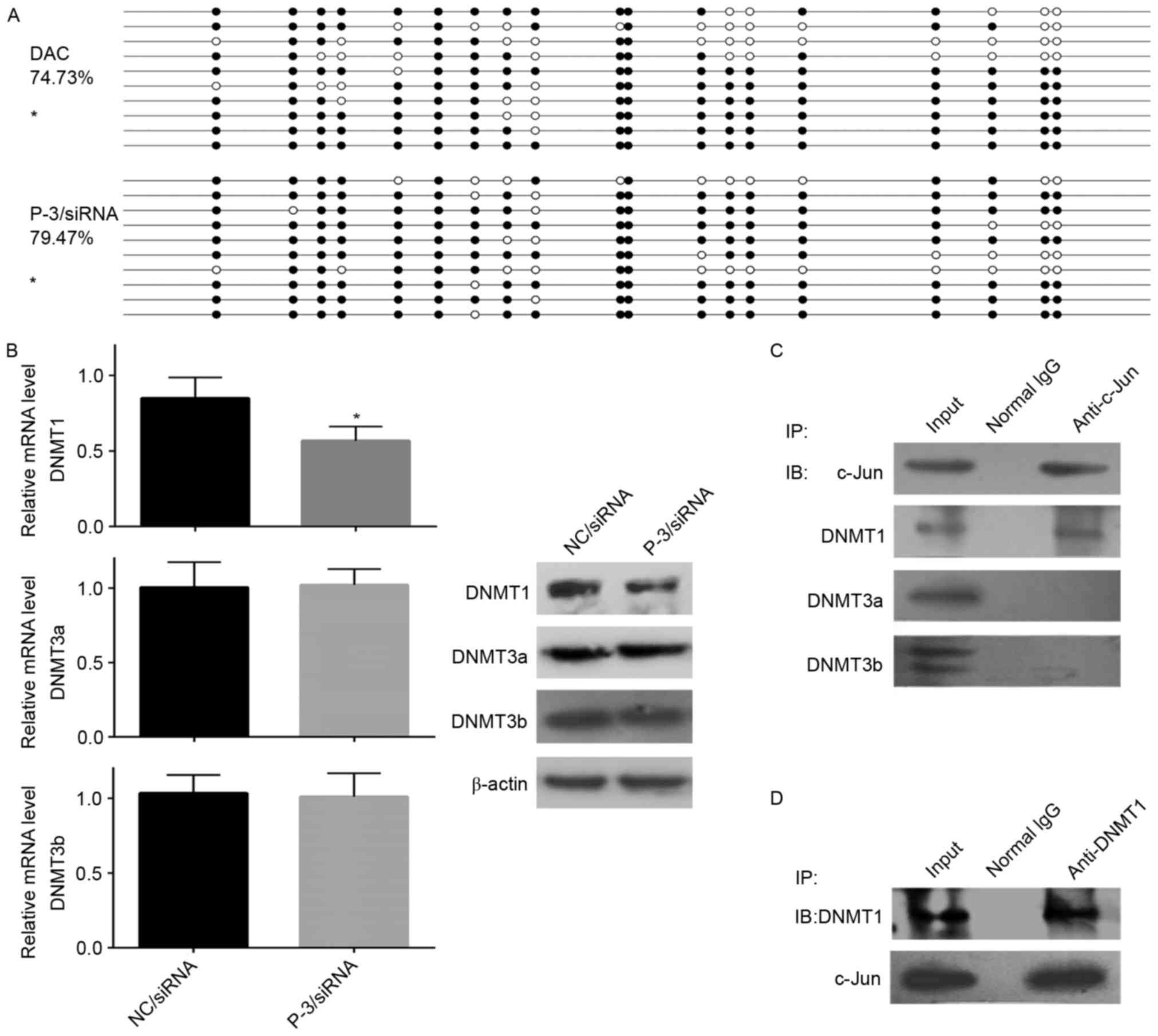
The results of the BSP analysis of the WIF-1
promoter in P-3/siRNA-transfected and DAC-treated NOZ cells were
consistent with the expression data. Hypermethylation was observed
for the majority of CpG dinucleotides within the amplified 255 bp
region of the WIF-1 promoter in NOZ cells (98.94%; Fig. 1E), however, a low level of CpG
methylation was observed in the c-Jun/siRNA (79.47%) group
(P<0.05; Fig. 4A). Of note, the
patterns of DNA methylation in the c-Jun/siRNA NOZ cells were most
similar to those in the DAC treatment group (74.73%; Fig. 4A), suggesting that c-Jun repressed
the expression of WIF-1 by inducing the hypermethylation of its
promoter region.
c-Jun effects the methylation of WIF-1
through transcriptional regulation and interaction with DNMT1
Methyl groups are added to CG dinucleotides by
DNMT3a/3b, and DNMT1 then maintains these DNA methylation patterns
during DNA replication to ensure epigenetic gene silencing
(8). In the present study, RT-qPCR
and western blot analyses revealed that the knockdown of c-Jun in
NOZ cells inhibited the expression of DNMT1, but had minimal effect
on the expression of DNMT3a or DNMT3b (Fig. 4B).
In addition to the regulation of DNMTs, it was
hypothesized that an alternative mechanism exists, whereby
c-Jun can physically modulate epigenetically associated
proteins to trigger epigenetic modifications around the regulatory
elements of WIF-1. Therefore, a co-immunoprecipitation assay
was designed using NOZ cells. As shown in Fig. 4C, c-Jun was immunoprecipitated with
DNMT1, but not DNMT3a or DNMT3b. Reverse immunoprecipitation with
DNMT1 antibodies was then performed to confirm the interaction
(Fig. 4D). These results indicated
that the methylation of WIF-1 associated with c-Jun
was predominantly through DNMT1.
Discussion
WIF-1 is a key gene, which encodes a secreted
protein that antagonizes the Wnt pathway and is involved in early
embryonic development (25). As an
important tumor suppressor, the abnormal expression of WIF-1
can result in carcinogenesis through the dysregulation of cell
proliferation and differentiation. It has been demonstrated that
the expression of WIF-1 is decreased in several types of
tumor (10–13). The findings of the present study
demonstrated a loss of the expression of WIF-1 in GBC cell
lines (NOZ, GBC-SD and SGC-996). The results also showed that
treatment with DAC, a potent demethylating drug, which binds to DNA
methylation enzymes to inhibit their activity, restored the mRNA
expression of WIF-1. In addition, the methylation-specific
PCR and BSP assays showed that the hypermethylation of WIF-1
DNA may explain its loss of expression in GBC cells.
Due to the relatively low incidence, there have been
few investigations of the role of WIF-1 in the pathogenesis
of GBC. In the present study, the protein expression of WIF-1 was
examined in 50 GBC and 20 cholecystitis specimens using
imunohistochemical techniques. A reduced expression of WIF-1 was
detected in the GBC specimens, which indicated that WIF-1
was important in the pathogenesis of GBC. However, in contrast to
previous reports of other tumors, subgroup analysis showed no
correlation between the expression of WIF-1 and key
clinicopathological parameters in GBC, including age and sex, pT
stage, lymph node metastasis, distant metastasis and histological
grade. This suggested that WIF-1 may be involved in the
tumorigenesis of GBC only, and not the progression.
There have been few investigations to date regarding
the upstream molecular mechanism for regulating WIF-1. In
the present study, it was observed that the protein levels of WIF-1
and c-Jun were negatively correlated in serial sections of 50 GBC
specimens, suggesting an association between c-Jun and
WIF-1. RNA interference was used to for the in vitro
knockdown c-Jun, and an effective siRNA was obtained
(P-3/siRNA). The mRNA and protein expression levels of WIF-1
were then detected in NOZ cells transiently transfected with
P-3/siRNA, and the results demonstrated that silencing of the
c-Jun gene upregulated the expression of WIF-1.
Considering this evidence, it was hypothesized that c-Jun
may be involved in regulating the expression of WIF-1 in
GBC.
The results described above prompted further
examination of the specific molecular mechanism underlying the
c-Jun-induced downregulated expression of WIF-1. The
epigenetic modification of cellular genes via aberrant methylation
in promoter regions has been identified as a crucial mechanism for
inactivating tumor suppressor genes in tumors (4,5,26).
The present study showed silencing of WIF-1 through
hypermethylation of its promoter region in GBC cells. With these
previous findings in consideration, it was hypothesized that
c-Jun repressed the expression of WIF-1 by modifying
the promoter methylation status of the WIF-1 gene. To
confirm the factors involved, BSP assays were performed, and the
obtained data showed that CpG islands in the WIF-1 promoter
region were hypermethylated at a higher frequency in NOZ cells,
compared with those in the knockdown group (P-3/siRNA cells),
revealing a similar methylation ratio to that observed in the
DAC-treated group. These results suggested that the knockdown of
c-Jun caused the demethylation of CpG islands in the
WIF-1 promoter region. Taken together, these findings are
confirmed the hypothesis that c-Jun can repress the
expression of WIF-1 through epigenetic modification by
inducing hypermethylation of the WIF-1 promoter in GBC.
Methylation modifications are mediated by DNMTs
through various mechanisms. DNMT1 and DNMT3b have been shown to be
responsible for the aberrant hypermethylation of WIF-1 in
the HCT116 colon cancer cell line (10). The present study investigated
whether c-Jun affected DNMTs in GBC cells. The experimental
results demonstrated that c-Jun knockdown in the NOZ cells
inhibited the expression of DNMT1, but not that of DNMT3a or
DNMT3b. A co-immunoprecipitation assay was performed to examine the
mechanism involved in the chromatin remodeling induced by
c-Jun, and it was found that c-Jun interacted with DNMT1
only, and not with DNMT3a or DNMT3b. Taken together, these results
showed that c-Jun affects the methylation of WIF-1
through transcriptional regulation and interaction with DNMT1, a
critical DNA methyltransferase.
However, the results of the present study were based
on data obtained using GBC cells and partial GBC tissues. Unlike
the findings in the present study, a previous study reported that
WIF-1 was expressed in the majority of colorectal cancer
specimens (27). The
downregulation of c-Jun has been observed in specific types
of human cancer, including breast cancer; these discrepancies may
be due to the different types and developmental stages of the cells
examined (28,29). Therefore, the regulatory mechanisms
of c-Jun and WIF-1 are complicated, however, the
results of the present study assist in elucidating the mechanisms
linking these proteins in GBC.
In conclusion, the present study found that the
expression of WIF-1 was low in GBC cells due to aberrant
hypermethylation of its promoter region. Additionally, an
alternative pathogenesis of GBC was indicated in which c-Jun
causes hypermethylation of the WIF-1 promoter region, and
represses the expression of WIF-1 through transcriptional
regulation and interaction with DNMT1 as an early event in the
tumorigenesis of GBC.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81672468)
and the National Clinical Key Specialty Construction Project
(General Surgery) of China (grant no. 2012-649). This manuscript
was edited for English language by American Journal Experts
(Durham, NC, USA).
References
|
1
|
Lazcano-Ponce EC, Miquel JF, Muñoz N,
Herrero R, Ferrecio C, Wistuba II, de Ruiz Alonso P, Urista Aristi
G and Nervi F: Epidemiology and molecular pathology of gallbladder
cancer. CA Cancer J Clin. 51:349–364. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sheth S, Bedford A and Chopra S: Primary
gallbladder cancer: Recognition of risk factors and the role of
prophylactic cholecystectomy. Am J Gastroenterol. 95:1402–1410.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Levy AD, Murakata LA and Rohrmann CA Jr:
Gallbladder carcinoma: Radiologic-pathologic correlation.
Radiographics. 21:295–314. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schroeder M and Mass MJ: CpG methylation
inactivates the transcriptional activity of the promoter of the
human p53 tumor suppressor gene. Biochem Biophys Res Commun.
235:403–406. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rizvi MM, Alam MS, Ali A, Mehdi SJ, Batra
S and Mandal AK: Aberrant promoter methylation and inactivation of
PTEN gene in cervical carcinoma from Indian population. J Cancer
Res Clin Oncol. 137:1255–1262. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oshimo Y, Nakayama H, Ito R, Kitadai Y,
Yoshida K, Chayama K and Yasui W: Promoter methylation of cyclin D2
gene in gastric carcinoma. Int J Oncol. 23:1663–1670.
2003.PubMed/NCBI
|
|
7
|
Mazieres J, He B, You L, Xu Z, Lee AY,
Mikami I, Reguart N, Rosell R, McCormick F and Jablons DM: Wnt
inhibitory factor-1 is silenced by promoter hypermethylation in
human lung cancer. Cancer Res. 64:4717–4720. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen T and Li E: Structure and function of
eukaryotic DNA methyltransferases. Curr Top Dev Biol. 60:55–89.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kawano Y and Kypta R: Secreted antagonists
of the Wnt signalling pathway. J Cell Sci. 116:2627–2634. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ai L, Tao Q, Zhong S, Fields CR, Kim WJ,
Lee MW, Cui Y, Brown KD and Robertson KD: Inactivation of Wnt
inhibitory factor-1 (WIF1) expression by epigenetic silencing is a
common event in breast cancer. Carcinogenesis. 27:1341–1348. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mazieres J, He B, You L, Xu Z, Lee AY,
Mikami I, Reguart N, Rosell R, McCormick F and Jablons DM: Wnt
inhibitory factor-1 is silenced by promoter hypermethylation in
human lung cancer. Cancer Res. 64:4717–4720. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ramachandran I, Thavathiru E, Ramalingam
S, Natarajan G, Mills WK, Benbrook DM, Zuna R, Lightfoot S, Reis A,
Anant S and Queimado L: Wnt inhibitory factor 1 induces apoptosis
and inhibits cervical cancer growth, invasion and angiogenesis in
vivo. Oncogene. 31:2725–2737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang Y, Du Q, Wu W, She F and Chen Y:
Rescued expression of WIF-1 in gallbladder cancer inhibits tumor
growth and induces tumor cell apoptosis with altered expression of
proteins. Mol Med Rep. 14:2573–2581. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dérijard B, Hibi M, Wu IH, Barrett T, Su
B, Deng T, Karin M and Davis RJ: JNK1: A protein kinase stimulated
by UV light and Ha-Ras that binds and phosphorylates the c-Jun
activation domain. Cell. 76:1025–1037. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Behrens A, Sibilia M and Wagner EF:
Amino-terminal phosphorylation of c-Jun regulates stress-induced
apoptosis and cellular proliferation. Nat Genet. 21:326–329. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Behrens A, Jochum W, Sibilia M and Wagner
EF: Oncogenic transformation by ras and fos is mediated by c-Jun
N-terminal phosphorylation. Oncogene. 19:2657–2663. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schreiber M, Kolbus A, Piu F, Szabowski A,
Möhle-Steinlein U, Tian J, Karin M, Angel P and Wagner EF: Control
of cell cycle progression by c-Jun is p53 dependent. Genes Dev.
13:607–619. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ryan KM, Phillips AC and Vousden KH:
Regulation and function of the p53 tumor suppressor protein. Curr
Opin Cell Biol. 13:332–337. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vogt PK: Jun, the oncoprotein. Oncogene.
20:2365–2377. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rice P, Longden I and Bleasby A: EMBOSS:
The european molecular biology open software suite. Trends Genet.
16:276–277. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li LC and Dahiya R: MethPrimer: Designing
primers for methylation PCRs. Bioinformatics. 18:1427–1431. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bock C, Reither S, Mikeska T, Paulsen M,
Walter J and Lengauer T: BiQ Analyzer: Visualization and quality
control for DNA methylation data from bisulfite sequencing.
Bioinformatics. 21:4067–4068. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hsieh JC, Kodjabachian L, Rebbert ML,
Rattner A, Smallwood PM, Samos CH, Nusse R, Dawid IB and Nathans J:
A new secreted protein that binds to Wnt proteins and inhibits
their activities. Nature. 398:431–436. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li HP, Leu YW and Chang YS: Epigenetic
changes in virus-associated human cancers. Cell Res. 15:262–271.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Byun T, Karimi M, Marsh JL, Milovanovic T,
Lin F and Holcombe RF: Expression of secreted Wnt antagonists in
gastrointestinal tissues: Potential role in stem cell homeostasis.
J Clin Pathol. 58:515–519. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Smith LM, Birrer MJ, Stampfer MR and Brown
PH: Breast cancer cells have lower activating protein 1
transcription factor activity than normal mammary epithelial cells.
Cancer Res. 57:3046–3054. 1997.PubMed/NCBI
|
|
29
|
Kharman-Biz A, Gao H, Ghiasvand R, Zhao C,
Zendehdel K and Dahlman-Wright K: Expression of activator protein-1
(AP-1) family members in breast cancer. BMC cancer. 13:4412013.
View Article : Google Scholar : PubMed/NCBI
|