Introduction
Von Hippel-Lindau (VHL) syndrome (Online Mendelian
Inheritance in Man:193300) is an autosomal dominant neoplastic
disorder. VHL syndrome is characterized by retinal and central
nervous system haemangioblastomas, clear cell renal cell carcinoma
(RCC), pheochromocytoma (PCC), pancreatic islet tumors and
endolymphatic sac tumors (1).
Furthermore, renal and pancreatic cysts as well as epididymal or
broad ligament cystadenomas are commonly associated with these
tumors. VHL syndrome can be classified into four groups based on
phenotypic characteristics: Types 1, 2A, 2B and 2C. PCCs are not
present in type 1; types 2A and 2B may exhibit PCCs and are
distinguishable by the risk of additional accompanying RCCs; and
type 2C presents with RCCs alone (2–4).
The prevalence of VHL Syndrome is 1 in every 36,000
live births (5). It has previously
been established that mutations in the VHL tumor suppressor
(VHL) gene are causative of VHL syndrome. Latif et al
(6) cloned VHL in 1993 and
Seizinger et al (7) located
it to chromosome 3p25-p26. The penetrance of VHL syndrome in
affected populations is estimated to be >90% by 60 years old
(8).
VHL encodes a 4.7 kb mRNA, which produces two
VHL tumor suppressor protein (pVHL) products: pVHL19 and pVHL30
(9). pVHL contains two functional
domains: α-domain and a β-domain. The α-domain binds to elongation
factors B and C (10). The
β-domain functions as the pVHL recognition domain, and has an
important role in cellular oxygen sensing via targeting of
hypoxia-inducible factors (HIFs) for ubiquitination and proteasomal
degradation; a mechanism that has been previously demonstrated to
be involved in the pathogenesis of cancer (10). HIFs have an important function in
the transcriptional response to changes in oxygen availability.
When associated with an E3 ligase complex, pVHL regulates the
stability of HIFs via its recognition domain. HIF-1α binding to
pVHL is dependent on a 20-residue oxygen-dependent degradation
(ODD) domain (11,12). Hydroxylation of a core proline
residue (HIF-1α-P564) within the ODD domain is necessary for this
binding (10–13).
Studies have demonstrated that mutant VHL proteins
exhibit abnormal interactions with HIF-α proteins: The pVHL-R167Q
mutant retains the ability to bind strongly to HIF-1α, whereas
pVHL-W88S and pVHL-L158P exhibit marked decreases in HIF-1α binding
activity (10,14). This illustrates the importance of
wild-type pVHL in HIF-1α-induced carcinogenesis.
In the present study, Sanger sequencing was
performed to investigate all exons of VHL in a Chinese
family suffering from VHL syndrome, and the results revealed a
heterozygous missense mutation responsible for a functional deficit
in pVHL.
Materials and methods
Subject recruitment and clinical
examination
A family with VHL syndrome from a Han Chinese
population was recruited for the present study in Hunan Cancer
Hospital (Changsha, China) between May 2014 and June 2014. There
were originally 23 patients in this pedigree; however, 6 patients
succumbed to mortality prior to recruitment. In total, 17 subjects
[including 8 males and 9 females; aged 4–65 years old; one patient
suffering from VHL syndrome (proband)] were enrolled and underwent
detailed clinical examinations, including the Romberg sign test, in
which the standing patient is asked to his/her close eyes with
their heels together and if the patient loses balance, a positive
result is recorded (15). In
addition, finger-nose tests were performed, which requires the
patient to alternately touch his/her nose and the examiner's finger
as quickly as possible. If the patient fails to touch his/her nose
accurately, a positive result is recorded (15). All members of the family were
examined by two experienced surgeons. Diagnoses of VHL syndrome
were determined via investigation of medical histories and
performance of physical examinations, including a complete
neurological examination and brain magnetic resonance imaging
(MRI). In addition, 200 unrelated volunteers (aged 35–65 years old;
116 males and 84 females) that did not have VHL syndrome or present
any associated phenotypes were recruited as controls in the Health
Management Center, Xiangya Hospital of Central South University
(Changsha, China) between September 2014 and October 2014. The
present study was approved by the Institutional Review Board of the
Xiangya Hospital of Central South University, and all subjects
enrolled in the present study provided written informed consent
prior to examination.
Immunostaining
Biopsy tissues isolated from the intracranial cyst
of the proband were obtained via surgical excision, fixed in 4%
paraformaldehyde at room temperature for one week and then embedded
in paraffin. Histopathological analysis of paraffin-embedded biopsy
tissue sections (6 µm) from the proband was performed using a
Hematoxylin and Eosin Staining Kit (cat. no. M020; GEFAN
Biotechnology, Co., Ltd., Shanghai, China) (16). Sections were blocked with 5% horse
serum (cat. no. HQ90041; Shanghai Bangyi Biotechnology Co., Ltd.,
Shanghai, China) at room temperature for 30 min and subsequently
incubated at 4°C overnight with antibodies against glial fibrillary
acidic protein (GFAP; 1:200; cat. no. KIT-0031), epithelial
membrane antigen (EMA; 1:80; cat. no. KIT-0011), S-100 protein, E3
SUMO-protein ligase nse (NSE; 1:100; cat. no. MAB-0584),
hematopoietic progenitor cell antigen CD34 (CD34; 1:50; cat. no.
KIT-0004), Factor VIII-related Antigen (F8; 1:100; cat. no.
RAB-0070) and proliferation marker protein Ki-67 (Ki-67; 1:100;
cat. no. MX006) (16). All primary
antibodies were purchased from Fuzhou Maixin Biotech Co., Ltd.
(Fuzhou, China). Following this, sections were incubated with
horseradish peroxidase-conjugated anti-mouse/rabbit secondary
antibodies (working dilution was provided by the kit; cat. no.
KIT-5010; Fuzhou Maixin Biotech Co., Ltd.) at room temperature for
2 h. Images of the sections were captured using Photographs were
taken using a light microscope Olympus BX51 (Olympus Corporation,
Tokyo, Japan; magnification ×400) and a Nikon N6006 camera (Nikon
Corporation, Tokyo, Japan). The number of positive cells was
calculated as the total area of positive staining divided by the
area of an individual positive cell using Image Pro Plus software
(version 6.0; Media Cybernetics, Inc., Rockville, MD, USA). The
percentage of positive cells was calculated as the number of
positive cells divided by the total number of cells.
Genomic DNA preparation and molecular
analysis
Genomic DNA was isolated from peripheral blood
lymphocytes of all subjects using the standard phenol-chloroform
method (17). VHL was amplified
via polymerase chain reaction (PCR). Three pairs of primers were
designed to cover all of the exons and splicing junctions of VHL
(Table I). The thermocycling
conditions of PCR used were as follows: Initial Denaturation at
95°C for 30 sec; followed by 30 cycles of denaturation at 95°C for
30 sec, annealing at 58°C for 30 sec and elongation 72°C for 30
sec; and a final extension at 75°C for 5 min. PCR amplification was
performed using an ABI 2720 PCR system (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) using a 10 µl mixture containing premixed
DNA polymerase (5 µl; Takara Biotechnology Co., Ltd., Dalian,
China), primers (2 µl), genomic DNA (1 µl) and double distilled
H2O (2 µl). PCR products were detected using 6%
polyacrylamide gel electrophoresis with silver staining. Sanger
sequencing was performed using an ABI 3100/3130 Genetic Analyzer
automated sequencer (Applied Biosystems; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. The sequencing
results were analyzed using SeqMan 3.0 (DNASTAR, Inc., Madison, WI,
USA), and compared with the reference sequences listed in the
National Center for Biotechnology Information database (https://www.ncbi.nlm.nih.gov/). Mutation naming
followed the nomenclature recommended by the Human Genomic
Variation Society (http://www.hgvs.org/).
 | Table I.Primers used for amplification of the
Von Hippel-Lindau disease tumor suppressor gene. |
Table I.
Primers used for amplification of the
Von Hippel-Lindau disease tumor suppressor gene.
| Primer | Primer sequences |
|---|
| VHL-1F |
AGCGCGTTCCATCCTCTAC |
| VHL-1R |
AAGATTGGATAACGTGCCTGA |
| VHL-2F |
GGAGAAAATAGGTGCCCTGAC |
| VHL-2R |
AAGATTGGATAACGTGCCTGA |
| VHL-3F |
GCAAAGCCTCTTGTTCGTTC |
| VHL-3R |
GCCACCACCTTCTCCTGATA |
RNA preparation and RNA stability
investigation
Total RNA was isolated from lymphocytes obtained
from the patient using a GeneJET RNA Purification Kit (Fermentas;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. First strand complementary (c)DNA was synthesized
from total RNA at 42°C for 60 min and then at 70°C for 5 min using
a RevertAid First Strand cDNA Synthesis Kit (Fermentas; Thermo
Fisher Scientific, Inc.), according to the manufcaturer's
instructions. Primer sequences for semi-quantitative (sq)PCR were
designed using Primer 3 (version 4.1.0; http://primer3.ut.ee/) and were as follows: pVHL
forward, 5′-CTCCCAGGTCATCTTCTGCA-3′ and reverse,
5′-CTTGACTAGGCTCCGGACAA-3′. sqPCR procedures were performed
according to the following protocol: Initial denaturation at 95°C
for 30 sec; followed by 10 cycles at 95°C for 30 sec, 62°C for 30
sec and 72°C for 30 sec; a further 23 cycles at 95°C for 30 sec,
59°C for 30 sec and 72°C for 30 sec; and a final extension at 75°C
for 5 min. Following this, 5% agarose gel electrophoresis was
performed to verify the PCR products. Proteins were visualized
using ethidium bromide. Normality of distributions was determined
using the Shapiro-Wilk test. Statistical significance was assessed
with an unpaired two-tailed Student's t-test, using GraphPad Prism
6.0 (Graphpad Software, Inc., La Jolla, CA, USA). Data are
presented as mean ± standard deviation.
Bioinformatics analysis
Lasergene MegAlign 9.0 (DNAStar, Inc.) was used to
investigate whether the identified mutation of pVHL was conserved
between different species. MutationTaster (http://mutationtaster.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml)
and SIFT (http://sift.jcvi.org) software, all
available online, were selected to investigate the effect of the
mutated amino acid on the protein's structure and function.
SWISS-MODEL (https://swissmodel.expasy.org) and PyMol software
(version 1.5.0.3; DeLano Scientific, San Carlos, CA, USA) were used
to predict and visualize three-dimensional protein models.
Results
Clinical evaluations
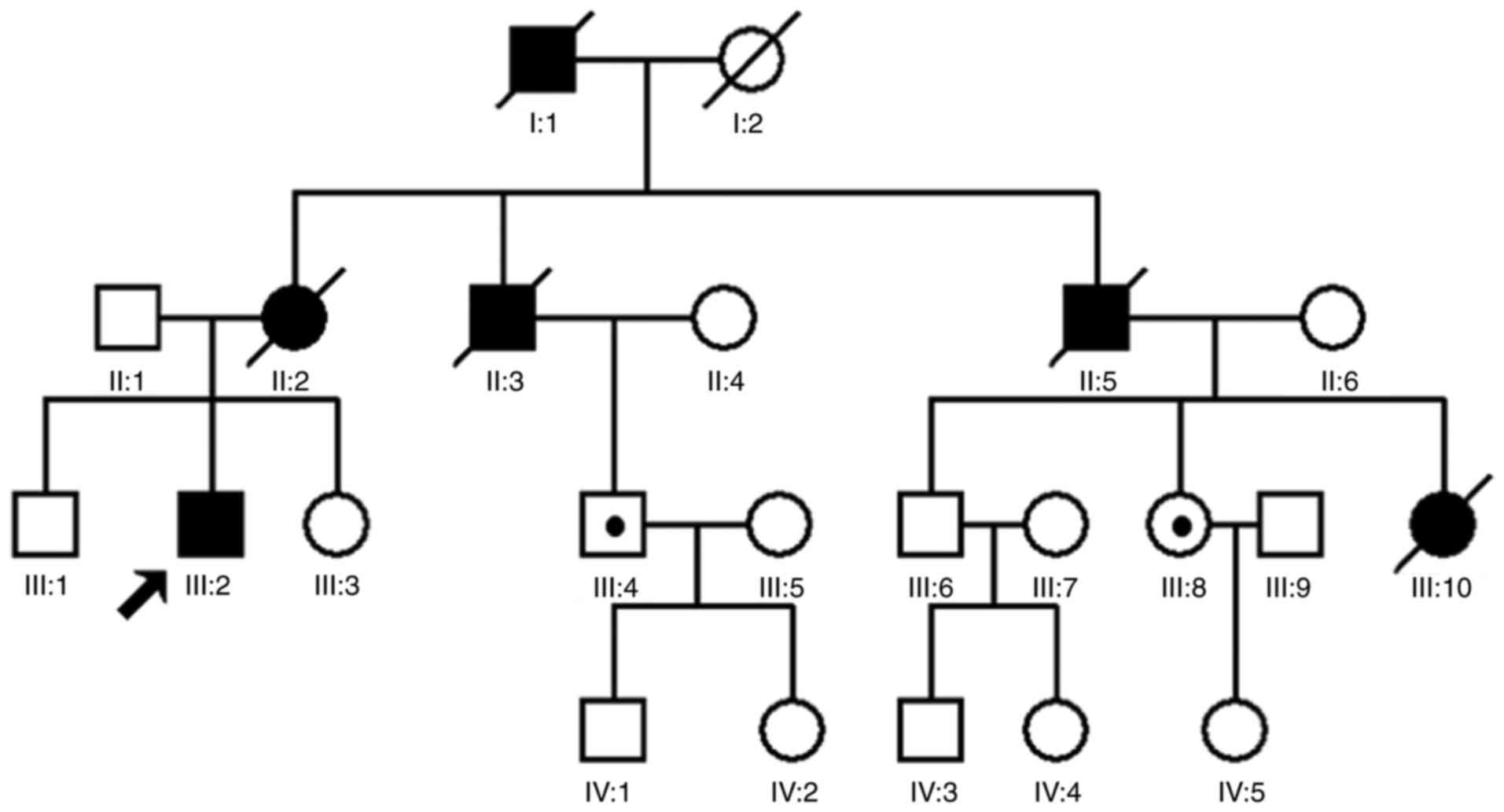
A four-generation family with diagnosed VHL syndrome
was recruited for the present study. The family consists of 23
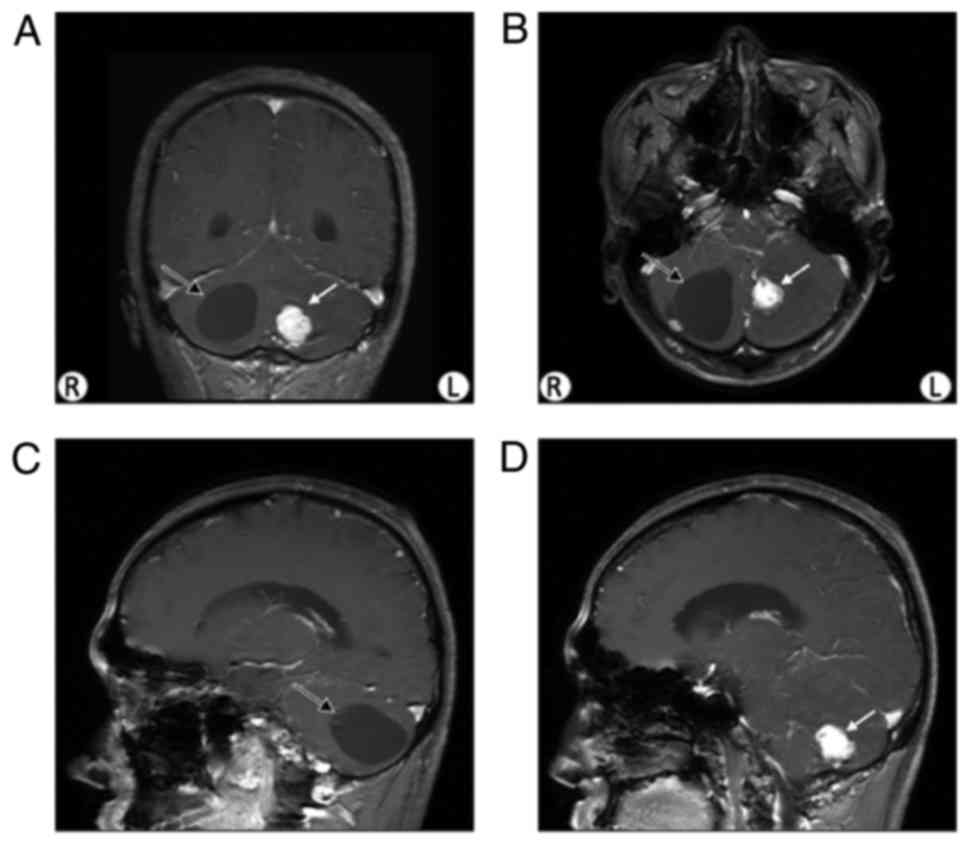
individuals, including 11 males and 12 females (Fig. 1). The proband was a 28-year-old
male, who presented with an episodic headache for 10 years. Both
the finger-nose test and the Romberg's sign were recorded as
positive in the physical examination. A cyst and a nodule were
revealed via brain MRI (Fig. 2).
The cyst was demonstrated to be a hemangioblastoma tumor following
surgical excision followed by histopathological analysis of the
biopsy tissues. H&E staining revealed that the lesion contained
a large number of thin-walled capillaries (Fig. 3). The sole neoplastic element,
known as the ‘stromal cell’, inhabited the interstices between
ramifying vascular arcades. Stromal cells stained for S-100 and NSE
exhibited dense-core intracytoplasmic granules, however did not
demonstrate expression of GFAP or EMA (Fig. 3), which has previously been
detected immunohistochemically in most nonneoplastic epithelia
(18). Stromal cells did not
exhibit positive staining for endothelial markers such as CD34,
whereas the thin-walled capillaries were positive; stromal cells
occasionally exhibited the expression of vascular marker F8
(Fig. 3). The expression of Ki-67,
which is a proliferation marker (19), was <1% (Range of normal value
1–2%; Fig. 3). Therefore, the
proband was diagnosed with VHL syndrome at the Hunan Cancer
Hospital (Changsha, China). A further five affected members of the
family had previously died from this syndrome.
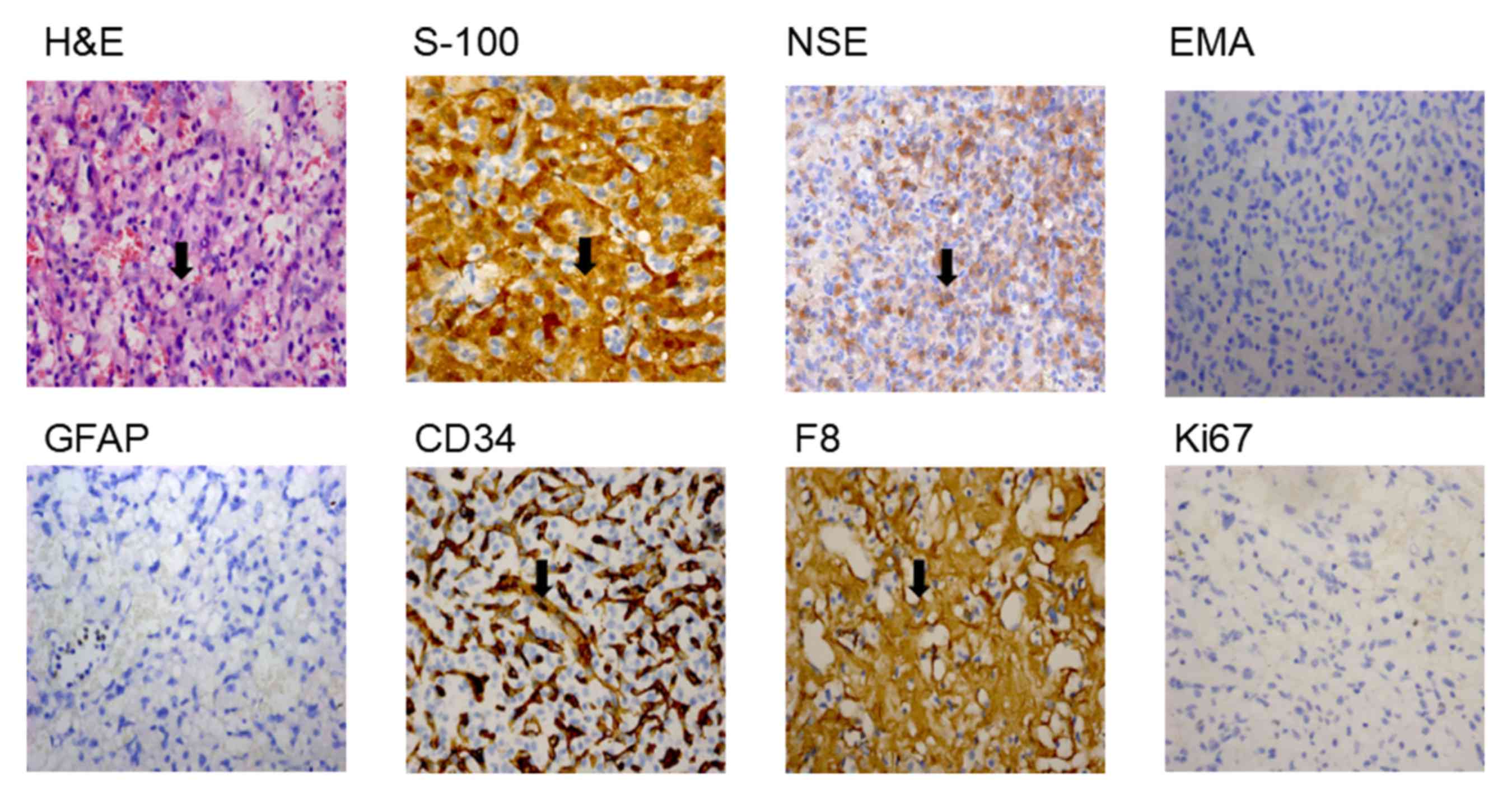 | Figure 3.Histopathology of the biopsy tissues
from the patient with VHL. Pathological analysis results from the
cerebellum cyst tissue sections, examined via staining. Numerous
small blood vessels, and a number of stroma cells, fat-like cells
and fibroblasts were observed (magnification, ×400).
Immunohistochemical analysis of the tissue biopsy samples from the
patient with VHL. HE, S-100, NSE and F8 were revealed to be present
in cells; and EMA, GFAP, CD34 and Ki67 were revealed to be absent
in cells (magnification, ×400). Black arrows indicate
histopathological changes. VHL, Von Hippel-Lindau; HE,
hemagglutinin-esterase; S-100, protein S-100; NSE, E3 SUMO-protein
ligase nse; F8, coagulation factor VIII; EMA, epithelial membrane
antigen; GFAP, glial fibrillary acidic protein; CD34, hematopoietic
progenitor cell antigen CD34. |
Mutation analysis
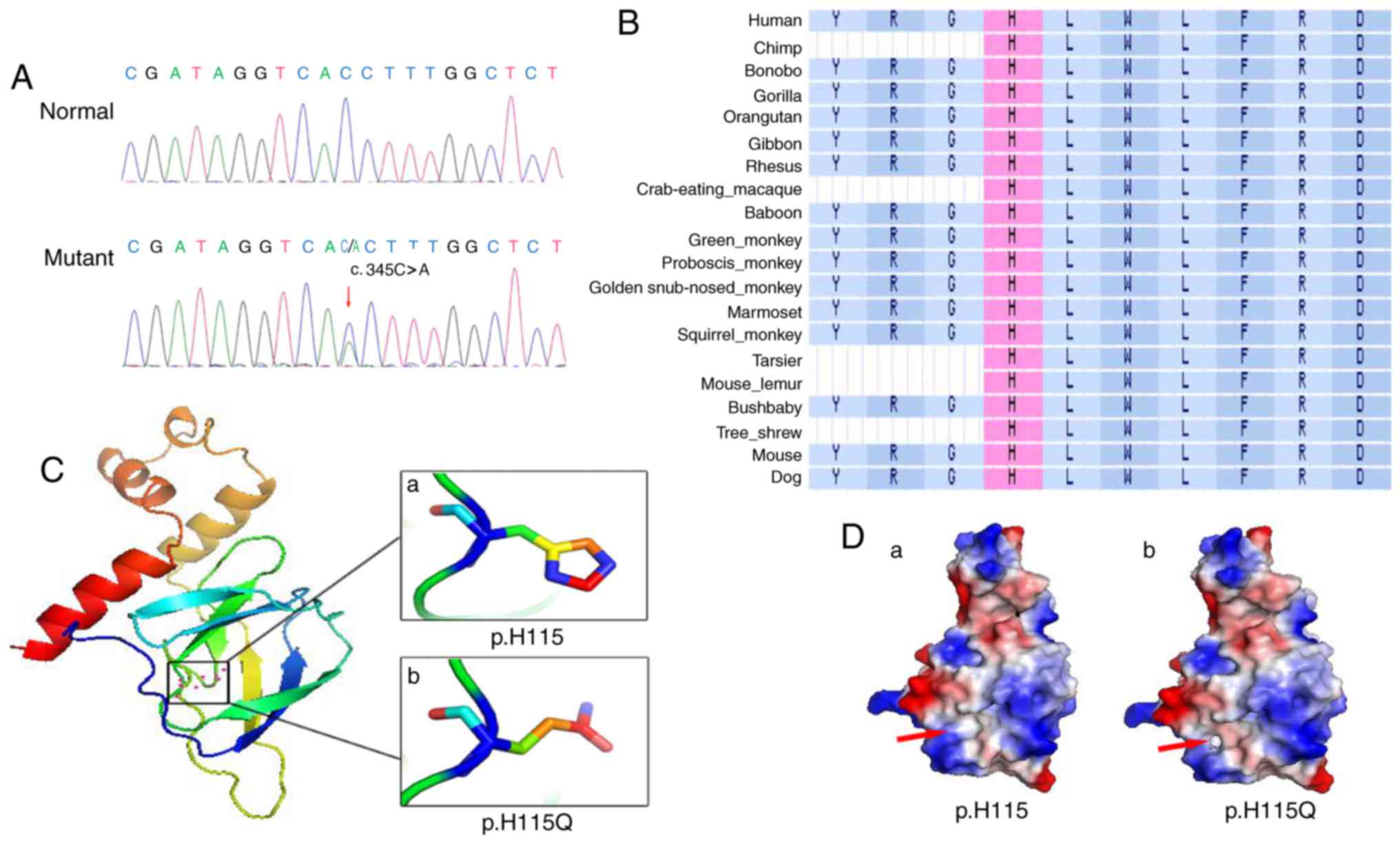
A heterozygous mutation c.345C>A (p.H115Q) in
exon 2 of the VHL gene was revealed to be present in the
proband III:2 (Fig. 4A). In
addition, two healthy individuals (III:4 and III:8) in the pedigree
were demonstrated to carry the mutation, as revealed by Sanger
sequencing (Fig. 1). The mutation
was not present in the 200 healthy patients.
Bioinformatics analysis
Alignment of VHL amino acid sequences in numerous
species revealed that the histidine at the 115th amino acid site is
highly conserved (Fig. 4B). The
functional prediction software packages Polyphen-2, SIFT and
MutationTaster; demonstrated that the variant is likely to be
deleterious (data not shown). The structures of pVHL and p.H115Q,
as predicted using SWISS-MODEL, were markedly different from one
another: The wild type pVHL exhibited an imidazole ring structure
(Fig. 4Ca), and the p.H115Q mutant
exhibited a non-imidazole ring structure (Fig. 4Cb). Protein surface analysis
revealed marked changes between the wild type pVHL and the p.H115Q
mutant pVHL. In wild type pVHL, the area highlighted by the red
arrow is non-polar charged and forms a continuous hydrophobic
cleft, which facilitates its interaction with HIF-α (Fig. 4D). However, the H115Q mutant
introduces a negative charge in this area (right panel),
subsequently disrupting the hydrophobic environment required for
binding with HIF-α (Fig. 4D).
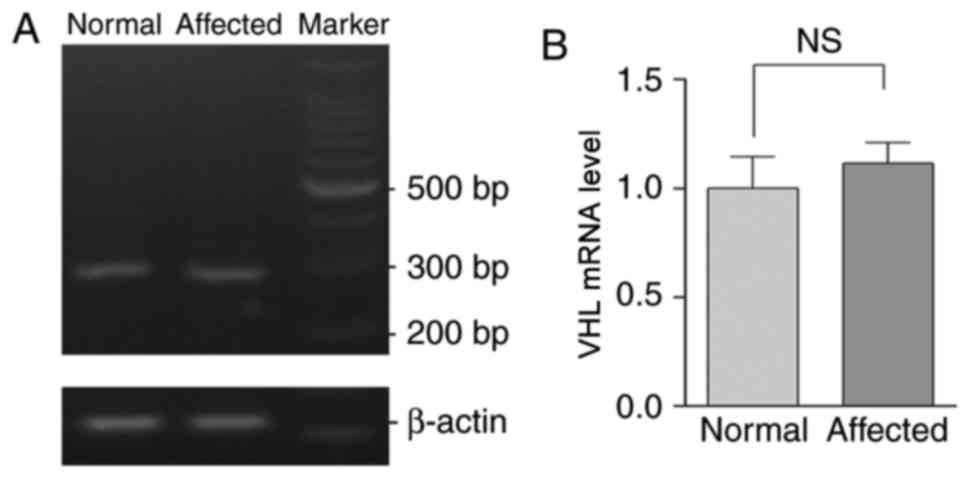
Levels of VHL mRNA in the proband and
healthy controls
cDNA was generated by reverse transcribing RNA
extracted from peripheral blood samples obtained from patients in
the pedigree and the healthy controls. No significant difference in
the expression of pVHL was demonstrated between the mutated and
wild type cDNA, as demonstrated by RT-sqPCR analysis (Fig. 5).
Discussion
VHL is located on chromosome 3p25-p26 and
encodes a single 4.7 kb mRNA. To date, >506 mutations in the
VHL gene associated with the development of VHL syndrome
have been reported in the Human Gene Mutation Database (http://www.hgmd.org), the majority of which are
deletion and missense mutations. The VHL gene and pVHL are
widely expressed in tissues, thus tissue-specific expression cannot
explain the complex tumor pattern in VHL syndrome (2).
VHL proteins exhibit numerous functions. A previous
study demonstrated that the functions of pVHL include regulating
the expression of HIFs, mediating the invasion and metastasis of
tumor cells via the regulation of microtubules, and regulating the
proliferation and apoptosis of cells (20). pVHL contains two domains, the
α-domain and the β-domain (20).
The α-domain is responsible for binding to elongation factor C,
which contains three α helices located at amino acid residues
155–192; the β-domain is the substrate recognition region for pVHL,
and is located at amino acid residues 63–154 (20). The α-domain binds to elongation
factors B and C, and Cullin 2, and has an important role in the
maintenance of the spatial conformation stability of pVHL (20). The β domain binds to the subunit of
HIF and participates in the degradation of the HIF subunit under
aerobic conditions (21).
According to previous studies, the most prevalent mutations present
in VHL syndrome are R167, R161, V155, Y98, G114, F76, P86, P81,
L158 and C162 (20,22,23).
The majority of the mutation sites are located within the HIF-1α
and elongation factor C domains (24).
The stability of HIF-1α binding to pVHL may affect
the degradation of the HIF protein, which may lead to
tumorigenesis. Previous studies have demonstrated that
hydroxylation of the P564 residue of HIF-1α has an important role
in its binding to pVHL, and pVHL-S111 and pVHL-H115 are important
regulators of this mechanism (10,12,13).
In the present study, a heterozygous c.345C>A
(p.H115Q) mutation in VHL was successfully revealed via
Sanger sequencing in the affected proband and gene carriers within
the pedigree; however, the mutation was revealed to not be present
within the non-affected members of the pedigree and the 200 healthy
control patients. The RT-sqPCR results revealed that the expression
levels of the mutant VHL were not significantly different
compared with the control group and the patients with VHL.
Bioinformatics analysis using SIFT, PolyPhen-2 and
MutationTaster revealed that the c.345C>A (p.H115Q) mutation is
deleterious and thus suggested that it may be pathogenic.
SWISS-MODEL software was used to investigate the three-dimensional
structure of the protein and to compare wild-type VHL with the
p.H115Q mutant, and it was revealed that the substitution of an
alkaline histidine with a neutral glutamine changes the structure
of pVHL from an imidazole ring to a non-imidazole ring.
Furthermore, the mutant H115Q introduces a negative charge in this
area, which subsequently disrupts the viral hydrophobic
environment. This may explain why the interaction between HIF-α and
VHL decreases dramatically following the H115Q mutation (14,25).
The bioinformatics results in the present study suggested that the
mutant protein may have also affected the binding ability of pVHL
with other interacting proteins, thus affecting the biological
function of the protein.
A previous study demonstrated that two steps are
required for the initiation of carcinogenesis following gene
mutation: The first step being the inheritance of a mutation via
germinal cells, and the second step being a mutation arising in
somatic cells (24,26). Thus, it was speculated that the
development of VHL syndrome may follow this hypothesis.
Furthermore, it can be suggested that the mutations in the somatic
cells of two carriers (III:4 and III:8) in the pedigree
investigated in the present study have not yet been activated, and
thus may not produce an associated phenotype.
In addition, studies of VHL knockout mice
have revealed an important role of pVHL in developmental biology.
Homozygous VHL−/− mice die at 10.5–12.5 days
post-birth due to placental angiogenesis dysfunction (27). VHL+/−
heterozygous mice develop vascular tumors of the liver (28). Further studies have confirmed that
VHL-heterozygous mice tend to develop tumors with blood
vessel hyperplasia phenotypes, and that pVHL is involved in
regulating the expression of angiogenesis-associated genes, which
are important for tumorigenesis (21,28).
In conclusion, a heterozygous p.H115Q mutation in
the VHL gene was screened in a Han Chinese family with VHL
syndrome. Despite this mutation having previously been revealed to
cause VHL syndrome (28,29), it also has a great significance for
validating the spectrum of gene mutations involved in the
development of VHL syndrome.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of Hunan Province, China (grant. no.
2017JJ3507), and by the Postdoctoral Science Foundation of Xiangya
Hospital, Central South University, China.
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZH recruited the family with VHL syndrome and
diagnosed the proband with Von Hippel-Lindau syndrome. ZD collected
peripheral blood lymphocytes from all individuals belonging to the
family with Von Hippel-Lindau syndrome. LX and AL extracted DNA
from peripheral blood lymphocytes, designed the primers and
performed the Sanger sequencing. ZH and BL screened for the
mutation site and performed the bioinformatics analysis. BL and ZH
drafted the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of the Xiangya Hospital of Central South University
(Changsha, China), and all subjects enrolled in the present study
provided written informed consent prior to examination.
Consent for publication
Written informed consent was provided.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Maher ER, Neumann HP and Richard S: von
Hippel-Lindau disease: A clinical and scientific review. Eur J Hum
Genet. 19:617–623. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hes FJ, Höppener JW, Luijt RB and Lips CJ:
Von hippel-lindau disease. Hered Cancer Clin Pract. 3:171–178.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen F, Kishida T, Yao M, Hustad T, Glavac
D, Dean M, Gnarra JR, Orcutt ML, Duh FM, Glenn G, et al: Germline
mutations in the von Hippel-Lindau disease tumor suppressor gene:
Correlations with phenotype. Hum Mutat. 5:66–75. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M
and Kaelin WG Jr: von Hippel-Lindau protein mutants linked to type
2C VHL disease preserve the ability to downregulate HIF. Hum Mol
Genet. 10:1019–1027. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Neumann HP and Wiestler OD: Clustering of
features of von Hippel-Lindau syndrome: Evidence for a complex
genetic locus. Lancet. 337:1052–1054. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Latif F, Tory K, Gnarra J, Yao M, Duh FM,
Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L, et al:
Identification of the von Hippel-Lindau disease tumor suppressor
gene. Science. 260:1317–1320. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seizinger BR, Smith DI, Filling-Katz MR,
Neumann H, Green JS, Choyke PL, Anderson KM, Freiman RN, Klauck SM,
Whaley J, et al: Genetic flanking markers refine diagnostic
criteria and provide insights into the genetics of Von Hippel
Lindau disease. Proc Natl Acad Sci USA. 88:2864–2868. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maher ER, Yates JR, Harries R, Benjamin C,
Harris R, Moore AT and Ferguson-Smith MA: Clinical features and
natural history of von Hippel-Lindau disease. Q J Med.
77:1151–1163. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Iliopoulos O, Ohh M and Kaelin WG Jr:
pVHL19 is a biologically active product of the von Hippel-Lindau
gene arising from internal translation initiation. Proc Natl Acad
Sci USA. 95:11661–11666. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Min JH, Yang H, Ivan M, Gertler F, Kaelin
WG Jr and Pavletich NP: Structure of an HIF-1alpha-pVHL complex:
Hydroxyproline recognition in signaling. Science. 296:1886–1889.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ivan M, Kondo K, Yang H, Kim W, Valiando
J, Ohh M, Salic A, Asara JM, Lane WS and Kaelin WG Jr: HIFalpha
targeted for VHL-mediated destruction by proline hydroxylation:
Implications for O2 sensing. Science. 292:464–468. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jaakkola P, Mole DR, Tian YM, Wilson MI,
Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji
M, Schofield CJ, et al: Targeting of HIF-alpha to the von
Hippel-Lindau ubiquitylation complex by O2-regulated prolyl
hydroxylation. Science. 292:468–472. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu F, White SB, Zhao Q and Lee FS:
HIF-1alpha binding to VHL is regulated by stimulus-sensitive
proline hydroxylation. Proc Natl Acad Sci USA. 98:9630–9635. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun W, Kato H, Kitajima S, Lee KL, Gradin
K, Okamoto T and Poelllinger L: Interaction between von
Hippel-Lindau protein and fatty acid synthase modulates hypoxia
target gene expression. Sci Rep. 7:71902017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stephen H: Harrison's Neurology in
Clinical Medicine. 2nd editon. McGraw-Hill; London: 2010
|
|
16
|
Rosai J: Rosai and Ackerman's surgical
pathology-2 Volume Set. 10th Edition. Mosby: 2011, View Article : Google Scholar
|
|
17
|
Sambrook J and Russell D: Molecular
cloning: A laboratory manual (Third Edition). Cold Spring Harb Lab.
2016.
|
|
18
|
Enriquez ML, Lal P, Ziober A, Wang L,
Tomaszewski JE and Bing Z: The use of immunohistochemical
expression of SF-1 and EMA in distinguishing adrenocortical tumors
from renal neoplasms. Appl Immunohistochem Mol Morphol. 20:141–145.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li LT, Jiang G, Chen Q and Zheng JN: Ki67
is a promising molecular target in the diagnosis of cancer
(review). Mol Med Rep. 11:1566–1572. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Barry RE and Krek W: The von Hippel-Lindau
tumour suppressor: A multi-faceted inhibitor of tumourigenesis.
Trends Mol Med. 10:466–472. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kleymenova E, Everitt JI, Pluta L, Portis
M, Gnarra JR and Walker CL: Susceptibility to vascular neoplasms
but no increased susceptibility to renal carcinogenesis in Vhl
knockout mice. Carcinogenesis. 25:309–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jia D, Tang B, Shi Y, Wang J, Sun Z, Chen
Z, Zhang L, Xia K and Jiang H: A deletion mutation of the VHL gene
associated with a patient with sporadic von Hippel-Lindau disease.
J Clin Neurosci. 20:842–847. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang Z, Yang Z, Xiong L, Huang S, Liu J,
Yang L and Miao X: Expression of VHL and HIF-1α and their
clinicopathologic significance in benign and malignant lesions of
the gallbladder. Appl Immunohistochem Mol Morphol. 19:534–539.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kibel A, Iliopoulos O, DeCaprio JA and
Kaelin WG Jr: Binding of the von Hippel-Lindau tumor suppressor
protein to Elongin B and C. Science. 269:1444–1446. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li L, Zhang L, Zhang X, Yan Q, Minamishima
YA, Olumi AF, Mao M, Bartz S and Kaelin WG Jr: Hypoxia-inducible
factor linked to differential kidney cancer risk seen with type 2A
and type 2B VHL mutations. Mol Cell Biol. 27:5381–5392. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Knudson AG Jr: Mutation and cancer:
Statistical study of retinoblastoma. Proc Natl Acad Sci USA.
68:820–823. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gnarra JR, Ward JM, Porter FD, Wagner JR,
Devor DE, Grinberg A, Emmert-Buck MR, Westphal H, Klausner RD and
Linehan WM: Defective placental vasculogenesis causes embryonic
lethality in VHL-deficient mice. Proc Natl Acad Sci USA.
94:9102–9107. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma W, Tessarollo L, Hong SB, Baba M,
Southon E, Back TC, Spence S, Lobe CG, Sharma N, Maher GW, et al:
Hepatic vascular tumors, angiectasis in multiple organs, and
impaired spermatogenesis in mice with conditional inactivation of
the VHL gene. Cancer Res. 63:5320–5328. 2003.PubMed/NCBI
|
|
29
|
Corrò C, Hejhal T, Poyet C, Sulser T,
Hermanns T, Winder T, Prager G, Wild PJ, Frew I, Moch H and
Rechsteiner M: Detecting circulating tumor DNA in renal cancer: An
open challenge. Exp Mol Pathol. 102:255–261. 2017. View Article : Google Scholar : PubMed/NCBI
|