Introduction
Natural killer (NK) cells and cytotoxic T
lymphocytes (CTLs) provide crucial defense against virus-infected
cells and tumor cells; NK cells and CTLs target tumor cells and
induce apoptosis (1). Granzyme B
(GrB) belongs to a family of serine proteases that are expressed in
the granules of activated CTLs and NK cells that induce apoptosis
(2,3). GrB is also expressed in non-lymphoid
lineage cells, including chondrocytes, neutrophils, keratinocytes
and macrophages (4). It has been
reported that the intracellular serpin proteinase inhibitor 9 may
protect monocytes and macrophages from misdirected GrB in the
lipopolysaccharide (LPS)-induced inflammatory process (5). GrB is considered a ‘natural born
killer’ that functions at multiple points, including directing
proteolytic processing and activation of procaspase-3 and −7, to
initiate the death of the harmful target cells, including
allogeneic, virus-infected and tumor cells (6). GrB directly targets caspase-3 and
initiates the caspase cascade, leading to DNA fragmentation and
apoptosis. Alternately, in the absence of caspase activity, GrB may
still be able to initiate mitochondrial events through the cleavage
of the B-cell lymphoma 2 homology domain 3-interacting domain death
agonist protein (7), which is
important to cell death and is susceptible to proteolytic cleavage
by GrB as well as caspases, calpains and cathepsins (8). A recent report suggested that GrB may
also possess non-cytotoxic roles under inflammatory circumstances
as well as possible functions in the extracellular space (9). In addition, previous studies have
reported that GrA and GrB may influence the production of
pro-inflammatory cytokines; however, the molecular targets of these
proteases in inflammation remain unknown (10,11).
Whether GrB is involved in and promotes the development of
inflammation also remains to be elucidated.
LPS is a component of the Gram-negative bacteria
cell wall that is known to induce inflammation in a number of
physiological and experimental settings. A previous study reported
that inhibition of endoplasmic reticulum (ER) stress alleviated
LPS-induced lung inflammation through modulation of the nuclear
factor-κB (NF-κB) signaling pathway (12). Serine protease inhibitor A3N
(serpin A3N; SA3N) is an extracellular inhibitor of GrB that has
been demonstrated to possess multiple biological functions,
including the attenuation of muscular dystrophy in mice (13), neuropathic pain (14) and GrB-mediated decorin cleavage and
rupture (15); it also induces
neuroprotection in vitro and in vivo (16).
The present study established an inflammatory cell
model by using NK92 cells stimulated with LPS, and demonstrated
that the LPS-induced inflammatory response was prevented by the
inhibition of GrB activity.
Materials and methods
Chemicals
LPS (Escherichia coli 0111:B4) was purchased
from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany); α-minimum
essential medium (MEM) and antibodies against glucose-regulated
protein 78 (GRP78; cat. no. sc-376768), C/EBP homologous protein
(CHOP; cat. no. sc-4066) and β-actin (cat. no. sc-8432) were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Antibodies against GrB (cat. no. 4275S), NF-κB (cat. no. 8242S) and
inhibitor of NF-κB (IκBα; cat. no. 9242S) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA).
Cell culture
NK92 cells were supplied from Kunming Institute of
Zoology (Kunming, China; the human NK92 cell were established from
a donor who suffered from malignant non-Hodgkin's lymphoma and who
gave written informed consent). NK92 cells were maintained in α-MEM
medium with 5% heat-inactivated fetal bovine serum (FBS) and 10%
heat-inactivated fetal horse serum (both Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and antibiotics (100 U/ml penicillin and
100 µg/ml streptomycin) at 37°C in a humidified 5% CO2
atmosphere.
LPS-induced inflammation
Concentration dependence of LPS
NK92 cells in logarithmic growth phase were
incubated in 6-well culture plates and divided into 6 groups, each
treated with 1, 10, 50, 100, and 500 ng/ml LPS, along with an
untreated control group. Following medium exchange, 100 µl of the
different concentrations of LPS were added to the 6 different LPS
treatment groups, and 100 µl α-MEM medium containing 5%
heat-inactivated FBS, 10% heat-inactivated fetal horse serum and
antibiotics (100 U/ml penicillin and 100 µg/ml streptomycin) were
added to the control group. The cells were then incubated at 37°C
in a humidified 5% CO2 atmosphere for 24 h. The cell
culture medium was collected and precipitated by centrifugation for
5 min (750 × g, 4°C) and the clear supernatant extract was analyzed
using ELISA. Experiments were performed in triplicate.
Time dependence of LPS
According to the optimal concentration determined by
the previous LPS concentration dependence test, 100 ng/ml LPS was
added to every test group. NK92 cells in logarithmic growth phase
were incubated in 6-well culture plates and divided into 5 groups,
which were incubated with LPS for 0 (control), 12, 24, 48 or 72 h.
The cell culture medium was collected and precipitated by
centrifugation for 5 min (750 × g, 4°C) and the clear supernatant
extract analyzed using ELISA. Experiments were performed in
triplicate.
Role of SA3N on LPS-induced
inflammation
NK92 cells in logarithmic growth phase were
incubated in 6-well culture plates and divided into 4 groups:
control, LPS, LPS + SA3N (GeneCopoeia, Inc., Rockville, MD, USA)
and SA3N groups. The cells were treated with SA3N (20 µM) for 30
min at 37°C followed by stimulation with 100 ng/ml LPS for 24 h at
37°C. Cell culture medium was collected and precipitated by
centrifugation for 5 min (750 × g, 4°C) the clear supernatant
extract was analyzed using ELISA, and the sedimented cells were
analyzed by western blotting. Experiments were performed in
triplicate.
ELISA analysis
The levels of TNF-α, IL-β and GrB in the clear
supernatant extract were quantified using Quantikine ELISA kits
(R&D Systems, Inc., Minneapolis, MN, USA; TNF-α, cat. no.
DTA00C; IL-β, cat. no. DLB50; GrB, cat. no. DY2906-05), according
to the manufacturer's protocols. Plates were read in an ELISA
reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA) at 450 nm.
The values obtained were plotted onto the standard plot prepared by
using serial dilutions of the standard provided with the kit and
TNF-α, IL-β and GrB concentrations were calculated.
Western blot analysis
Cells were plated in 6-well plates at a density of
1×105 cells/well. Total protein was extracted using
lysis buffer and incubated on ice for 1 h. Protein lysates were
prepared using a solubilizing solution [20 mM Tris-HCl (pH 7.4),
150 mM NaCl, 1% NP-40, 1 mM EDTA, 1 mM phenylmethanesulfonyl
fluoride, 1 mM ethylene-bis(oxyethylenenitrilo)-tetraacetic acid,
1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM
Na3VO4, 1 mM β-glycerol phosphate, and 1
mg/ml leupeptin]. Protein concentrations were determined using a
Bio-Rad protein assay reagent (Bio-Rad Laboratories, Inc.). Samples
were separated on 12% SDS-PAGE and transferred to a polyvinylidene
difluoride membrane (EMD Millipore, Billerica, MA, USA). The
membrane was rinsed with PBS and nonspecific sites were blocked by
incubating the membrane with blocking buffer (PBS + 0.1% Tween-20,
containing 10% nonfat milk) overnight at 4°C or for 2 h at room
temperature. Membranes were subsequently incubated with primary
antibodies to GrB (1:1,500), GRP78 (1:1,000), CHOP (1:1,500), NF-κB
(1:1,000), IκBα (1:1,500), and β-actin (1:1,500) for 1 h at room
temperature, followed by incubation with horseradish
peroxidase-conjugated anti-mouse (1:10,000; cat. no. MA5-16308) or
anti-rabbit (1:10,000; cat. no. 42-6600) immunoglobulin G (KPL,
Inc., Gaithersburg, MD, USA) for 1 h at room temperature. Protein
bands were visualized using an ECL Western Blot Detection kit (EMD
Millipore). ImageJ V1.8.0 software (National Institutes of Health,
Bethesda, MD, USA) was used for the densitometric analysis. All
protein bands are normalized to β-actin. Three independent
experiments were performed.
Statistical analysis
Data were analyzed by using SPSS software 16.0
(SPSS, Inc., Chicago, IL, USA) and were expressed as the mean ±
standard error of the mean. One-way analysis of variance followed
by a Bonferroni post hoc multiple comparison test was used to
compare control and treated groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
LPS-induced inflammatory response in
NK92 cells
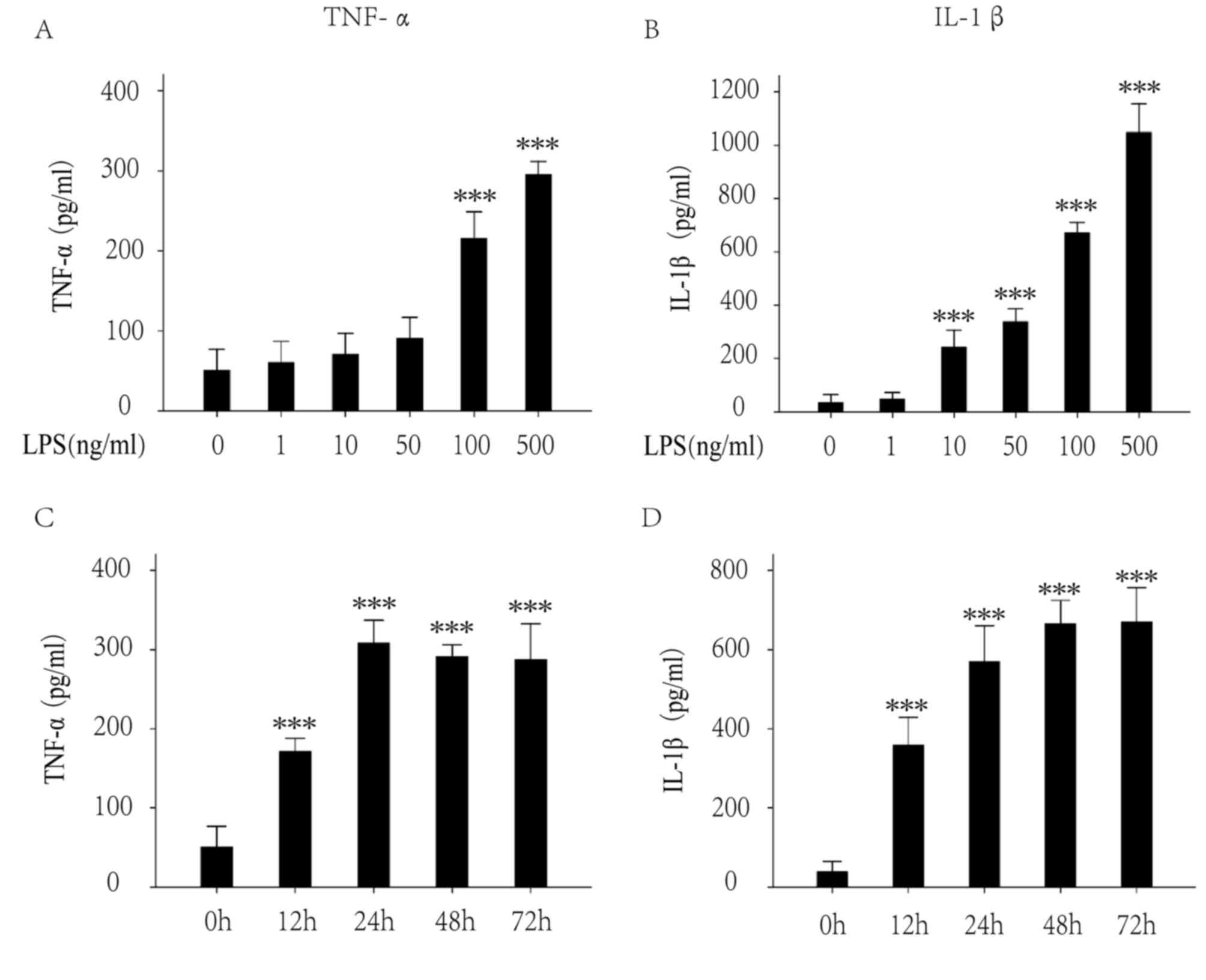
TNF-α and IL-1β are key cytokines involved in
inflammation. The extracellular levels of TNF-α and IL-1β were
investigated by ELISA following LPS treatment (1, 10, 50, 100 and
500 ng/ml) for 24 h. The results demonstrated that 100 ng/mg LPS
significantly increased the levels of TNF-α (Fig. 1A) and IL-1β (Fig. 1B). The extracellular levels of
TNF-α and IL-1β following LPS treatment (100 ng/ml) for various
times (0, 12, 24, 48 and 72 h) were subsequently investigated. The
results demonstrated that LPS significantly increased the levels of
TNF-α (Fig. 1C) and IL-1β
(Fig. 1D) following treatment for
12 h, and the levels become stable at 24 h. Thus, 100 ng/ml for 24
h was set as the best condition for the following experiments.
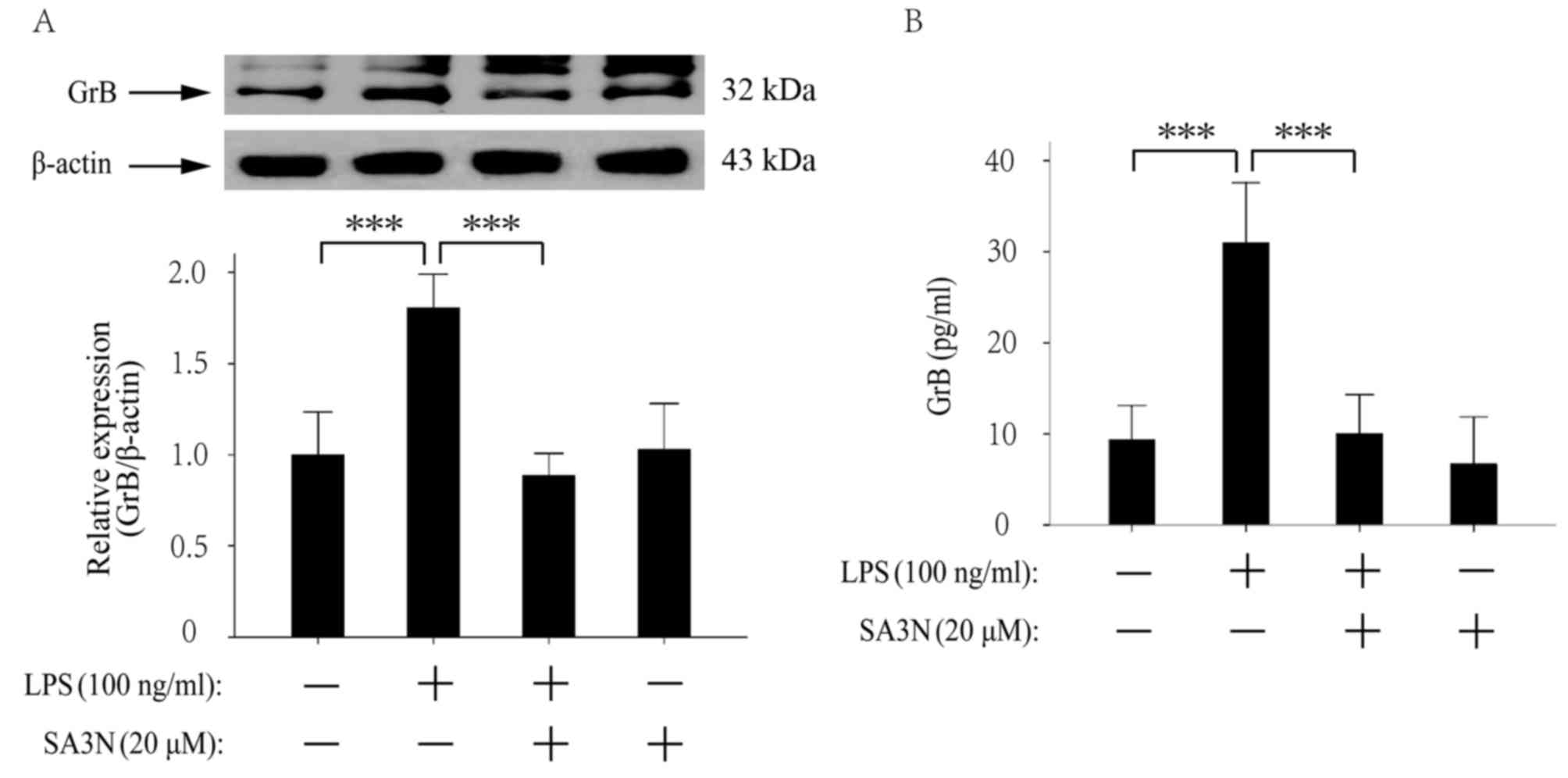
SA3N pretreatment prevents the
expression and exocytosis of GrB by LPS
The expression of GrB was examined by western
blotting and the results demonstrated that LPS increased the
expression of GrB, whereas SA3N (20 µM) pretreatment suppressed the
increase by LPS (Fig. 2A). In
Fig. 2A, the bands that appear
above GrB may be a larger fragment of GrB or may also be a
nonspecific binding, as the active GrB is derived from the
precursor GrB by deglycosylation. Generally, the molecular weight
of the precursor GrB is 35 kDa, and the active GrB is 32 kDa. The
extracellular levels of GrB were examined by ELISA and the results
demonstrated that LPS increased the extracellular levels of GrB,
whereas SA3N pretreatment suppressed the increase by LPS
stimulation (Fig. 2B).
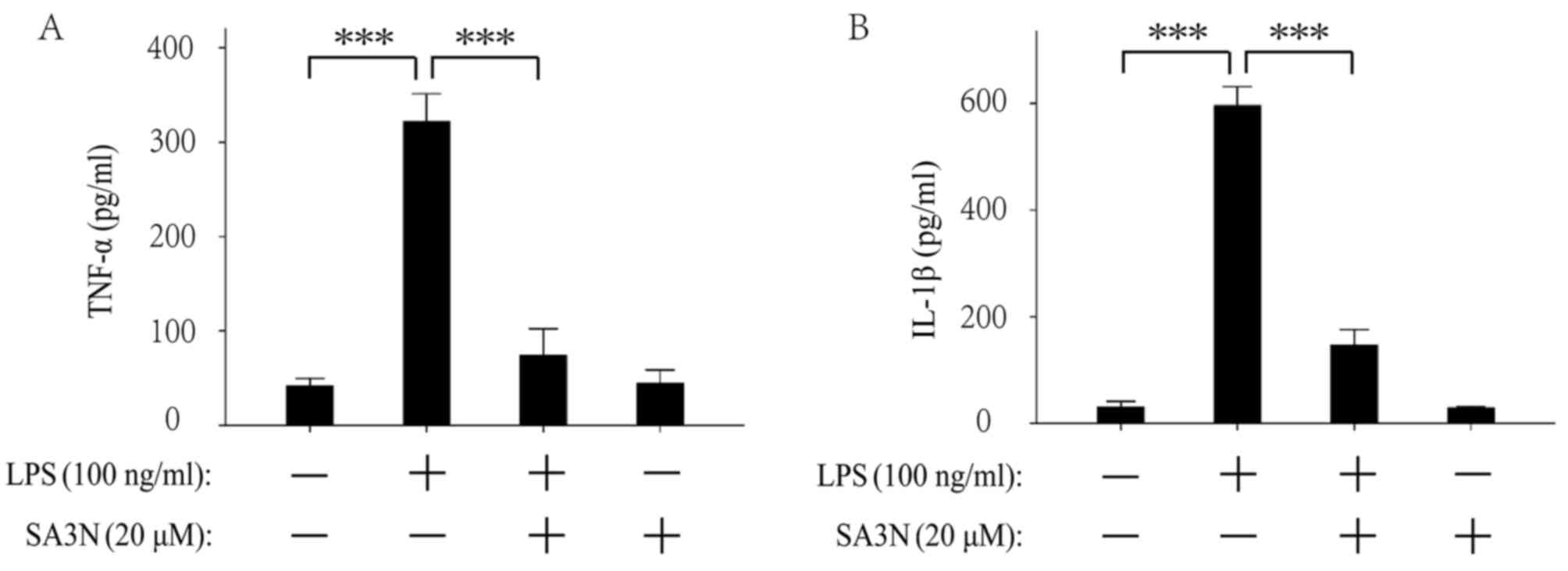
SA3N pretreatment prevents the
exocytosis of TNF-α and IL-1β induced by LPS
The extracellular levels of TNF-α and IL-1β were
examined by ELISA. The results demonstrated that LPS increased the
extracellular levels of TNF-α (Fig.
3A) and IL-1β (Fig. 3B),
whereas SA3N pretreatment suppressed the increases by LPS
treatment. The results indicated that reduction of GrB activity may
contribute to alleviate the LPS-induced inflammatory response.
SA3N pretreatment prevents the
LPS-induced changes in expression levels of GRP78, CHOP, NF-κB and
IκBα proteins
GRP78, CHOP, NF-κB and IκBα were involved in
LPS-induced ER stress (12,17).
It has been demonstrated that inhibition of ER mediated by NF-κB
pathway may alleviate LPS-induced lung inflammation (12). Thus, the NF-κB pathway was
examined. The western blotting results demonstrated that LPS
stimulation increased the expression levels of GRP78, CHOP and
NF-κB, and decreased the expression of IκBα, and SA3N pretreatment
suppressed the changes in expression levels induced by LPS
(Fig. 4A-D). The results indicated
that the reduction of GrB activity alleviated LPS-induced
inflammatory response may occur through the regulation of the NF-κB
pathway.
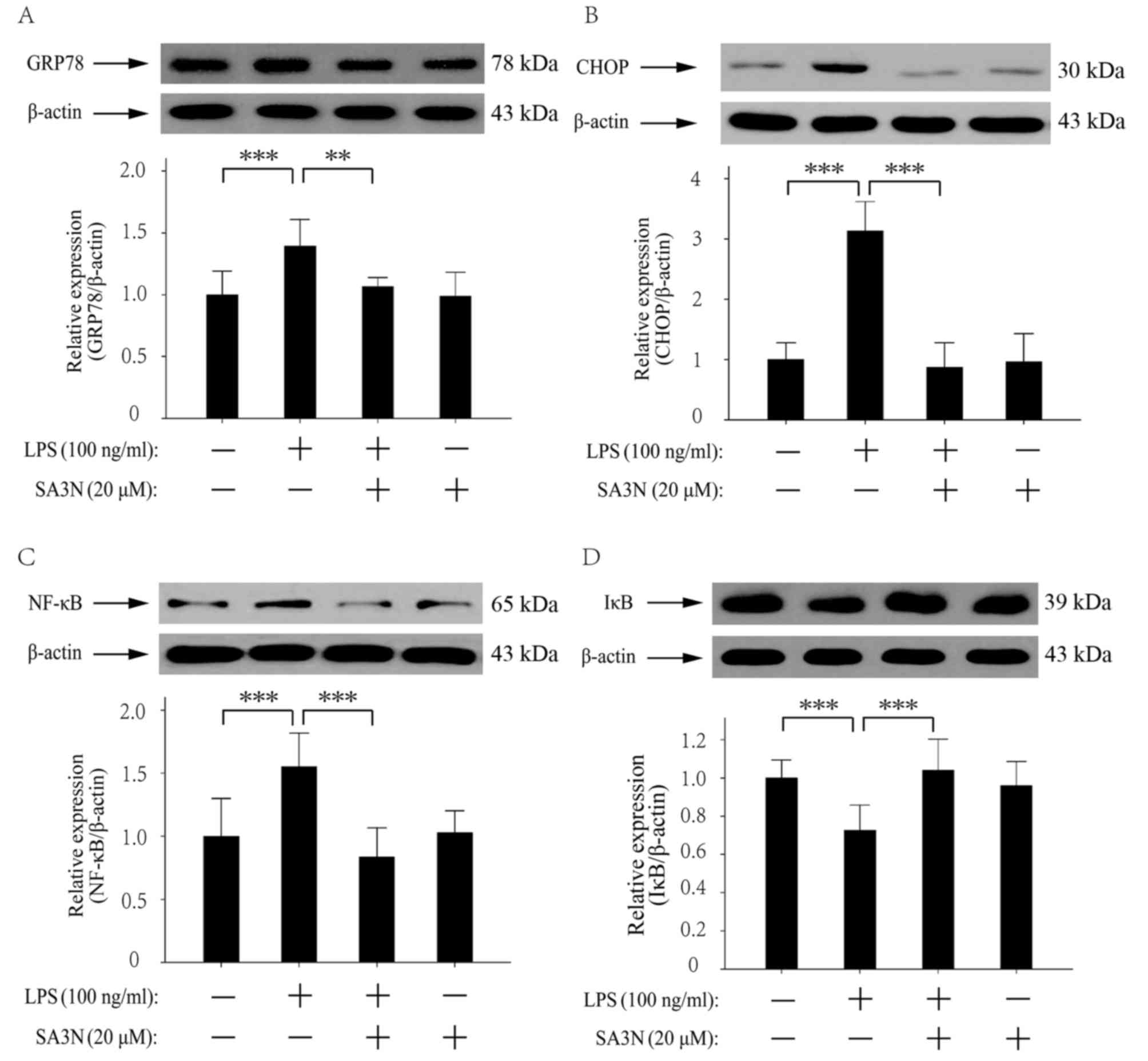 | Figure 4.Role of SA3N pretreatment on the
LPS-induced changes of GRP78, CHOP, NF-κB and IκBα protein
expression levels. LPS increased the expression levels of (A)
GRP78, (B) CHOP and (C) NF-κB, whereas SA3N pretreatment suppressed
the increases induced by LPS. (D) LPS treatment decreased the
expression of IκBα, and SA3N pretreatment suppressed the reduction
induced by LPS. **P<0.01 and ***P<0.001. CHOP, C/EBP
homologous protein; GRP78, glucose-regulated protein 78; IκBα,
inhibitor of NF-κB; LPS, lipopolysaccharide; NF-κB, nuclear
factor-κB; SA3N, serpin A3N. |
Discussion
GrB is a major constituent of CTL and NK cell
granules, and the mechanism of GrB-mediated cell death has been
well studied (9). Recent evidence
has begun to uncover possible non-cytotoxic roles for GrB (18). Elevated levels of circulating GrB
are a characteristic feature of a number of inflammatory diseases
(19,20). In addition, a previous study
reported that GrB may be involved in LPS-induced toxic shock, which
suggested that, not only does GrB promote cell death upon delivery
to target cells, but it may also function upon release into the
extracellular space (21).
However, it remains to be elucidated if GrB is involved in the
development of inflammation. Exploring the role of GrB in the
development of inflammation will provide a new strategy for the
treatment of inflammatory diseases.
LPS activates innate immune cells, which leads to
the production of pro-inflammatory cytokines (22). It has been reported that citrate is
able to modulate LPS-induced monocyte inflammatory responses
(23) and that suppression of
NF-κB signaling in BV-2 microglial cells inhibits LPS-induced
inflammatory responses (24). The
present study demonstrated that LPS treatment significantly
increased the release of inflammatory cytokines TNF-α and IL-1β,
which indicated that LPS was able to induce the inflammatory
response in NK92 cells. The present results also demonstrated that
LPS stimulation increased the expression and release of GrB in NK92
cells, which indicated that GrB may be involved in the development
of inflammation; however, SA3N pretreatment suppressed this
LPS-induced increase. SA3N forms a complex and stable covalent bond
with GrB that results in the inhibition of the enzymatic activity
of the protease (25). In the
present study, treatment with SA3N alone did not change the
expression of GrB, whereas pretreatment with SA3N prior to LPS
stimulation inhibited the expression of GrB in NK92 cells, which
suggested that pretreatment with SA3N may block the LPS-induced
cytotoxicity mediated by GrB by suppressing the activity of GrB;
this may be the reason that the expression of GrB in LPS + SA3N
treated cells was not increased compared with the control group. It
was also identified that LPS treatment significantly increased the
levels of TNF-α and IL-1β, which were also suppressed in cells
pretreated with SA3N pretreatment. To elucidate how GrB served its
role on LPS-induced inflammation, ER stress was investigated.
ER stress has been reported to be induced by LPS
(26). The NF-κB signaling pathway
is involved in ER stress (27),
and GRP78 and CHOP are markers of ER stress and are involved in the
unfolding protein reaction and in the protection mechanism during
ER stress (28–30). It has been reported that the
increased expression of CHOP during the development of ER stress
was inhibited when ER stress was blocked (30). Consistent with these reports, the
results of the present study demonstrated that LPS treatment
significantly induced the expression levels of GRP78 and CHOP,
which were inhibited by SA3N pretreatment. NF-κB activation
regulates the expression of >100 genes involved in diverse cell
processes, including cell proliferation, differentiation, apoptosis
and the inflammation and immune responses (31,32).
A previous study demonstrated that CHOP induced cell inflammatory
responses by activating NF-κB (33). Results from the present study
indicated that LPS treatment induced the expression of NF-κB and
decreased the expression of IκBα, which indicating that LPS induced
inflammatory response may be through initiating ER stress. However,
inhibition of GrB activity suppressed the changes of NF-κB and IκBα
expression levels induced by LPS. Thus, it was hypothesized that
inhibition of GrB activity may have suppressed ER stress by
blocking the NF-κB pathway, which may indicate that inhibition of
GrB activity blocked the development of inflammation. In addition,
it has been reported that TNF-α mediated the activation of NF-κB in
various disease processes, including inflammation and cell
apoptosis (34). In the present
study, inhibition of GrB activity blocked the exocytosis of TNF-α,
and it is therefore suggested that the reduction of TNF-α levels
may be the key point that blocked the activation of the NF-κB
pathway.
SA3N is the only known GrB activity inhibitor that
is secreted extracellularly (35).
It has been previously reported that SA3N attenuates GrB-mediated
decorin cleavage and rupture in a mouse model of aortic aneurysm
(15) and that SA3N induces
neuroprotection in vitro and in vivo, which has been
reported to be a potentially novel therapeutic approach for
inflammation-mediated neurodegenerative diseases such as multiple
sclerosis (23). However, SA3N is
not a GrB specific inhibitor. It has been reported that SA3N may
accelerate the wound healing process by inhibiting the activity of
the serine proteases cathepsin G and GrB that are associated with
inflammation, as well as matrix metalloproteinase 9 that is
associated with extracellular matrix breakdown and remodeling
(13,15,36).
In addition, SA3N also inhibits T cell-derived leukocyte elastase,
which helps attenuate neuropathic pain (14). Taken together, SA3N as a GrB
activity inhibitor may serve various protective roles by acting on
different targets. Thus, it was suggested that SA3N may serve its
protective role on LPS-induced inflammatory response as the result
of the concurrent effects of various factors and mechanisms.
In conclusion, the results of the present study
indicated that GrB may be a potential pro-inflammatory factor that
is induced by LPS, and the inhibition of GrB activity may block
LPS-induced inflammatory response. It is hypothesized that GrB may
be involved in the development of inflammation through regulation
of the NF-κB pathway, mediated by TNF-α. Therefore, GrB has
potential as a therapeutic agent for inflammatory diseases.
However, the connection between GrB and TNF-α, and the molecular
mechanism of their interaction requires further study.
Acknowledgements
Not applicable.
Funding
This study was funded by The National Natural
Science Foundation of China (grant no. 81202331).
Availability of data and material
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JT and LW were responsible for the study concept and
design. LW and SJ performed the western blot analysis. LX and LC
performed the ELISA analysis. YZ performed the MTT assay. JT and LW
assisted with data analysis and interpretation of results. LW
drafted the manuscript. JT and LW provided critical revision of the
manuscript for important intellectual content. All authors
critically reviewed the content and approved final version for
publication.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bladergroen BA, Meijer CJ, ten Berge RL,
Hack CE, Muris JJ, Dukers DF, Chott A, Kazama Y, Oudejans JJ,
Berkum O, et al: Expression of the granzyme B inhibitor, protease
inhibitor 9, by tumor cells in patients with non-Hodgkin and
Hodgkin lymphoma: A novel protective mechanism for tumor cells to
circumvent the immune system? Blood. 99:232–237. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thomas DA, Scorrano L, Putcha GV,
Korsmeyer SJ and Ley TJ: Granzyme B can cause mitochondrial
depolarization and cell death in the absence of BID, BAX, and BAK.
Proc Natl Acad Sci USA. 98:14985–14990. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alimonti JB, Shi L, Baijal PK and
Greenberg AH: Granzyme B induces BID-mediated cytochrome c release
and mitochondrial permeability transition. J Biol Chem.
276:6974–6982. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mattila JT, Maiello P, Sun T, Via LE and
Flynn JL: Granzyme B-expressing neutrophils correlate with
bacterial load in granulomas from Mycobacterium
tuberculosis-infected cynomolgus macaques. Cell Microbiol.
17:1085–1097. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Classen CF, Bird PI and Debatin KM:
Modulation of the granzyme B inhibitor proteinase inhibitor 9
(PI-9) by activation of lymphocytes and monocytes in vitro and by
Epstein-Barr virus and bacterial infection. Clin Exp Immunol.
143:534–542. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rousalova I and Krepela E: Granzyme
B-induced apoptosis in cancer cells and its regulation (review).
Int J Oncol. 37:1361–1378. 2010.PubMed/NCBI
|
|
7
|
Lord SJ, Rajotte RV, Korbutt GS and
Bleackley RC: Granzyme B: A natural born killer. Immunol Rev.
193:31–38. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yin XM: Bid, a BH3-only multi-functional
molecule, is at the cross road of life and death. Gene. 369:7–19.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Afonina IS, Cullen SP and Martin SJ:
Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme
B. Immunol Rev. 235:105–116. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Froelich CJ, Pardo J and Simon MM:
Granule-associated serine proteases: Granzymes might not just be
killer proteases. Trends in Immunol. 30:117–123. 2009. View Article : Google Scholar
|
|
11
|
Afonina IS, Tynan GA, Logue SE, Cullen SP,
Bots M, Lüthi AU, Reeves EP, McElvaney NG, Medema JP, Lavelle EC
and Martin SJ: Granzyme B-dependent proteolysis acts as a switch to
enhance the proinflammatory activity of IL-1α. Mol Cell.
44:265–278. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim HJ, Jeong JS, Kim SR, Park SY, Chae HJ
and Lee YC: Inhibition of endoplasmic reticulum stress alleviates
lipopolysaccharide-induced lung inflammation through modulation of
NF-κB/HIF-1α signaling pathway. Sci Rep. 3:11422013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tjondrokoesoemo A, Schips T, Kanisicak O,
Sargent MA and Molkentin JD: Genetic overexpression of Serpina3n
attenuates muscular dystrophy in mice. Hum Mol Genet. 25:1192–1202.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vicuña L, Strochlic DE, Latremoliere A,
Bali KK, Simonetti M, Husainie D, Prokosch S, Riva P, Griffin RS,
Njoo C, et al: The serine protease inhibitor SerpinA3N attenuates
neuropathic pain by inhibiting T cell-derived leukocyte elastase.
Nat Med. 21:518–523. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ang LS, Boivin WA, Williams SJ, Zhao H,
Abraham T, Carmine-Simmen K, McManus BM, Bleackley RC and Granville
DJ: Serpina3n attenuates granzyme B-mediated decorin cleavage and
rupture in a murine model of aortic aneurysm. Cell Death Dis.
2:e2092011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Haile Y, Carmine-Simmen K, Olechowski C,
Kerr B, Bleackley RC and Giuliani F: Granzyme B-inhibitor serpina3n
induces neuroprotection in vitro and in vivo. 12:1572015.
|
|
17
|
Duan L, Zhang X, Panpan YU, Feng YU, Liu W
and Song N: Effects of cholecystokinin-octopeptide on endoplasmic
reticulum stress induced by LPS in mouse monocyte RAW264.7 cells.
Journal of Shandong University. 53:16–20. 2015.
|
|
18
|
Afonina IS, Cullen SP and Martin SJ:
Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme
B. Immunol Rev. 235:105–116. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tak PP, Kummer JA, Hack CE, Daha MR,
Smeets TJ, Erkelens GW, Meinders AE, Kluin PM and Breedveld FC:
Granzyme-positive cytotoxic cells are specifically increased in
early rheumatoid synovial tissue. Arthritis Rheum. 37:1735–1743.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Goldbach-Mansky R, Suson S, Wesley R, Hack
CE, El-Gabalawy HS and Tak PP: Raised granzyme B levels are
associated with erosions in patients with early rheumatoid factor
positive rheumatoid arthritis. Ann Rheum Dis. 64:715–721. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Metkar SS, Menaa C, Pardo J, Wang B,
Wallich R, Freudenberg M, Kim S, Raja SM, Shi L, Simon MM and
Froelich CJ: Human and mouse granzyme A induce a proinflammatory
cytokine response. Immunity. 29:720–733. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rossol M, Heine H, Meusch U, Quandt D,
Klein C, Sweet MJ and Hauschildt S: LPS-induced cytokine production
in human monocytes and macrophages. Crit Rev Immunol. 31:379–446.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ashbrook MJ, McDonough KL, Pituch JJ,
Christopherson PL, Cornell TT, Selewski DT, Shanley TP and Blatt
NB: Citrate modulates lipopolysaccharide-induced monocyte
inflammatory responses. Clin Exp Immunol. 180:520–530. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kwon SH, Ma SX, Hong SI, Lee SY and Jang
CG: Lonicera japonica THUNB. Extract Inhibits
Lipopolysaccharide-Stimulated Inflammatory Responses by Suppressing
NF-κB Signaling in BV-2 Microglial Cells. J Med Food. 18:762–775.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Haile Y, Carmine-Simmen K, Olechowski C,
Kerr B, Bleackley RC and Giuliani F: Granzyme B-inhibitor serpina3n
induces neuroprotection in vitro and in vivo. J Neuroinflammation.
12:1572015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu H, Yin JJ, Cao MM, Liu GD, Su Y and Li
YB: Endoplasmic reticulum stress induced by lipopolysaccharide is
involved in the association between inflammation and autophagy in
INS-1 cells. Mol Med Rep. 16:5787–5792. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Prell T, Lautenschläger J, Weidemann L,
Ruhmer J, Witte OW and Grosskreutz J: Endoplasmic reticulum stress
is accompanied by activation of NF-κB in amyotrophic lateral
sclerosis. J Neuroimmunol. 270:29–36. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu H, Qian J, Wang F, Sun X, Xu X, Xu W
and Zhang X and Zhang X: Expression of two endoplasmic reticulum
stress markers, GRP78 and GADD153, in rat retinal detachment model
and its implication. Eye (Lond). 24:137–144. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee AS: The ER chaperone and signaling
regulator GRP78/BiP as a monitor of endoplasmic reticulum stress.
Methods. 35:373–381. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen G, Li X, Huang M, Li M, Zhou X, Li Y
and Bai J: Thioredoxin-1 increases survival in sepsis by
inflammatory response through suppressing endoplasmic reticulum
stress. Shock. 46:67–74. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin WC, Chuang YC, Chang YS, Lai MD, Teng
YN, Su IJ, Wang CC, Lee KH and Hung JH: Endoplasmic reticulum
stress stimulates p53 expression through NF-κB activation. PLoS
One. 7:e391202012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ghosh S and Karin M: Missing pieces in the
NF-kappaB puzzle. Cell. 109 Suppl:S81–S96. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Willy JA, Young SK, Stevens JL, Masuoka HC
and Wek RC: CHOP links endoplasmic reticulum stress to NF-κB
activation in the pathogenesis of nonalcoholic steatohepatitis. Mol
Biol Cell. 26:2190–2204. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Legler DF, Micheau O, Doucey MA, Tschopp J
and Bron C: Recruitment of TNF Receptor 1 to Lipid Rafts Is
Essential for TNFalpha-Mediated NF-kappaB Activation. Immunity.
18:655–664. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Palacios MM and Bleackley C:
Characterization of Critical Residues of the Granzyme B Inhibitor,
Serpina3n (50.43). J Immunol. 182:43–50. 2009.
|
|
36
|
Han YP, Yan C and Garner WL: Proteolytic
activation of matrix metalloproteinase-9 in skin wound healing is
inhibited by alpha-1-Antichymotrypsin. J Invest Dermatol.
128:2334–2342. 2008. View Article : Google Scholar : PubMed/NCBI
|