Introduction
Bone marrow-derived mesenchymal stem cells (BM-MSCs)
are capable of self-renewal and multilineage differentiation into
various cell types (1–3), including endothelial cells, neural
cells, smooth muscle cells, skeletal myoblasts, and cardiac
myocytes (4,5). Considerable experimental and clinical
evidence has demonstrated that infracted myocardium regeneration
with BM-MSCs is safe and feasible (2). Transplantation of BM-MSCs leads to
the improved neovascularization of ischemic myocardium and
myocardial fibrosis inhibition, as well as an increase in
prosurvival growth factor secretion, including vascular endothelial
growth factor, hepatocyte growth factor, and insulin-like growth
factor (5–8). Despite these advantages, the poor
survival rate of BM-MSCs within the first few days following
engraftment in the infarcted heart results in only marginal
functional improvement (9,10). Therefore, it is necessary to
protect the cells against the hostile microenvironment created by
ischemia, hypoxia, the inflammatory response and pro-apoptotic
factors, in order to improve the efficacy of BM-MSC transplantation
therapy.
microRNAs (miRNAs/miRs) are endogenous RNAs ~22
nucleotides in length that negatively regulate gene expression by
targeting mRNAs for cleavage or translational repression, which
occurs primarily through base pairing to the 3′ untranslated region
(UTR) of target mRNAs (11,12).
Previous studies have reported that miRNA-regulated translational
repression appears to occur on the post-transcriptional level via
inhibition of protein translation from mRNAs or by promotion of
mRNA degradation (13,14). Furthermore, bioinformatics analyses
have predicted that each miRNA regulates hundreds of targets,
suggesting that miRNAs may have a central role in an increasing
number of biological processes, including cell proliferation,
migration, differentiation and apoptosis (11,14,15).
Additionally, it has been demonstrated that miRNAs are involved in
the self-renewal and differentiation of MSCs (16).
Among the known miRNAs, miR-16 has been reported to
regulate apoptosis and the cell cycle by targeting cyclin D3
(CCND3), cyclin E1 (CCNE1), CDK6 and B-cell lymphoma (Bcl)-2 in
tumor cells (15,17–20).
Bcl-2, originally found to be overexpressed in B-cell lymphoma
(21), is a critical inhibitor in
the apoptotic pathway (22).
Previous studies have demonstrated that Bcl-2 is an integral
membrane protein that resides on the outer membrane of
mitochondria. It prevents the initiation of the cellular apoptotic
program by blocking cytochrome c release from mitochondria in
response to a variety of stimuli (23,24).
Cytochrome c is a key mediator of apoptosis that may bind to
apoptotic protease activating factor-1 (APAF-1), which subsequently
activates caspase-9 (25).
Accumulating evidence suggests that miR-16 targets
Bcl-2 at the posttranscriptional level (18–20).
Additionally, miR-16 downregulation in various cell types coincides
with Bcl-2 upregulation (19,20).
However, if miR-16 functions as a regulator of apoptosis in stem
cells remains unknown. In the current study, it was hypothesized
that miR-16 inhibition may protect BM-MSCs from apoptosis. It was
investigated whether miR-16, which is among the upregulated miRNAs
in a hypoxic/serum deprived (SD) environment, regulated the
apoptosis of BM-MSCs by targeting Bcl-2. The data indicated that
inhibition of miR-16 resulted in increased Bcl-2 protein expression
and a reduced rate of apoptosis. Additionally, it was demonstrated
that miR-16 regulated mitochondrial apoptosis through
APAF-1/caspase-9/poly (ADP ribose) polymerase (PARP) pathway.
Materials and methods
Animals and ethics statement
Male Sprague-Dawley rats weighing 60–80 g (8–10
weeks old) were housed under standardized conditions at 22°C in a
12-h light/dark cycle and fed a laboratory diet with water ad
libitum. They were obtained from the Laboratory Animal Science
Department of the Second Affiliated Hospital of Harbin Medical
University (Harbin, China). All experimental animal procedures were
approved by the Ethical Committee on Animal Care and Use of Harbin
Medical University (Harbin, China).
BM-MSC culture and treatment
BM-MSCs were cultured using the whole bone marrow
adherent method, as previously reported (4). Briefly, total bone marrow was
harvested from the tibia and femur of rats. BM-MSCs were placed in
25 cm2 culture flasks at a density of 106
cells/ml in Dulbecco's modified Eagle's medium/nutrient mixture F12
(DMEM/F12; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 15% fetal bovine serum (FBS; Thermo Fisher
Scientific, Inc.) and 1% penicillin/streptomycin. Cells were
incubated at 37°C in a humidified tissue culture incubator with 5%
CO2. After 3 days of incubation, the medium was changed
and then replaced every 2 days thereafter. Passage 3 BM-MSCs were
used in all experiments.
Hypoxia and SD treatment
Apoptosis was induced by hypoxia and SD in
vitro. The method was designed to mimic the in vivo
conditions of ischemia in the myocardium and was performed as
previously described (26). In
brief, BM-MSCs were washed with serum-free DMEM/F12 and incubated
in a 5% CO2/95% N2 incubator (Controlled
Atmosphere Chamber, PLAS-Labs, Inc., Lansing, MI, USA) for 3–24 h.
BM-MSCs incubated in a 5% CO2/95% O2
incubator were used as the normoxic control and were cultured in
DMEM/F12 supplemented with 15% FBS and 1%
penicillin/streptomycin.
Cell transfection
Small interfering RNAs (siRNAs) are small
double-stranded RNAs that target mRNA to silence its expression. A
Bcl-2 siRNA duplex was synthesized by Thermo Fisher Scientific,
Inc. (sense, 5′-GCUGCACCUGACGCCCUUCTT-3′ and antisense,
3′-TTCGACGUGGACUGCGGGAAG-5′). Cells were transfected using
X-tremeGENE™ siRNA Transfection Reagent (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) as previously described (27). Cells were seeded in a 6-well plate
(2×105 cells/well) and incubated at 37°C for 24 h and
subsequently transfected with miR-16 mimics, miR-16 mimic
inhibitor, scrambled miRNA, or Bcl-2 siRNA (100 nM). siRNA
(GCUGCACCUGACGCCCUUCTT; TTCGACGUGGACUGCGGGAAG); Scramble
(UUCUCCGAACGUGUCUCG; TTAAGAGGCUUGCACAGUGCA; all from Invitrogen;
Thermo Fisher Scientific, Inc.) and incubated in 2 ml FBS-free
Opti-MEM (Invitrogen; Thermo Fisher Scientific, Inc.) for 6–8 h.
All cells were subjected to hypoxia and SD treatment prior to
transfection. Following this, the medium was replaced with fresh
complete medium and the cells were incubated for an additional 24
h. Transfected cells were subjected to analysis 72 h
post-transfection.
Cell viability and proliferation
assays
BM-MSC viability and proliferation was determined
using an MTT assay (Sigma-Aldrich; Merck KGaA) and an EdU
incorporation assay (Guangzhou RiboBio Co., Ltd., Guangzhou,
China), respectively, according to the manufacturers' protocols.
For the MTT assay, cells were seeded into a 96-well plate (3,000
cells/well), and viability was detected with the addition of 20 µl
MTT (5 mg/ml), dissolved in DMSO, to the culture medium. The
absorbance of each well was quantified at 490 nm using the Infinite
M200 PRO plate reader (Tecan, Morrisville, NC, USA). All data were
calculated from triplicate samples and are presented as the mean ±
standard deviation.
For the EdU incorporation assay, BM-MSCs were
cultured in 96-well plates at a density of 4×103
cells/well for 24 h at 37°C. Following this, 50 µM EdU was added to
each well and cells were cultured for additional 2 h at 37°C. Cells
were fixed with 4% formaldehyde for 15 min at room temperature and
subsequently treated with 0.5% Triton X-100 for 20 min for
permeabilization. Following three washes with PBS, 100 µl 1X Apollo
reaction cocktail was added to each well and the cells were
incubated for 30 min at room temperature prior to staining with 100
µl Hoechst 33342 (10 µg/ml) at room temperature (24°C) for 30 min
and visualization under a fluorescent microscope (magnification, ×
100; Leica Microsystems GmbH, Wetzlar, Germany). The positive
staining rate (%) was counted as positive cells (green
cells)/overall cells (blue cells). DAPI (50 µg/ml) stain was
conducted in 37°C for 2 h. Cells were counted from 6 random fields
in triplicate wells for each condition and expressed as percentage
of total number of cells in the field. All experiments were
performed in triplicate and three independent repeated experiments
were performed.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the BM-MSCs with TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and reverse
transcribed into cDNA with the miRcute miRNA first-strand cDNA
synthesis kit (Tiangen Biotech Co., Ltd., Beijing, China) according
to the manufacturer's instructions. PCR (Initial Denaturation,
98°C, 30 sec, 25–35 cycles at 98°C, 5–10 sec; 45–72°C, 10–30 sec;
72°C, 15–30 sec per kb. Final Extension, 72°C, 5–10 min. Hold,
4–10°C.) was performed to analyze the level of miR-16 using the
miRcute miRNA qPCR Detection kit (SYBR Green; Tiangen Biotech Co.,
Ltd.). All primers for miR-16 and U6 for the TaqMan miRNA assays
were purchased from Thermo Fisher Scientific, Inc. The miR-16
primer sequences were as follows: forward,
5′-GCTTCGGCAGCACATATACTAAAAT-3′ and reverse,
5′-CGCTTCACGAATTTGCGTGTCAT-3′. The relative expression level of
miR-16 was normalized to that of the internal control U6 using the
2−∆∆Cq cycle threshold method (28).
Protein extraction and western blot
analysis
Cells were washed twice with ice-cold PBS 72 h after
transfection and harvested for further analysis. Total protein was
obtained with RIPA protein extraction buffer (Thermo Fisher) and
the concentration was analyzed using the bicinchoninic acid protein
assay and 50 µg total protein per lane, extracted with ice-cold PBS
was resolved via 12% SDS-PAGE and subsequently transferred onto
polyvinylidene difluoride membranes. Non-specific binding was
inhibited by incubating the membranes with 5% skim milk and Tris
buffered saline with 0.5% Tween-20 (TBST) at 4°C for 12 h.
Membranes were incubated with the following primary antibodies
overnight at 4°C: Bcl-2, caspase-3 (1:1,000; cat no. ab49822),
caspase-9 (1:1,000; cat. no. ab61789; both Abcam, Cambridge, UK),
APAF-1 (1:1,000), cleaved PARP (1:1,000; Cell Signaling Technology;
Danvers, MA, USA) and β-actin (1:1,000; OriGene Technologies, Inc.,
Beijing, China). The membranes were washed with TBS-T and
subsequently incubated with horseradish peroxidase-conjugated
Affinipure goat anti-rabbit IgG and anti-mouse IgG secondary
antibodies (1:5,000; TA140003; OriGene Technologies, Inc.) for 1 h
at 37°C. Specific complexes were visualized on an X-ray film via
enhanced chemiluminescence (ECL) detection with BeyoECL Plus
(Beyotime Institute of Biotechnology, Beijing, China) according to
the manufacturer's protocol. Densitometric analysis was performed
using a GS-710 Imaging Densitometer (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) and Image Lab™ Software (170–9690) to measure
protein levels normalized to that of internal control β-actin. All
data were obtained in triplicate, independent experiments.
Flow cytometric evaluation of
apoptosis
Cell apoptosis was examined using an Annexin
V-fluorescein isothiocyanate (FITC) Apoptosis Detection kit (BD
Biosciences, Franklin Lakes, NJ, USA). Briefly, cells were
incubated with 5 µl Annexin V-FITC solution in the dark at room
temperature for 15 min, followed by staining with 5 µl propidium
iodide (PI) at room temperature for 5 min, according to the
manufacturer's protocol. Stained cells were distinguished as viable
(Annexin V/PI−), dead (Annexin V/PI+), early
apoptotic (Annexin V+/PI−), or late apoptotic
(Annexin V+/PI+) using a FACSCalibur flow
cytometer. Data were analyzed with the BD FACSCanto II equipped
with BD FACSDiva software version 8.0 (BD Biosciences). Experiments
were done in triplicate.
Mitochondrial membrane potential (MMP)
assay
The MMP was analyzed using the JC-1 MMP Assay kit
(Beyotime Institute of Biotechnology) according to the
manufacturer's protocol (29).
Cells were collected, resuspended in culture medium (DMEM/F12
supplemented with 15% FBS and 1% penicillin/streptomycin) to a
concentration of 106 cells/ml and incubated with
staining dye at 37°C for 15 min. Following this, cells were washed
twice with cold JC-1 staining buffer and were observed under a
fluorescence microscope (magnification, ×100; DMI4000B; Leica,
Wetzlar, Germany).
Statistical analysis
All data are expressed as the mean ± standard
deviation. Differences between the mean values were calculated with
one way analysis of variance with Fisher's LSD used for the
post-hoc multiple comparisons test. Statistical analyses were
performed using SPSS software (version 22.0; IBM Corp., Armonk, NY,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
miR-16 expression after
hypoxia/SD
To determine if miR-16 was involved in
hypoxia/SD-induced apoptosis, BM-MSCs were exposed to hypoxic/SD
conditions for different periods of time (3–24 h). Apoptosis was
detected by Annexin V-FITC, which binds to phosphatidylserine, a
phospholipid that is redistributed from the inner to the outer
leaflet of the cell membrane in early apoptosis. Following membrane
integrity loss, PI may also enter the cell and intercalate into DNA
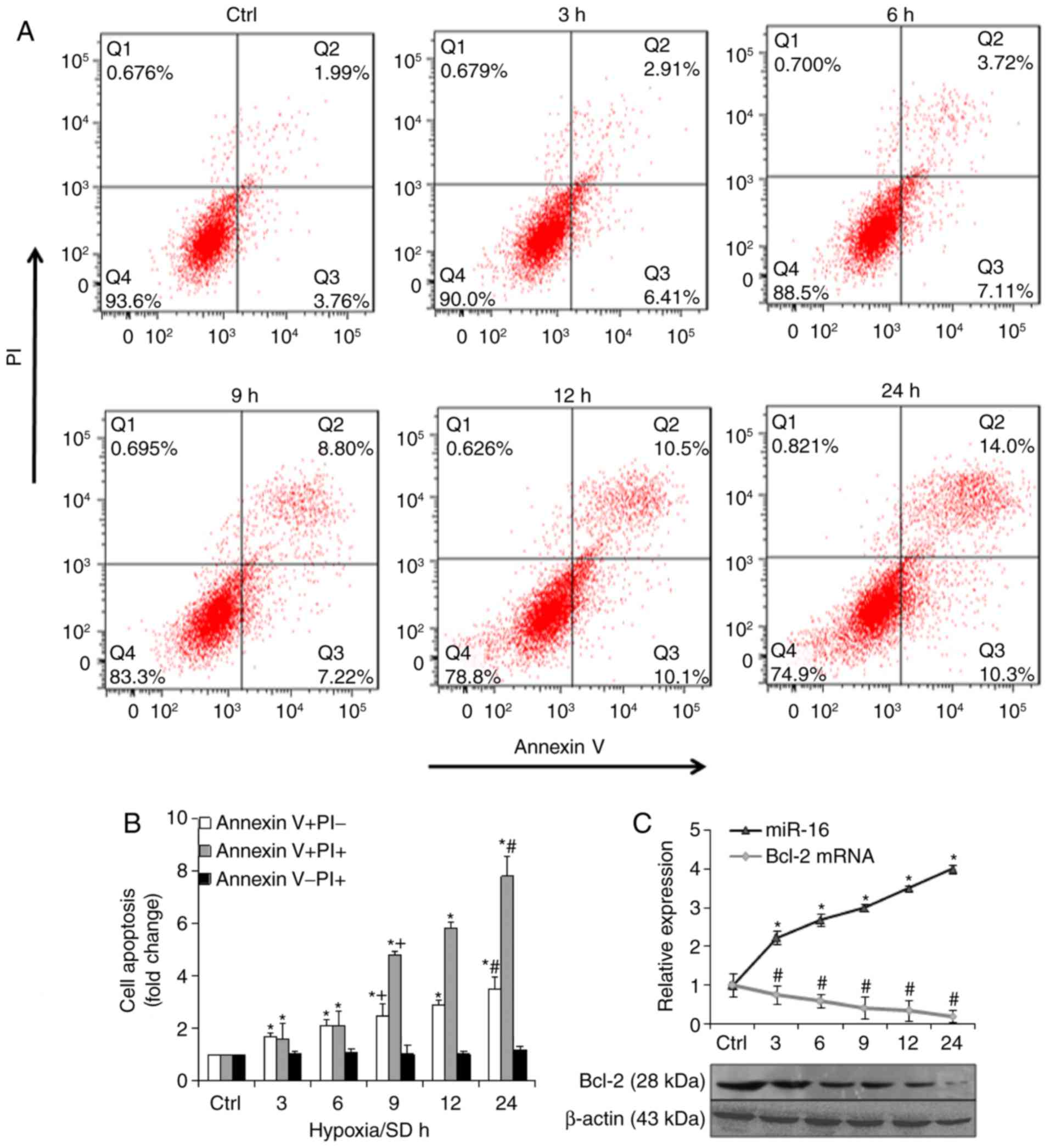
(30). The results revealed that
hypoxia/SD induced BM-MSCs to undergo apoptosis (Fig. 1A). Notably, apoptosis (Annexin
V+/PI) significantly increased with time (Fig. 1B). RT-qPCR and western blot
analyses were used to evaluate expression levels of miR-16 and
Bcl-2. The results indicated that in hypoxia/SD conditions, miR-16
expression was markedly upregulated and Bcl-2 mRNA and protein
expression was markedly downregulated, in a time-dependent manner
(Fig. 1C).
Bcl-2 is a direct target of
miR-16
Previous studies have indicated that miRNAs regulate
gene expression by decreasing mRNA translation, increasing mRNA
degradation, or both (13,14). To examine if miR-16 regulated Bcl-2
expression, RT-qPCR and western blot analyses were performed to
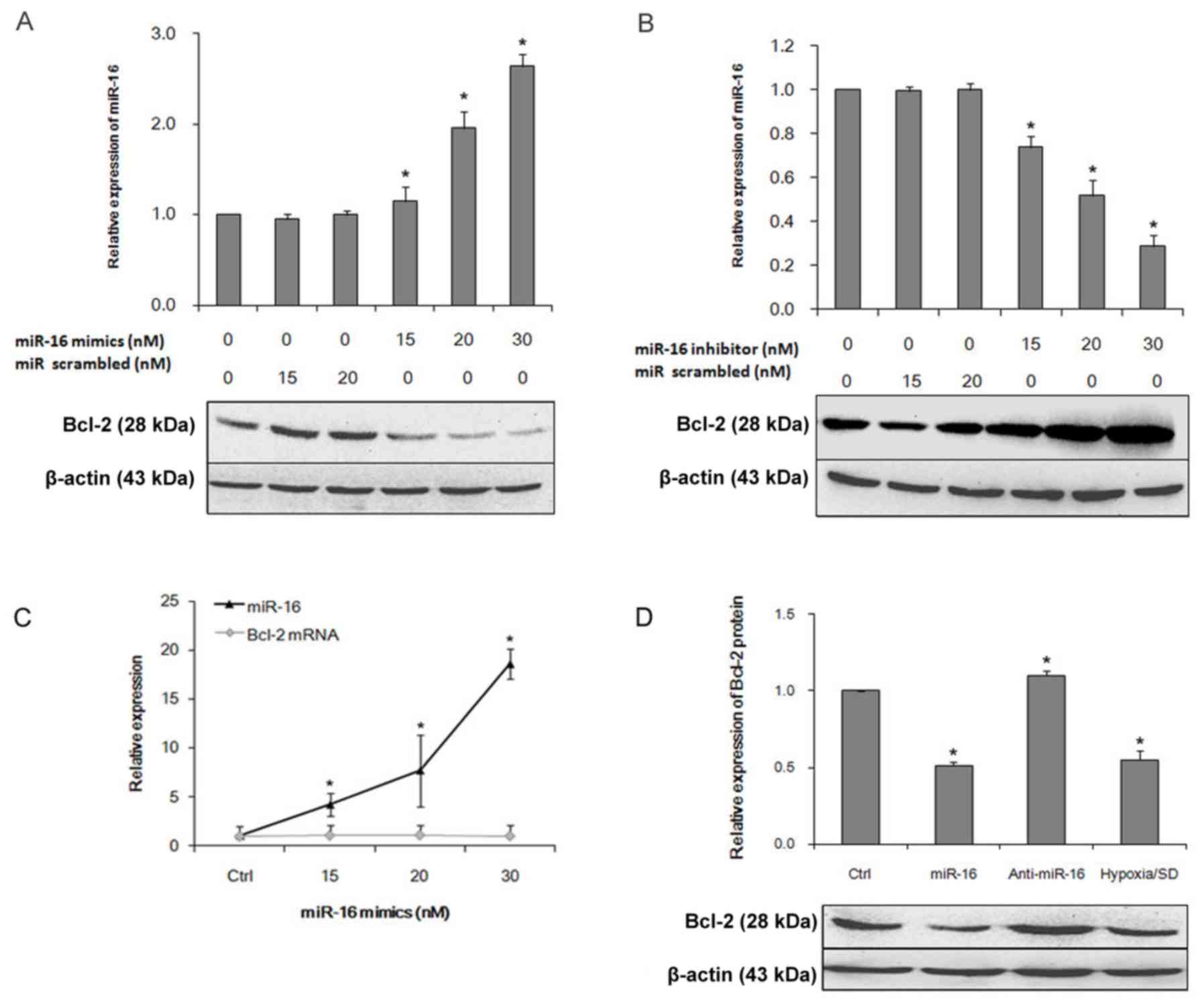
investigate the expression of Bcl-2 following miR-16 transfection.
The miR-16 mimic decreased Bcl-2 protein expression in a
dose-dependent manner (Fig. 2A).
By contrast, a miR-16 inhibitor increased Bcl-2 protein expression
(Fig. 2B).
miR-16 inhibits Bcl-2 expression in
hypoxia/SD-induced BM-MSC apoptosis
To confirm whether elevated miR-16 expression
decreased Bcl-2 expression in hypoxia/SD-induced BM-MSCs apoptosis,
miR-16 mimics were transfected. As a control, BM-MSCs were
transfected with a miR-16 inhibitor or were placed in hypoxic/SD
conditions with no transfection. The miR-16 mimic increased miR-16
expression in a dose-dependent manner; no significant difference in
Bcl-2 mRNA expression was detected (Fig. 2C). Western blot analysis confirmed
that Bcl-2 protein expression was significantly decreased in miR-16
mimic-transfected cells, compared with control cells (Fig. 2D). Notably, the decreased
expression of miR-16 in BM-MSCs resulted in increased expression of
Bcl-2 protein. Taken together, these results demonstrate that
increased miR-16 expression results in decreased Bcl-2 protein
expression.
Overexpression of miR-16 reduces cell
viability and inhibits cell proliferation
To examine the potential role of miR-16 in
regulating cell viability and proliferation, MTT and EdU assays
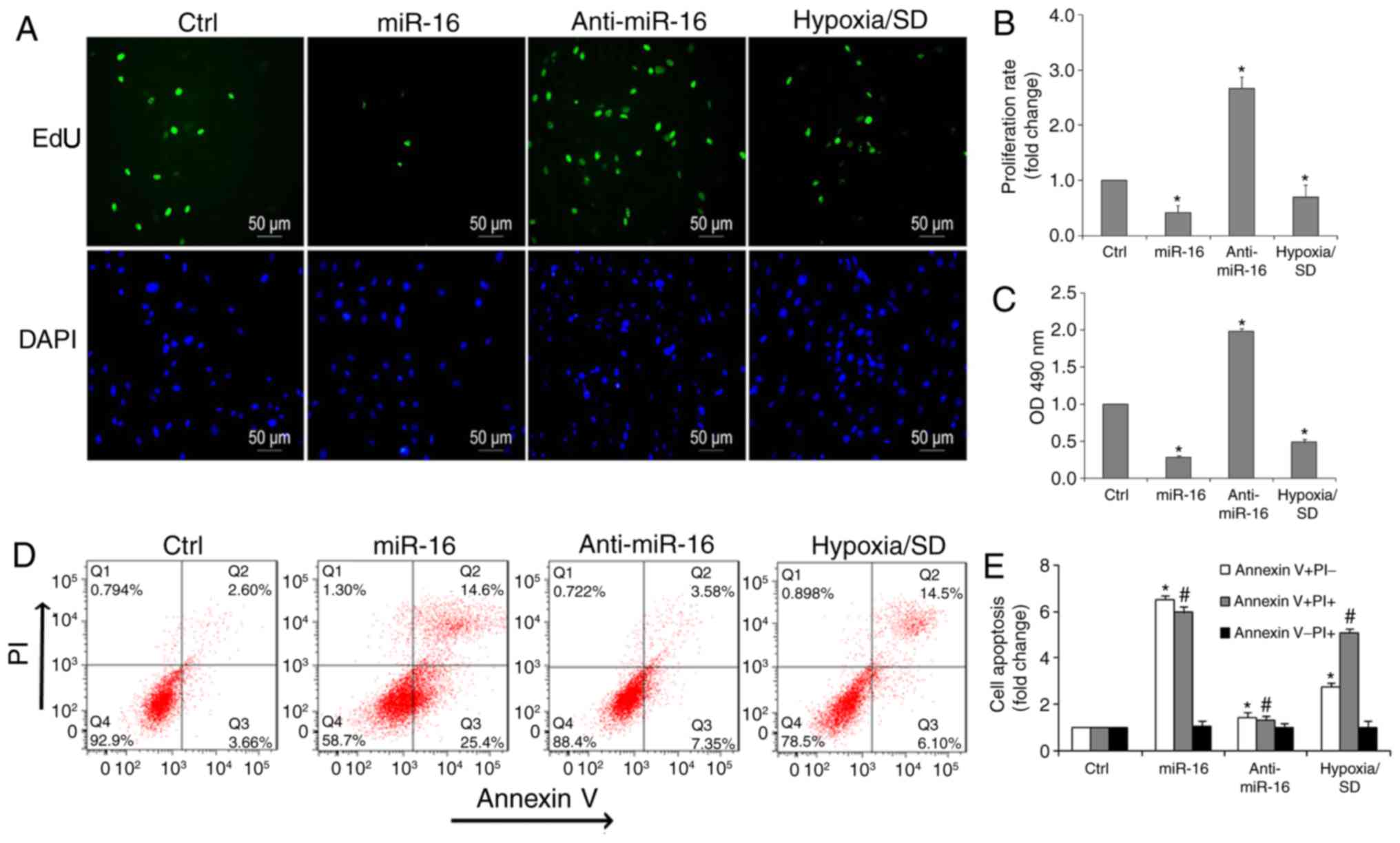
were performed. Notably, it was determined that the number of
EdU-positive cells (green) in the miR-16 mimic group were reduced
compared with the inhibitor group (Fig. 3A and B). Additionally, cell
viability significantly increased in the presence of the miR-16
inhibitor, compared with the miR-16 mimic or hypoxia/SD (Fig. 3C).
Overexpression of miR-16 promotes the
apoptosis of BM-MSCs
To examine the role of miR-16 in apoptosis
promotion, miR-16 expression was increased or decreased in BM-MSCs
by transfecting cells with a miR-16 mimic, or a miR-16 inhibitor
for 72 h, respectively. After 24 h of incubation under hypoxic/SD
conditions, miR-16 led to a marked pro-apoptotic effect. Flow
cytometry also demonstrated that there was a significant decrease
in the percentage of apoptotic cells in the anti-miR-16 group
(Fig. 3D and E). Exposure of
BM-MSCs to hypoxia/SD resulted in a 7.88±0.46-fold increase in
apoptotic rate when compared with controls, whereas 40.1±0.32% of
cells transfected with miR-16 were apoptotic. In contrast, only
10.9±0.23% of cells transfected with anti-miR-16 were apoptotic
(Fig. 3E).
miR-16 exerts it pro-apoptotic effects
via activation of the mitochondrial pathway of apoptosis
Cytochrome c is a key mediator of apoptosis that is
released from mitochondria, and its release is inhibited by the
presence of Bcl-2 incorporated into the outer membrane of
organelles (28,29). Therefore, to investigate if the
intrinsic apoptotic pathway was activated by miR-16, cell lysates
fractions were subjected to western blot analysis. The results
revealed that cytoplasmic cytochrome c was significantly increased
in the miR-16 mimic transfected group. By contrast, its expression
was markedly decreased in the miR-16 inhibitor transfected group,
compared with the controls (Fig.
4A). Caspase-9 and APAF-1 demonstrated a similar expression
pattern, with significantly increased levels observed in the miR-16
mimic transfected group (Fig. 4B)
Furthermore, cleaved caspase-3 and cleaved PARP were also
significantly upregulated in the miR-16 mimic group and reduced in
the miR-16 inhibitor group, compared with the control (Fig. 4C). The effect of miR-16 was further
ascertained by examining the MMP (Fig.
4D). Compared with the control group, cells transfected with
the miR-16 mimic displayed noticeable alterations in MMP. These
results suggest that miR-16 may be involved in the apoptotic
process of BM-MSCs induced by hypoxia/SD through activation of the
mitochondrial pathway of apoptosis, via APAF-1/caspase-9/PARP.
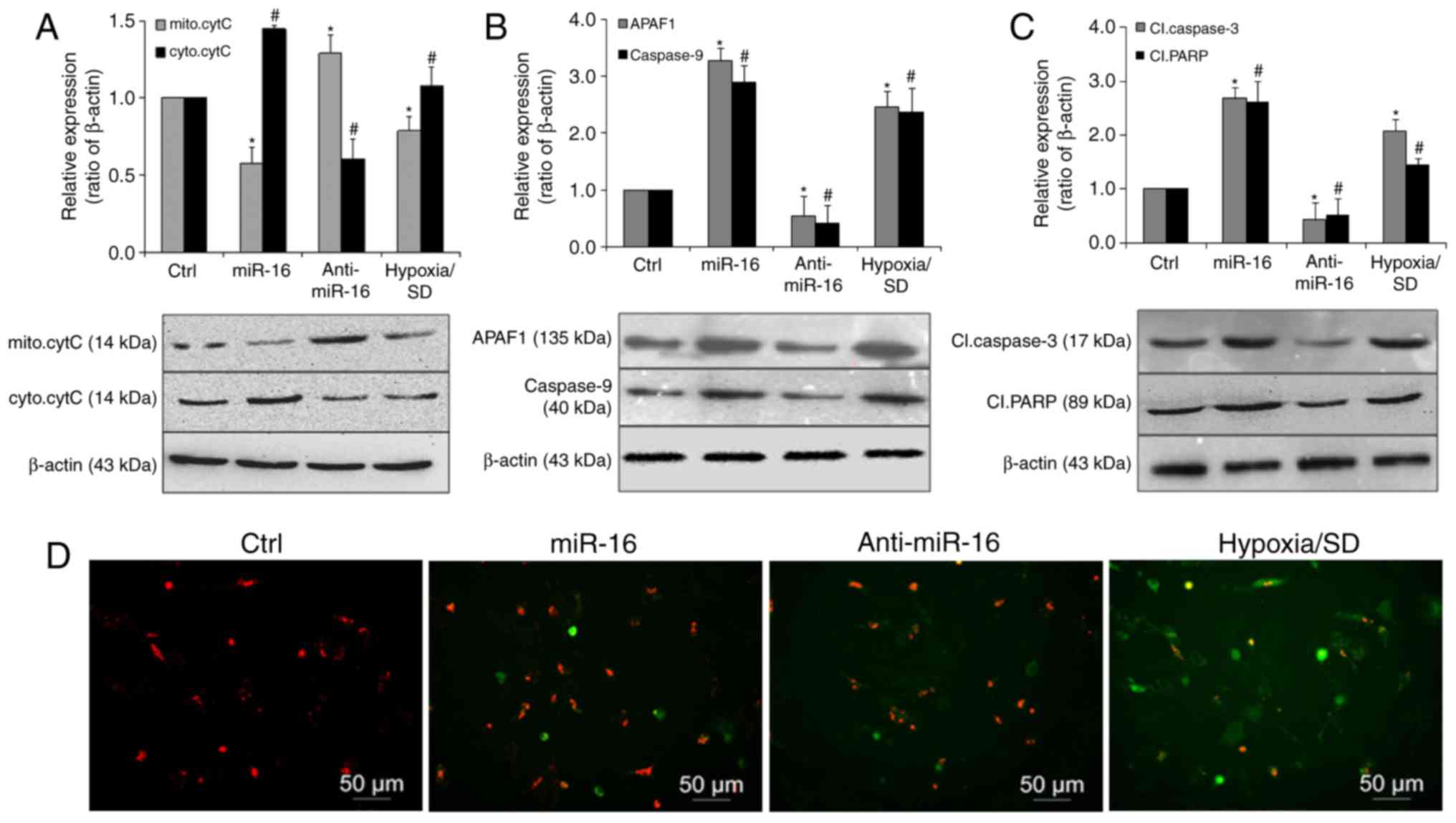 | Figure 4.miR-16 promoted apoptosis via
activation of a mitochondrial pathway involving
APAF-1/caspase-9/PARP. (A) Representative immunoblots and
quantitative analysis of mitochondrial or cytosolic cytochrome c
levels in BM-MSCs transfected with miR-16 mimics or inhibitor. The
expression of (B) APAF-1 and caspase-9, and (C) cleaved caspase-3
and cleaved PARP was significantly increased in cells transfected
with miR-16 mimics. (D) BM-MSCs (red) transfected with miR-16
mimics or inhibitor were then exposed to hypoxia/SD for 24 h, and
mitochondrial membrane (green) potential was assessed using a JC-1
MMP Assay. Magnification, ×100. Data are presented as the mean ±
standard deviation of three independent experiments. *P<0.05 and
#P<0.05 vs. the respective Ctrl group. miR-16,
microRNA-16, APAF-1, apoptotic protease activating factor-1; PARP,
poly (ADP ribose) polymerase; BM-MSC, bone marrow-derived
mesenchymal stem cell; SD, serum deprivation; cytC, cytochrome c;
mito, mitochondrial; cyto, cytoplasmic; Cl, cleaved; MMP,
mitochondrial membrane potential. |
Bcl-2 knockdown promotes apoptosis and
suppresses proliferation
To explore miR-16-mediated regulation of Bcl-2 in
BM-MSCs apoptosis and growth, Bcl-2 siRNA was used to knockdown
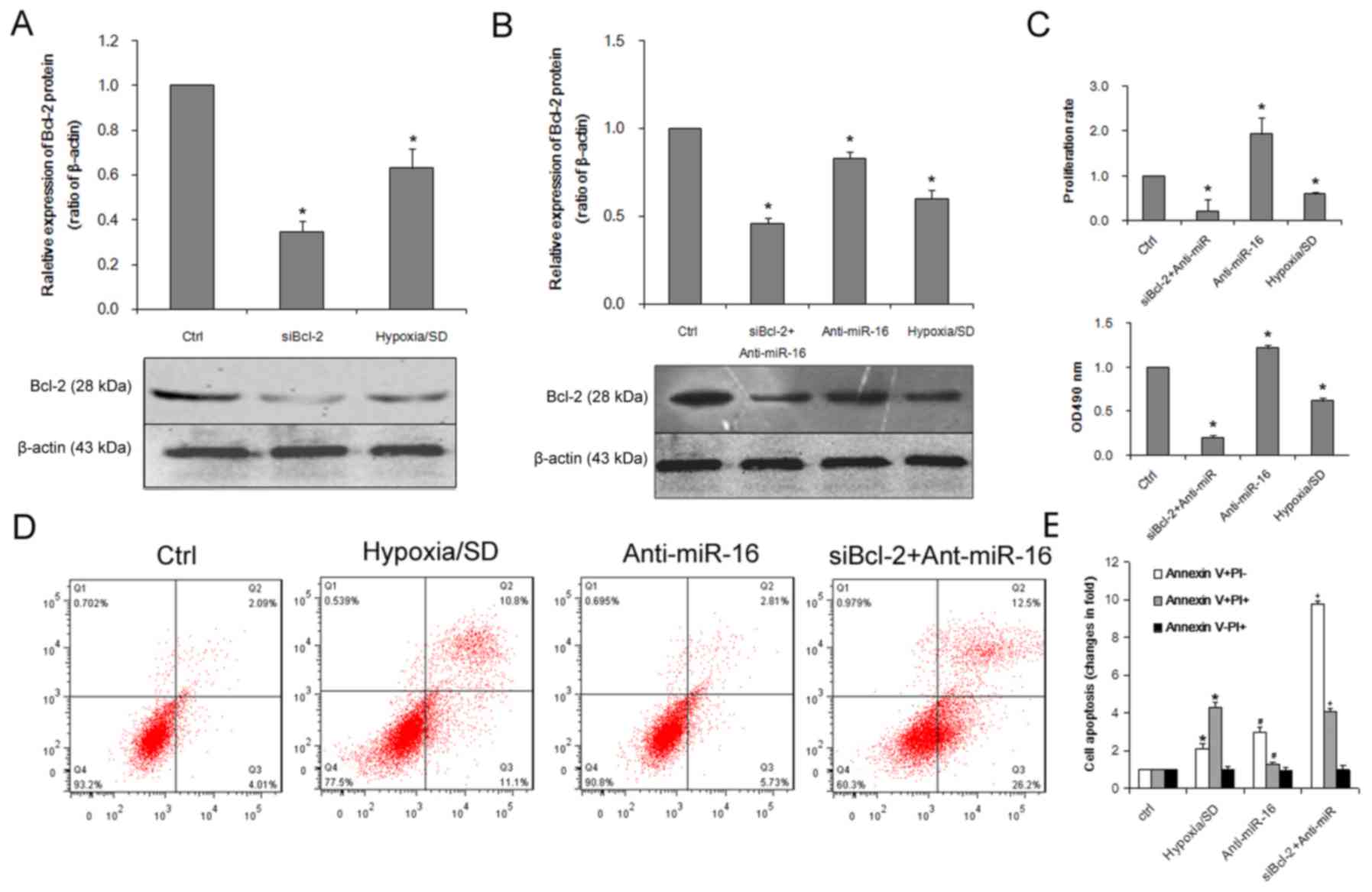
Bcl-2 expression. As presented in Fig.
5A, treatment with Bcl-2 siRNA significantly reduced the
expression of Bcl-2 protein. To determine if dysregulation of Bcl-2
expression was involved in the regulation of miR-16-induced cell
apoptosis, BM-MSCs were co-transfected with a miR-16 inhibitor and
Bcl-2 siRNA. The effect of Bcl-2 siRNA on Bcl-2 expression was
confirmed by western blot analysis (Fig. 5B). MTT and EdU assays revealed that
Bcl-2 siRNA inhibited proliferation (Fig. 5C) and cell viability (Fig. 5D) to an extent that resembled the
inhibitory effects of miR-16 overexpression on BM-MSCs (Fig. 3). It was demonstrated that the
apoptosis-promoting effects of miR-16 were partially elevated by
Bcl-2 siRNA and Bcl-2 siRNA mimicked the effects of miR-16
(Fig. 5E) suggesting that Bcl-2
may be involved in miR-16-induced apoptosis under conditions of
hypoxia/SD.
Discussion
The results of the present study revealed that
miR-16 expression was significantly upregulated under hypoxic/SD
conditions in BM-MSCs. Downregulation of miR-16 resulted in
decreased apoptosis and enhanced cell proliferation. Bcl-2 was
identified as a direct and functional target of miR-16. Further
experiments indicated that miR-16 induced an intrinsic apoptosis
pathway via APAF-1/caspase-9/PARP. The present study may lead to
the discovery of a more optimized and effective target in MSC-based
therapy for myocardial infarction. BM-MSC transplantation is a
potential therapeutic approach to improve cardiac function in
patients following myocardial infarction (4–6).
However, the repair and regeneration of cardiomyocytes and the
restoration of heart function are limited by the poor survival of
engrafted BM-MSCs in the infarcted area. In the present study, it
was revealed that under conditions of hypoxia/SD, designed to mimic
the in vivo conditions occurring in ischemic myocardium,
apoptosis of BM-MSC was triggered, as detected by
phosphatidylserine translocation to the cell surface and loss of
membrane integrity. This was demonstrated by Annexin V/PI stain,
positive Annexin V staining demonstrated phosphatidylserine
translocation to the cell surface and positive PI staining
indicated loss of membrane integrity. This finding was confirmed by
flow cytometry analysis. The results of the present study were in
agreement with the results obtained by Zhu et al (26), who reported that transplanted
BM-MSCs may be lost due to apoptosis induced by hypoxia/SD. In
addition to producing ATP, mitochondria are also key regulators of
apoptosis in a variety of cell types (29). The results of the current study
indicated that the release of cytochrome c from mitochondria was
involved in subsequent caspase activation during apoptosis.
Recent evidence has revealed significant roles of
miRNAs, including miR-16, in numerous forms of cardiovascular
disease (31). In miRNA-induced
apoptosis, expected targets include genes that encode proteins with
apoptotic activity or targets that act as negative regulators of
cell proliferation and survival (16,32).
miR-16 has been demonstrated to be involved in apoptosis in other
cell types (16–18) and to inhibit various key regulators
of cell cycle progression (33,34).
For example, miR-16 contributes to BM-MSC differentiation into
myocardial cells in a cardiac niche by enhancing G1 phase arrest
(35). Accumulating data have
presented miR-16 as an appealing target for further investigation
(19,20). In the present study, the effect of
miR-16 on BM-MSC apoptosis and proliferation under conditions of
hypoxia/SD was focused on. Previous studies have demonstrated that
miR-16 negatively regulates Bcl-2 expression via imperfect
complementarity with Bcl-2 mRNA, and, therefore promotes apoptosis
(19,36). Bcl-2 blocks the mitochondrial
release of cytochrome c and inhibits the activation of caspase-9 by
the cytoplasmic scaffolding protein APAF-1 (18). In the current study, Bcl-2
knockdown with siRNA mimicked the effects of miR-16 overexpression
in suppressing cell proliferation and inducing cell apoptosis.
These data indicate the specific role of Bcl-2 in miR-16 regulation
of apoptosis. miR-16 has been reported to target a number of genes
(18–20). In future studies, it is planned to
identify if other apoptotic regulatory genes are involved in
miR-16-induced apoptosis.
To elucidate the association between the
upregulation of miR-16 expression under hypoxic/SD conditions and
downregulation of its target Bcl-2 protein, RT-qPCR and western
blot analysis was performed in the present study. Overexpression of
miR-16 led to decreased Bcl-2 protein expression and increased
apoptosis. In BM-MSCs transfected with miR-16 mimics, increased
apoptosis, release of cytochrome c, and cleavage of pro-caspase-9
and PARP was observed, indicating that the reduction in Bcl-2
protein expression by miR-16 was sufficient to initiate the
apoptotic process. These results suggest that miR-16 induced
apoptosis in BM-MSCs through activation of the
APAF-1/caspase-9/PARP pathway.
In conclusion, the present study indicated that
exposure to hypoxic/SD conditions significantly increased the
expression of miR-16 in BM-MSCs. miR-16 overexpression may promote
apoptosis through downregulation of the anti-apoptotic Bcl-2
protein. Additionally, it was demonstrated that miR-16 suppressed
the expression of Bcl-2 at the post-transcriptional level and
induced apoptosis by activating the intrinsic APAF-1/caspase-9/PARP
pathway. Furthermore, inhibition of miR-16 provided protection to
BM-MSCs against hypoxia/SD-induced apoptosis. Therefore, miR-16 may
be a potential therapeutic target for supporting cellular
transplantation therapy in myocardial infarction.
Acknowledgements
The authors would like to thank Dr Wei Liu (The Key
Laboratory of Myocardial Ischemia Mechanism and Treatment, Harbin
Medical University, Ministry of Education, Harbin, Heilongjiang,
China) for her excellent technical assistance and helpful
discussions.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 3001-30400432).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JR contributed to the experimental design, performed
the molecular biology experiments and the statistical analysis, and
drafted the manuscript. SF participated in the design of the study
and performed the statistical analysis. YW revised the manuscript
and performed some of the experiments. BL was responsible for MSC
transfection and statistical analysis. BY participated in the
design of the study. SL conceived the study and participated in its
design and coordination, and helped to draft the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental animal procedures were approved by
the Ethical Committee on Animal Care and Use of Harbin Medical
University (Harbin, China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pittenger MF, Mackay AM, Beck SC, Jaiswal
RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S and
Marshak DR: Multilineage potential of adult human mesenchymal stem
cells. Science. 284:143–147. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Small EM and Olson EN: Pervasive roles of
microRNAs in cardiovascular biology. Nature. 469:336–342. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Afzal MR, Samanta A, Shah ZI, Jeevanantham
V, Abdel-Latif A, Zuba-Surma EK and Dawn B: Adult bone marrow cell
therapy for ischemic heart disease: Evidence and insights from
randomized controlled trials. Circ Res. 117:558–575. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Strauer BE and Steinhoff G: 10 years of
intracoronary and intramyocardial bone marrow stem cell therapy of
the heart: from the methodological origin to clinical practice. J
Am Coll Cardiol. 58:1095–1104. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li W, Ma N, Ong LL, Nesselmann C, Klopsch
C, Ladilov Y, Furlani D, Piechaczek C, Moebius JM, Lützow K, et al:
Bcl-2 Engineered MSCs Inhibited Apoptosis and Improved Heart
Function. Stem Cells. 25:2118–2127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stamm C, Westphal B, Kleine HD, Petzsch M,
Kittner C, Klinge H, Schümichen C, Nienaber CA, Freund M and
Steinhoff G: Autologousbone-marrow stem-cell transplantation for
myocardial regeneration. Lancet. 361:45–46. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Menasché P, Hagège AA, Vilquin JT, Desnos
M, Abergel E, Pouzet B, Bel A, Sarateanu S, Scorsin M, Schwartz K,
et al: Autologous skeletal myoblast transplantation for severe
postinfarction left ventricular dysfunction. J Am Coll Cardiol.
41:1078–1083. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Leistner DM, Fischer-Rasokat U, Honold J,
Seeger FH, Schächinger V, Lehmann R, Martin H, Burck I, Urbich C,
Dimmeler S, et al: Transplantation of progenitor cells and
regeneration enhancement in acute myocardial infarction
(TOPCARE-AMI): Final 5-year results suggest long-term safety and
efficacy. Clin Res Cardiol. 100:925–934. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Toma C, Pittenger MF, Cahill KS, Byrne BJ
and Kessler PD: Human mesenchymal stem cells differentiate to a
cardiomyocyte phenotype in the adult murine heart. Circulation.
105:93–98. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hare JM, Traverse JH, Henry TD, Dib N,
Strumpf RK, Schulman SP, Gerstenblith G, DeMaria AN, Denktas AE,
Gammon RS, et al: A randomized, double-blind, placebo-controlled,
dose-escalation study of intravenous adult human mesenchymal stem
cells (prochymal) after acute myocardial infarction. J Am Coll
Cardiol. 54:2277–2286. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bartel DP: MicroRNAs: Genomics,
Biogenesis, Mechanism, and Function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Carè A, Catalucci D, Felicetti F, Bonci D,
Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, et al:
MicroRNA-133 controls cardiac hypertrophy. Nat Med. 13:613–618.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Valencia-Sanchez MA, Liu J, Hannon GJ and
Parker R: Control of translation and mRNA degradation by miRNAs and
siRNAs. Genes Dev. 20:515–524. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Davis JA, Saunders SJ, Mann M and Backofen
R: Combinatorial microRNA target predictions. Nat Genet.
37:495–500. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Calin RI GA and Croce CM: miR-15a and
miR-16-1 in cancer: discovery, function and future perspectives.
Cell Death Differ. 17:215–220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
van Rooij E, Marshall WS and Olson EN:
Toward MicroRNA-based therapeutics for heart disease: The sense in
antisense. Circ Res. 103:919–928. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bandi N, Zbinden S, Gugger M, Arnold M,
Kocher V, Hasan L, Kappeler A, Brunner T and Vassella E: miR-15a
and miR-16 are implicated in cell cycle regulation in a
Rb-dependent manner and are frequently deleted or down-regulated in
non-small cell lung cancer. Cancer Res. 69:5553–5559. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cimmino A, Calin GA, Fabbri M, Iorio MV,
Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et
al: miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl
Acad Sci USA. 102:13944–13949. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo CJ, Pan Q, Li DG, Sun H and Liu BW:
miR-15b and miR-16 are implicated in activation of the rat hepatic
stellate cell: An essential role for apoptosis. J Hepatol.
50:766–778. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xia L, Zhang D, Du R, Pan Y, Zhao L, Sun
S, Hong L, Liu J and Fan D: miR-15b and miR-16 modulate multidrug
resistance by targeting BCL2 in human gastric cancer cells. Int J
Cancer. 123:372–379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bonci D, Coppola V, Musumeci M, Addario A,
Giuffrida R, Memeo L, D'Urso L, Pagliuca A, Biffoni M, Labbaye C,
et al: The miR-15a-miR-16-1 cluster controls prostate cancer by
targeting multiple oncogenic activities. Nat Med. 14:1271–1277.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of Apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kluck RM, Bossy-Wetzel E, Green DR and
Newmeyer DD: The release ofcytochrome c from mitochondria: A
primary site for Bcl-2 regulation of apoptosis. Science.
275:1132–1136. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhu W, Chen J, Cong X, Hu S and Chen X:
Hypoxia and serum deprivation-induced apoptosis in mesenchymal stem
cells. Stem Cells. 24:416–425. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu J, Wang Y, Du W, Liu W, Liu F, Zhang
L, Zhang M, Hou M, Liu K, Zhang S and Yu B: Wnt1 inhibits hydrogen
peroxide-induced apoptosis in mouse cardiac stem cells. PLoS One.
8:e588832013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real time quantitative PCR and
the 2(-Delta Delta C(t)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1311. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu WH, Liu JJ, Wu J, Zhang LL, Liu F, Yin
L, Zhang MM and Yu B: Novel mechanism of inhibition of dendritic
cells maturation by mesenchymal stem cells via interleukin −10 and
the JAK1/STAT3 Signaling Pathway. PLoS One. 8:e554872013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Han D, Huang W, Li X, Gao L, Su T, Li X,
Ma S, Liu T, Li C, Chen J, et al: Melatonin facilitates
adipose-derived mesenchymal stem cells to repair the murine
infarcted heart via the SIRT1 signaling pathway. J Pineal Res.
60:178–192. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Barwari T, Joshi A and Mayr M: MicroRNAs
in cardiovascular disease. J Am Coll Cardiol. 68:2577–2584. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yan X, Liang H, Deng T, Zhu K, Zhang S,
Wang N, Jiang X, Wang X, Liu R, Zen K, et al: The identification of
novel targets of miR-16 and characterization of their biological
functions in cancer cells. Mol Cancer. 12:922013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Roccaro AM, Sacco A, Thompson B, Leleu X,
Azab AK, Azab F, Runnels J, Jia X, Ngo HT, Melhem MR, et al:
MicroRNAs 15a and 16 regulate tumor proliferation in multiple
myeloma. Blood. 113:6669–6680. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu JL, Jiang L, Lin QX, Deng CY, Mai LP,
Zhu JN, Li XH, Yu XY, Lin SG and Shan ZX: MicroRNA 16 enhances
differentiation of human bone marrow mesenchymal stem cells in a
cardiac niche toward myogenic phenotypes in vitro. Life Sci.
90:1020–1026. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Calin GA, Dumitru CD, Shimizu M, Bichi R,
Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, et al:
Frequent deletions and down-regulation of micro-RNA genes miR15 and
miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci
USA. 99:15524–15529. 2002. View Article : Google Scholar : PubMed/NCBI
|