Introduction
Osteoporosis is an important public health issue,
especially for postmenopausal women and the elderly, and is caused
by unequilibrated bone remodeling resulting from decreased bone
formation and/or accelerated bone resorption (1). In osteoporotic patients, bone marrow
fat increases and bone mass decreases (2). Previous studies have confirmed that
increased bone marrow fat may have toxic effects on bone cells
through the release of saturated fatty acids (FA), which results in
reduced bone mass (3–5). Moreover, high saturated FA
consumption was shown to be associated with an increased risk of
osteoporotic fractures (6). As the
most common saturated FA in humans, palmitate (PA) has been widely
investigated to evaluate the effects of FAs on various cell types
(7,8). In bone cells, lipotoxicity has been
reported in osteoblasts after exposure to PA (5,9).
Autophagy is an evolutionarily conserved process for
the catabolism of damaged proteins and organelles in the cell to
maintain intracellular environmental homeostasis by ‘self-clearing’
(10,11). Studies suggest that moderate
autophagy is required for coping with cellular stress, inhibition
of apoptosis, and is conducive to cell survival; however, the
persistence of adverse factors can lead to excessive autophagy,
which induces cell death (12).
Autophagy can be found in all types of bone cells, and plays an
important role in osteogenesis (13,14).
Bone marrow mesenchymal stem cells (BMSCs) are multipotent cells
that can differentiate into osteoblasts and other cell types such
as adipocytes and chondrocytes (15). Previous studies have shown that PA
can cause apoptosis in human MSCs (15,16);
however, it is still unknown whether PA can cause autophagy, and
the effects of autophagy on PA-induced apoptosis remain
unclear.
Previous studies have confirmed that PA released by
adipocytes can induce apoptosis in osteoblasts by increasing
reactive oxygen species (ROS) production (17). In addition, ROS has been shown to
be a strong signal for the activation of c-Jun N-terminal kinases
(JNKs); JNK activation may mediate antioxidant responses including
the induction of autophagy and cell death (18). Furthermore, the p38
mitogen-activated protein kinase (MAPK) has been shown to regulate
autophagy in response to chemotherapy drugs (19). However, whether JNK and p38 MAPK
play a role in PA-induced autophagy in BMSCs remains unclear.
Therefore, we studied the correlation of ROS, JNK, and p38 MAPK
with autophagy induced by PA, as well as the role of autophagy in
apoptosis in BMSCs.
Materials and methods
Reagents
Antibodies against cleaved caspase-3 (9679S, 1:800),
p62 (39749S, 1:800), JNK (9252S, 1:1,000), phospho-JNK (4668S,
1:800), p38MAPK (8690S, 1:1,000), phospho-p38 MAPK (4511S, 1:800),
β-Actin (8457S, 1:3,000) and GAPDH (5174S, 1:3,000) were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA). Antibodies
for LC3B-II were purchased from Abcam (ab51520, 1:3,000; Cambridge,
MA, USA). Fetal bovine serum (FBS) was obtained from Gemini Bio
Products (900108; Woodland, CA, USA). FA-free bovine serum albumin
(BSA) was obtained from Solarbio (A8850; Beijing, China). PA
(P0500), SP600125 (S5567), and SB203580 (S8307) were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Rapamycin (RA,
S1039) and 3-methyladenine (3-MA, S2767) were obtained from Selleck
Chemicals (Houston, TX, USA).
Cell culture
BMSCs derived from Sprague-Dawley rats were
purchased from Cyagen Biosciences (RASMX-90011; Shanghai, China).
According to tests conducted by the supplier, the majority of BMSCs
were positive for CD29, CD44 and CD90; negative for CD11, CD34, and
CD45; and had the potential to differentiate into osteoblasts,
adipocytes, and chondrocytes. Cells were plated in growth media at
37°C with 5% CO2. Growth media was composed of DMEM/F12
(SH30026.01B; HyClone; GE Healthcare Life Sciences, Logan, UT, USA)
and 10% FBS. After reaching approximately 90% confluence, cells
were trypsinized, detached, and passaged. BMSCs were between
passages 3 and 8 for all experiments.
PA preparation
The PA stock solution was prepared, and treatment
was performed as previously described (5,9).
Briefly, the stock solution was prepared by dissolving PA (final
concentration: 10 mM) in 0.01 M NaOH at 70°C for 30 min with
FA-free BSA. The molar ratio of PA to BSA was 7:1. The stock
solution was added to a serum-free medium to achieve a final
concentration of 0.5 mM PA and 1% BSA. Control cells were treated
with 1% BSA only.
Cell viability assay
BMSCs were seeded in 96-well plates
(6×103 cells/well) for 24 h. When 80% confluence was
achieved, the culture medium was replaced with medium with or
without increasing concentrations of PA (0.125–0.50 mM).
Separately, BMSCs were also treated with 0.5 mM PA for different
durations (0, 3, 6, 12, 18 and 24 h). Following PA treatment, 20 µl
3-[4,5-dimethylthiazol-2-y]-2,5-diphenyltetrazolium bromide (MTT,
KGA311; KeyGEN, Nanjing, China) was added to each well and
incubated for 4 h at 37°C. Thereafter, the medium containing MTT
was discarded and 150 µl dimethyl sulfoxide (DMSO, ST038; Beyotime,
Nantong, China) was added to each well and incubated for 10 min in
the dark at room temperature. The absorbance at 490 nm (A490) was
measured using a microplate reader. The cell survival rate (%) was
calculated as follows: Experimental group A490/control group
A490.
Apoptosis analysis
BMSCs were seeded in 6-well plates (2×105
cells/well) and treated with PA (0.125–0.50 mM) for 24 h.
Separately, the BMSCs were also treated with 0.5 mM PA for 24 h
with or without 3-methyladenine (3-MA; 5 mM) or rapamycin (RA; 5
µM) pretreatment. Cells were then evaluated using the Annexin
V-FITC/PI apoptosis detection kit according to the manufacturer's
instructions (AD10; Dojindo Molecular Technologies, Inc., Kumamoto,
Japan). Briefly, BMSCs were harvested and washed three times with
PBS before being resuspended in 400 µl binding buffer, then 5 µl
Annexin V-FITC and PI were added to the samples and incubated for
15 min in the dark at room temperature. The samples were analyzed
by a flow cytometer equipped with Modfit LT 3.0 (BD Biosciences,
Franklin Lakes, NJ, USA), and results were analyzed using Cell
Quest software. Different subpopulation was distinguished using the
following criteria: Q1, necrotic cells (FITC-/PI+); Q2, late
apoptotic cells (FITC+/PI+); Q3, viable cells (TITC-/PI-); Q4,
early apoptotic cells (FITC+/PI-). The apoptotic rate was
determined as the percentage of Q2 + Q4.
Measurement of caspase-3/9
activity
The detection of caspase-3 and caspase-9 activity
was performed using caspase colorimetric assay kits according to
the manufacturer's instructions (KGA202 and KGA402; Keygen Biotech,
Nanjing, China). Briefly, BMSCs were harvested and washed twice
with PBS after treatment for 24 h, and then resuspended in lysis
buffer and incubated on ice for 1 h. Subsequently, lysates were
centrifuged at 10,000 × g for 1 min and the concentration of each
sample was detected using a bicinchoninic acid (BCA) protein assay
kit (AR1110; Boster, Wuhan, China). Accordingly, 150 µg total
protein from each sample was combined with PBS to achieve a final
volume of 50 µl, then the samples were incubated with 5 µl caspase
substrate in the dark at 37°C for 4 h. The A405 was measured using
a microplate reader. Caspase-3 and caspase-9 activity were
calculated as follows: Experimental group A405/control group
A405.
Detection of intracellular ROS
BMSC ROS levels were detected using the ROS
detection kit (S0033; Beyotime). Cells (2×105
cells/well) in 6-well plates were treated with variable
concentrations of PA for 6 h at 37°C. Thereafter, BMSCs were
incubated with DCFH-DA (5 mM) at 37°C for 20 min, and washed three
times with serum-free medium. ROS levels were measured using an
inverted fluorescence microscope (IX71; Olympus Corporation, Tokyo,
Japan). In addition, cells were harvested and washed twice with
serum-free medium after treatment as indicated for 6 h at 37°C,
then resuspended in 500 µl PBS with 5 mM DCFH-DA; the samples were
then incubated for 20 min in the dark at 37°C. Intracellular ROS
was detected by flow cytometry (BD Biosciences) and analyzed using
Cell Quest software.
Western blotting
After treatment, BMSCs were extracted with lysis
buffer for 30 min and centrifuged at 14,000 × g for 30 min at 4°C.
The supernatant containing total protein was harvested. The protein
concentration in the extract was determined using the BCA protein
assay (AR1110; Boster). Equal amounts of protein were loaded into
each lane, separated by a 10 or 12% SDS-PAGE, and electroblotted
onto PVDF membranes (LC2002; EMD Millipore, Billerica, MA, USA) at
50 or 70 V for 100 min at 4°C. The membrane was then blocked in 5%
non-fat milk for 1 h at room temperature. Subsequently, proteins
were incubated using primary antibodies at 1:500 or 1:1,000
dilution overnight at 4°C. Next, the membrane was incubated with
goat anti-rabbit secondary antibody conjugated to horseradish
peroxidase (ZB-2306, 1:8,000; ZSGB-BIO, Beijing, China) for 2 h at
room temperature. The protein signal was enhanced using a
chemiluminescence system (DNR MF-ChemiBIS 3.2) and the band density
was measured using ImageJ.
Transmission electron microscopy
After treatment with or without PA (0.5 mM) for 6 h
at 37°C, the cells were collected and fixed with 5% (v/v)
glutaraldehyde. The cells were then conventionally dehydrated,
embedded, sectioned, and stained. Transmission electron microscopy
(TEM, H-7650; Hitachi, Ltd., Tokyo, Japan) was used to observe the
formation of autophagosomes. The number of intracellular
autophagosomes was counted in every ten fields.
Immunofluorescence staining
Cells (8×103 cells/well) were seeded in
24-well plates and treated with PA (0.50 mM) for 6 h with or
without 3-MA (5 mM) or N-Acetyl-cysteine (NAC; 1 mM, S0077;
Beyotime). Subsequently, cells were fixed in 4% paraformaldehyde
(P0099; Beyotime) for 15 min at room temperature and washed twice
with PBS, then permeabilized with 0.2% triton X-100 (P0096;
Beyotime) for 5 min at room temperature. Thereafter, cells were
washed with PBS, incubated with 5% BSA for 1 h, and incubated in
anti-LC3 antibody (ab51520, 1:200; Abcam) overnight at 4°C. The
cells were then incubated with goat anti-rabbit secondary antibody
labeled with fluorescein (ZF-0511, 1:400; ZSGB-BIO) for 1 h at room
temperature before staining with DAPI (C1006; Beyotime). Samples
were photographed using a wide field fluorescent microscope (IX71;
Olympus Corporation). The number of LC3 positive cells in each
microscopic field was divided by the number of nuclei in the same
field, and was regarded as the rate of LC3 positive cells.
Statistical analysis
All experiments were repeated three times.
Quantitative data are presented as the mean ± standard error mean.
SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA) was used for
analysis. Statistical analysis was performed using Student's t-test
or one-way analysis of variance followed by a Bonferroni post hoc
test for multiple comparisons. Differences between groups were
considered statistically significant at P<0.05.
Results
PA induces apoptosis in BMSCs
Considering that PA is the most ubiquitous FA in
humans (including their bone marrow), and since PA induces
apoptosis in other systems, in this study we tested whether it also
induces these processes in BMSCs (20–22).
Here we used a dose of PA that corresponds to the concentration
secreted by adipocytes into the media (5), was already used in previous studies
looking at PA-induced lipotoxicity in bone cells (5,9), and
closely corresponds to the levels of PA found in bone marrow of
human subjects (23).
To study the toxic effects of PA on BMSCs, cells
were treated with increasing concentrations of PA (0.125–0.50 mM).
PA caused a marked reduction of cell viability in BMSCs in a time-
and dose-dependent manner (Fig.
1A). In addition, we measured the levels of cleaved caspase-3
in BMSCs. As shown in Fig. 1B, PA
triggered the cleavage of caspase-3 in a dose-dependent manner.
Moreover, flow cytometry revealed a marked increase in apoptosis in
BMSCs treated with PA (Fig. 1C and
D). Collectively, these results indicated that PA can induce
apoptosis in BMSCs.
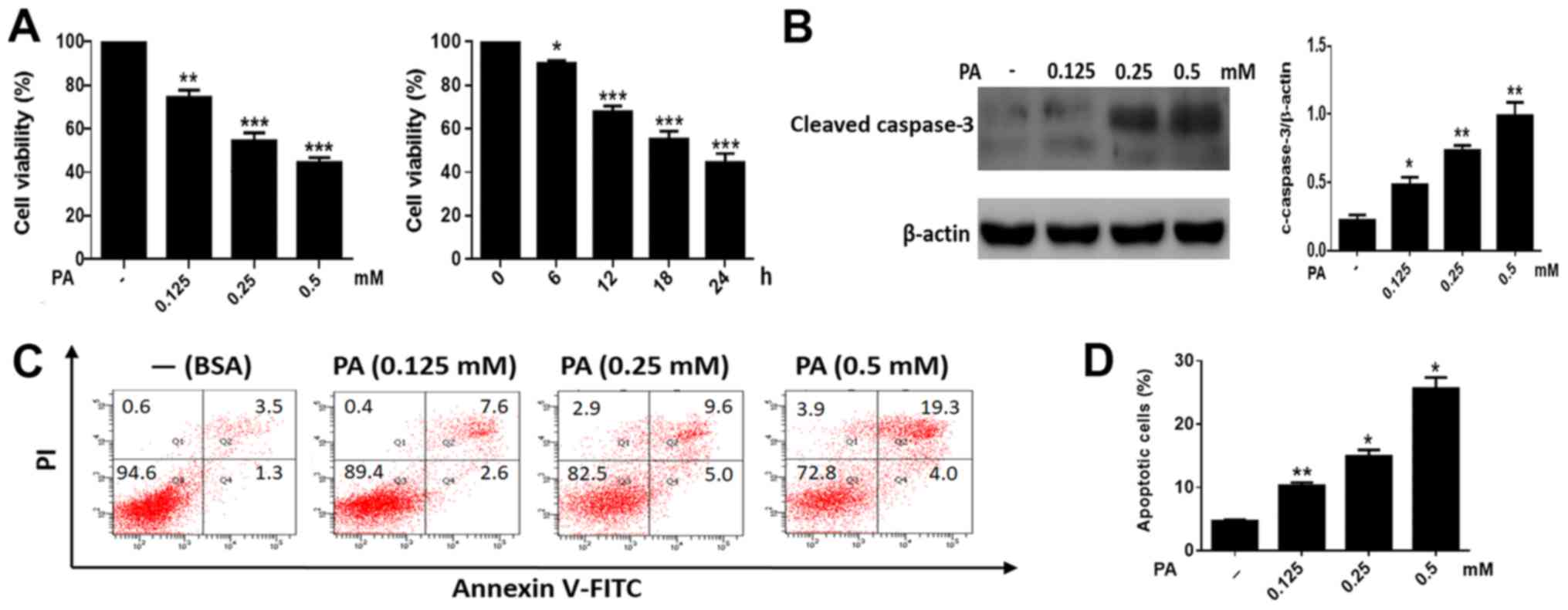 | Figure 1.PA promotes apoptosis in BMSCs. (A)
Cells were treated with PA (0.125, 0.25 and 0.5 mM) for 24 h. Cell
viability was measured using an MTT assay. The viability of BMSCs
treated with PA (0.5 mM) at different times (0, 3, 6, 12, 18 and 24
h) was detected using an MTT assay. (B) Western blotting was
performed to detect the cleaved caspase-3 levels in BMSCs treated
with PA (0.125, 0.25 and 0.5 mM) for 24 h. (C) BMSCs treated with
PA (0.125, 0.25 and 0.5 mM) for 24 h were stained with Annexin V/PI
and measured using flow cytometry. (D) The apoptosis ratio of
PA-treated BMSCs was obtained by flow cytometry. The apoptotic rate
was determined as the percentage of Q2 + Q4. BMSCs treated with BSA
were used as the control group. Data are shown as the mean ±
standard error mean from three independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs. control (0 mM/0 h). PA,
palmitate; BMSCs, bone marrow mesenchymal stem cells; PI, propidium
iodide; BSA, bovine serum albumin. |
PA induced autophagy in BMSCs, but
autophagic flux decreased over time
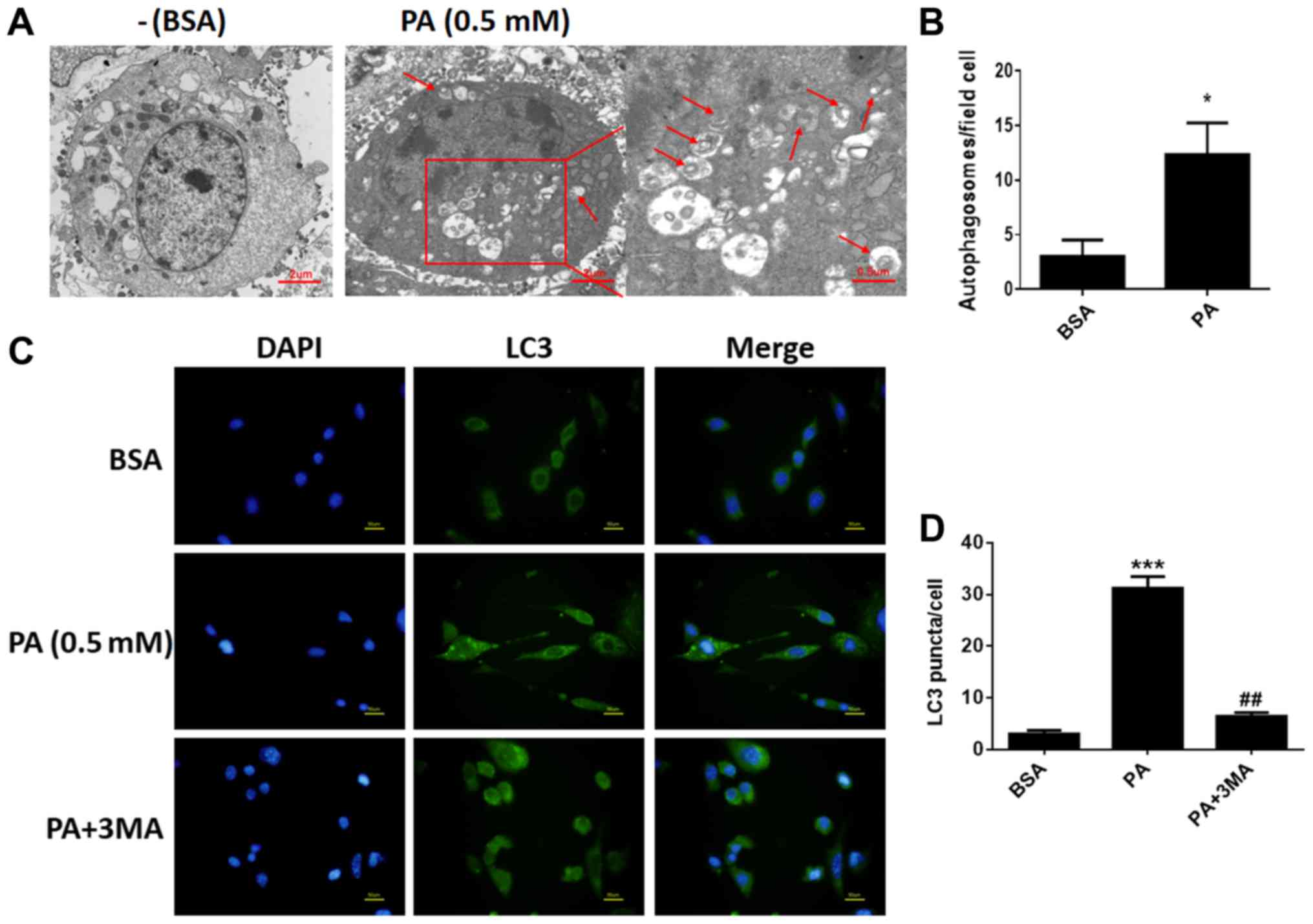
To determine whether PA stimulated autophagy in
BMSCs, we used TEM to observe and count autolysosomes. As shown in
Fig. 2A and B, after treatment
with PA (0.50 mM) for 6 h, the number of autolysosomes in BMSCs
increased remarkably compared to control groups. In addition,
fluorescence microscopy was used to observe cells with punctate
aggregation (autolysosomes) of internal LC3 (Fig. 2C). Punctate aggregation increased
substantially after treatment with PA for 6 h compared to the
control groups (Fig. 2D); this
result was consistent with the findings obtained via TEM. When
cells were treated with 3-MA, a classical inhibitor of autophagy,
punctate aggregation greatly decreased compared to the PA-treated
groups without 3-MA (Fig. 2C).
Autophagic flux decreased after longer
exposure to PA
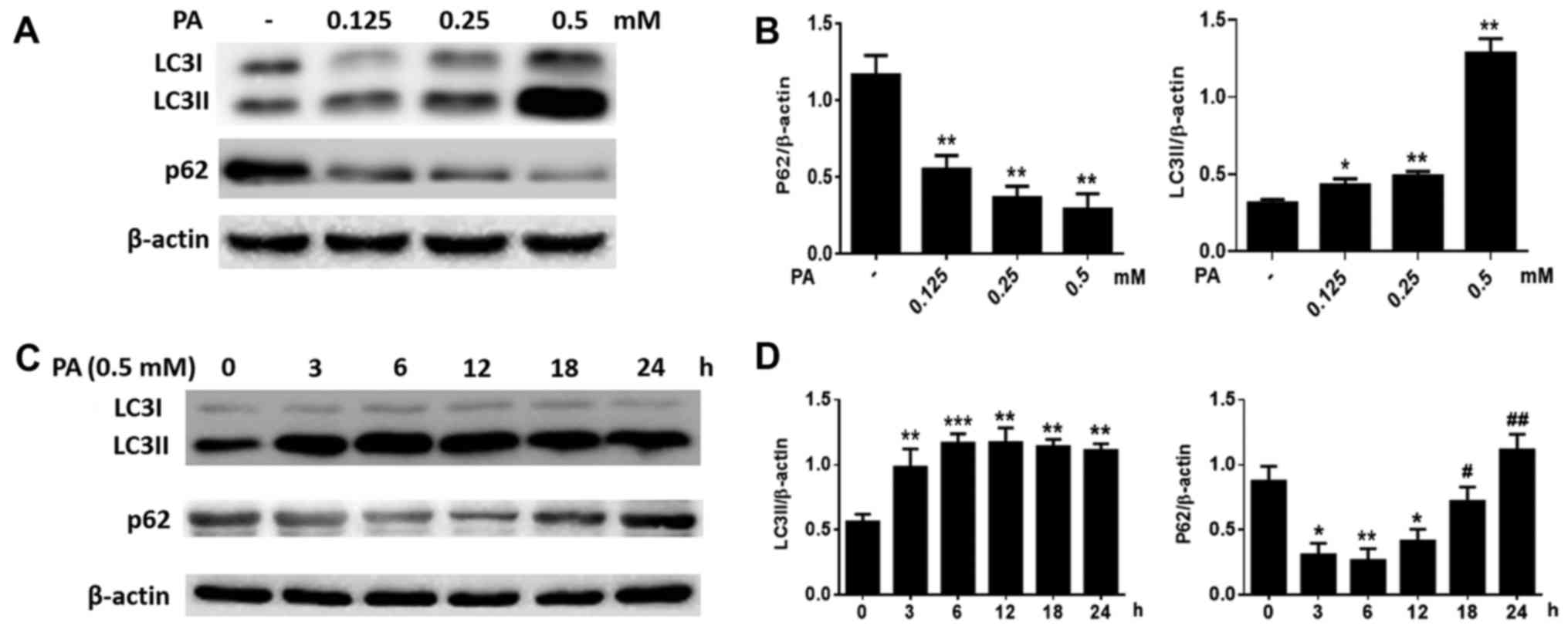
We also measured the levels of LC3 and p62. After
BMSCs were treated for 6 h with increasing concentrations of PA
(0.125–0.50 mM), the LC3-II levels increased considerably, while
p62 levels decreased (Fig. 3A and
B) in a dose-dependent manner. Furthermore, when BMSCs were
treated with PA (0.5 mM) at various time-points, LC3B-II levels
increased gradually starting from 3 h and peaked at 12 h, while p62
levels were greatly attenuated at 3 h and reached a minimum at 12 h
(Fig. 3C and D). However, p62
protein levels increased markedly at 18 and 24 h (Fig. 3C and D). These results suggest that
PA can induce autophagy in BMSCs at early stages, but autophagy
gradually reduced after longer exposure to PA.
PA-induced apoptosis is accelerated by
3-MA and inhibited by RA
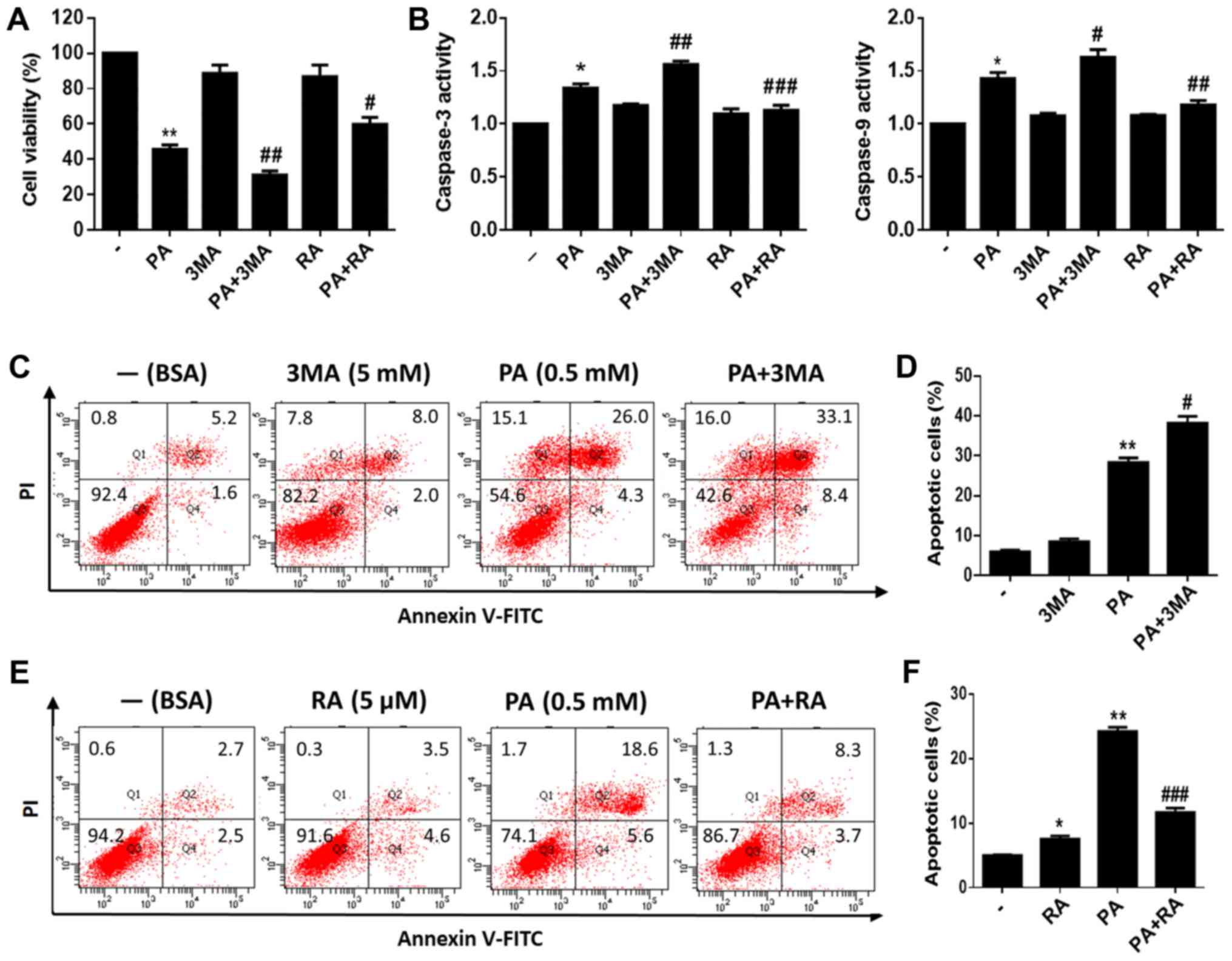
To investigate the role of autophagy in PA-induced
apoptosis of BMSCs, we used 3-MA, a classical inhibitor of
autophagy, to inhibit autophagy. Additionally, RA, a classical
autophagy inducer, was used to induce autophagy. We measured the
viability of PA-treated BMSCs with or without pretreatment with
3-MA or RA. The results showed that BMSCs pretreated with 3-MA for
2 h had substantially reduced viability. Furthermore, RA
pretreatment increased the viability of PA-treated BMSCs (Fig. 4A). In addition, caspase-3/9
activity was evaluated after PA treatment for 24 h, and was found
to be increased after pretreatment with 3-MA for 2 h and decreased
after RA pretreatment compared to cells treated with PA alone
(Fig. 4B). Moreover, flow
cytometry revealed that 3-MA pretreatment increased PA-induced
apoptosis (Fig. 4C and D), while
RA pretreatment reduced PA-induced apoptosis in BMSCs (Fig. 4E and F).
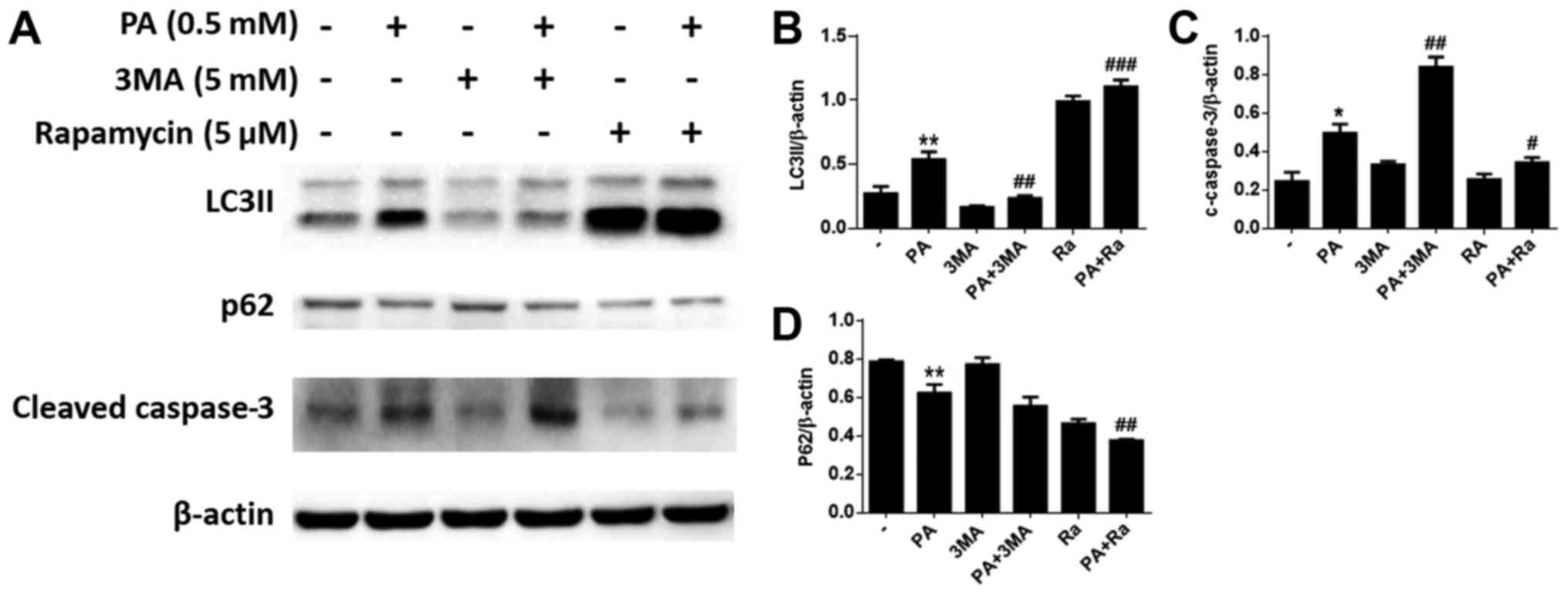
In addition, western blotting indicated that 3-MA
pretreatment decreased the LC3-II levels in BMSCs, and increased
cleaved caspase-3 expression levels compared to cells treated with
PA alone (Fig. 5A-C).
Correspondingly, we found that RA pretreatment increased the levels
of LC3-II in BMSCs, and reduced the levels of cleaved caspase-3
(Fig. 5A, C and D).
PA up-regulated ROS production and ROS
mediate PA-induced autophagy in BMSCs
After BMSCs were exposed to various concentrations
of PA (0.125, 0.25, 0.50 mM) for 6 h, intracellular ROS levels were
evaluated. Intracellular ROS levels increased as the dose of PA
increased (Fig. 6B and C),
suggesting that PA up-regulated ROS production in a dose-dependent
manner. In addition, NAC, a commonly used antioxidant, was used to
inhibit oxidative stress; cells incubated with NAC showed strong
inhibition of PA-induced ROS production (Fig. 6A). To explore whether ROS play a
role in the activation of PA-induced autophagy, we investigated the
role of NAC in autophagy. Western blot analysis showed that NAC
markedly decreased the levels of LC3B-II (Fig. 7A and B). Furthermore,
immunofluorescence staining revealed that the rate of LC3-positive
cells was lower in cells treated with NAC and PA than in those
treated with PA alone (Fig. 6D and
E). These results suggest that PA may induce autophagy by
up-regulating ROS levels in BMSCs.
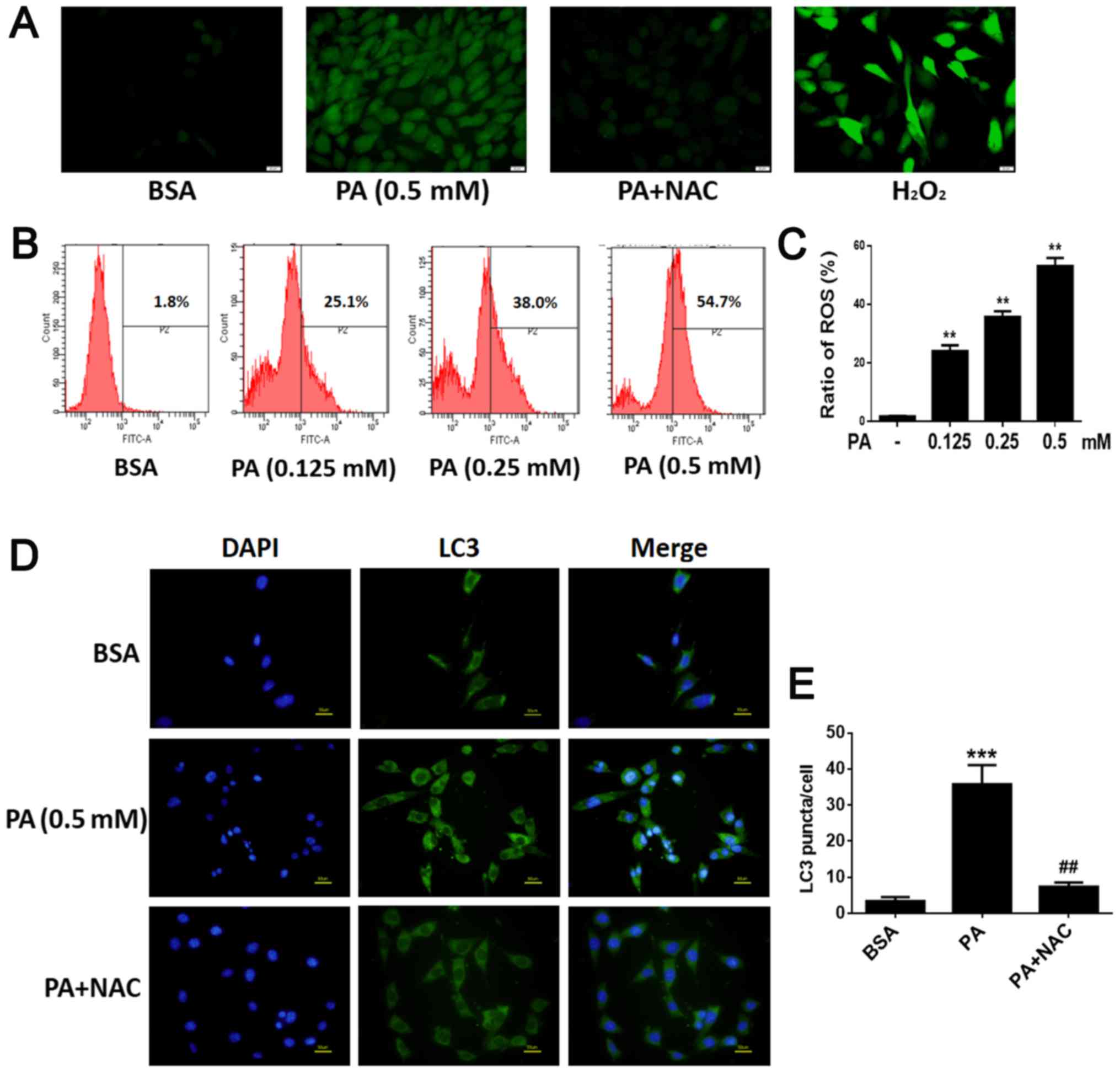 | Figure 6.PA upregulates ROS production and in
turn, ROS mediates PA-induced autophagy in BMSCs. (A) BMSCs were
treated with PA (0.5 mM) for 6 h with or without NAC (1 mM) and
incubated with 2′,7′-dichlorodihydrofluorescein diacetate.
Fluorescence microscopy was used to measure intracellular ROS
levels (magnification, ×400). Scale bars, 20 µm. (B) Following BMSC
exposure to PA (0.125, 0.25 and 0.50 mM) for 6 h, intracellular ROS
levels were determined. Intracellular ROS levels increased as the
dose of PA increased. (C) Quantification analysis of ROS. (D)
Fluorescence microscopy was used to observe internal LC3 (green)
and nuclei (blue) in BMSCs treated with PA (0.5 mM) for 6 h with or
without NAC (1 mM). Scale bars, 50 µm. (E) LC3 puncta were
quantified. Data are shown as the mean ± standard error mean from
three independent experiments. **P<0.01 and ***P<0.001 vs.
control group (0 mM/BSA); ##P<0.01 vs. PA
only-treated group. PA, palmitate; ROS, reactive oxygen species;
BMSCs, bone marrow mesenchymal stem cells; NAC, N-Acetyl-cysteine;
LC3, light chain 3; H2O2, hydrogen
peroxide. |
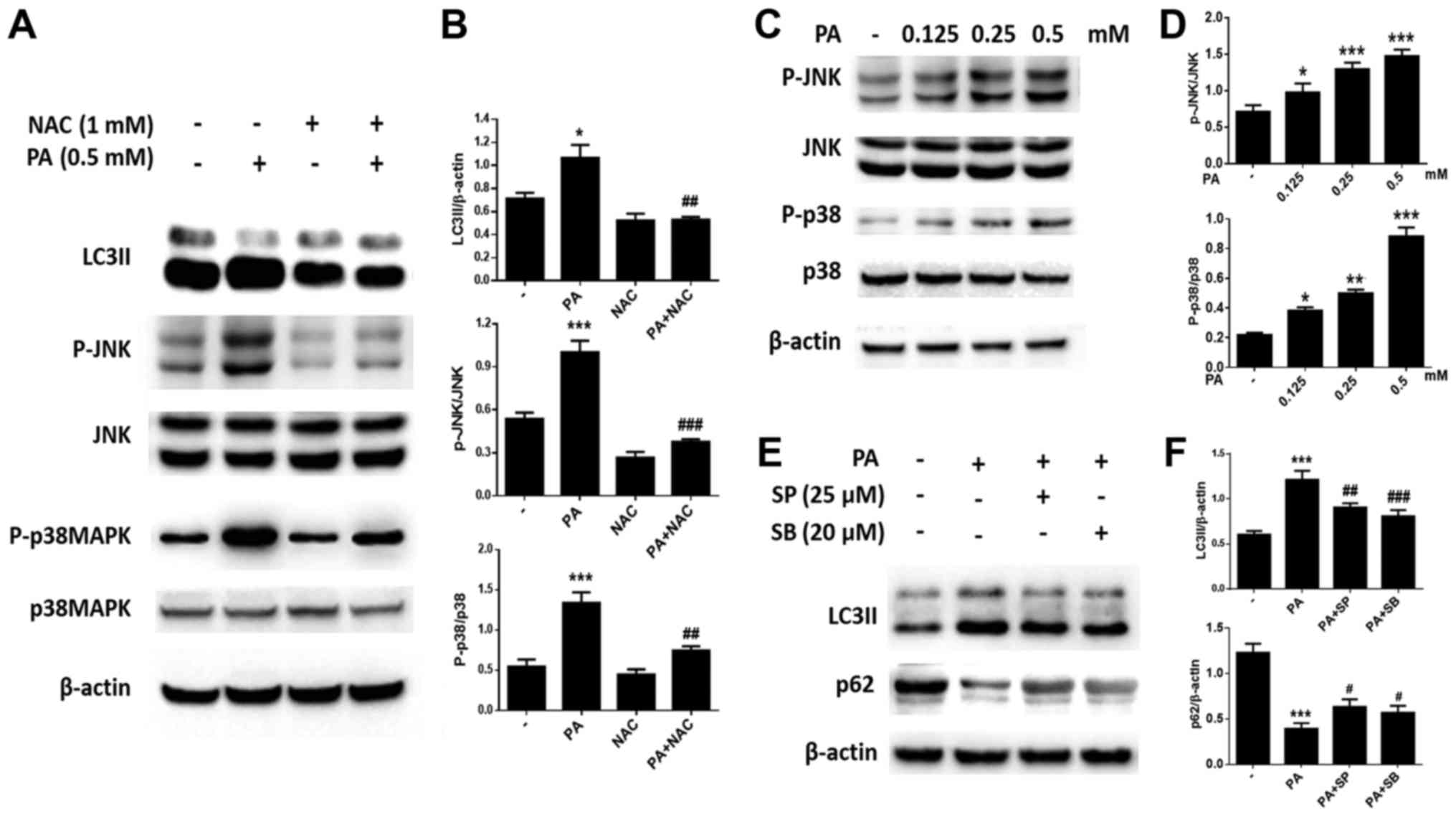 | Figure 7.PA induces autophagy through the
ROS-JNK/p38 MAPK signaling pathway in BMSCs. (A) Western blotting
was used to detect (B) LC3, p-JNK, JNK, p-p38 MAPK and p38 MAPK
levels in BMSCs treated with PA (0.5 mM) for 6 h with or without
NAC (1 mM). (C) Western blotting was also performed to determine
(D) the levels of p-JNK, JNK, p-p38 MAPK and p38 MAPK in BMSCs
treated with PA (0.125, 0.25 and 0.5 mM) for 6 h. (E and F) BMSCs
were pretreated with the inhibitor of JNK (SP600125), or the
inhibitor of p38 MAPK (SB203580) for 1 h, then treated with PA (0.5
mM) for 6 h. (E) Western blot to detect (F) LC3II and p62 levels in
different groups. Data are shown as the mean ± standard error mean
from three independent experiments. *P<0.05, **P<0.01 and
***P<0.001 vs. control group; #P<0.05,
##P<0.01 and ###P<0.001 vs. PA
only-treated group. PA, palmitate; ROS, reactive oxygen species;
JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein
kinase; BMSCs, bone marrow mesenchymal stem cells; LC3, light chain
3; p-, phospho-; NAC, N-Acetyl-cysteine. |
PA induces autophagy through the
ROS-JNK/p38 MAPK pathway in BMSCs
We further investigated the mechanism of PA-induced
autophagy in BMSCs. Western blotting revealed that the expression
of P-JNK and P-p38 MAPK increased with increasing PA concentrations
(Fig. 7C and D). Furthermore,
pretreatment with the JNK inhibitor SP600125 or the p38 MAPK
inhibitor SB203580 greatly reduced PA-induced autophagy (Fig. 7E and F). Furthermore, NAC was used
to observe whether ROS is involved in the activation of JNK and p38
MAPK. As expected, the phosphorylation of JNK and p38 MAPK were
inhibited by NAC (Fig. 7A and B).
Therefore, PA may induce autophagy in BMSCs via the ROS-JNK/p38
MAPK pathway.
Discussion
The inverse relationship between bone marrow adipose
tissue (MAT) and bone mass in the process of aging and the
development of osteoporosis has been well established (24,25).
Recent studies demonstrated that bone mass and MAT are inversely
correlated (26,27). Adipocytes are an important part of
the bone marrow, but their physiological function in the bone
marrow stem cell niche and the consequence of their excessive
accumulation in pathological entities remain unclear (28). Although they have long been
considered as inert cells, it is now widely accepted that
adipocytes constitute a metabolically active depot that may affect
skeletal metabolism (3,29–31).
Through the release of FAs, marrow adipocytes may generate a local
microenvironment that could impair the viability and function of
bone cells (31–33). Recent studies demonstrated that
adipocytes liberate factors that inhibit preosteoblast
proliferation and differentiation (34,35).
Moreover, MSC-derived adipocytes enhanced Ob apoptosis via the
release of saturated FA (5,36).
In fact, a higher consumption of saturated FA is negatively
correlated with bone mineral density and is associated with an
increased risk of osteoporotic fractures (6,37).
As multipotent cells that can differentiate into
osteoblasts and chondrocytes, BMSCs are crucial for maintaining
bone mass. In osteoporotic and aging patients, the quantity of MSCs
is reduced and is correlated with increased MSC apoptosis (38). PA is a highly prevalent saturated
FA that can induce apoptosis and autophagy (20,22).
We found that PA-treated BMSCs showed a considerable reduction in
cell viability in a time- and dose-dependent manner. This result
was validated by increased expression of the apoptotic factor
cleaved caspase-3. The Annexin V/PI staining assay further
confirmed the apoptotic effects of PA.
Both autophagy and apoptosis have been identified as
key determinants in skeletal maintenance (13,39).
PA-induced autophagy is considered to be a ubiquitous process, and
has been induced in other cell lines (40). When autophagy is induced, LC3
levels are up-regulated. In this study, western blotting revealed
that the levels of LC3-II increased greatly after incubation for
3–12 h. p62, a biomarker of the degradation of autolysosomes that
is negatively correlated with autophagy, was decreased in BMSCs
treated with PA from 3–12 h; this is in accordance with the change
in LC3 levels, implying the activation of autophagy. However, after
12 h, p62 began to accumulate, p62 is a substrate and regulatory
protein of mammalian autophagy (41). The cellular p62 level is inversely
proportional to the number of autophagy lysosomes and is an
important indicator of autophagosome clearance. The increase of p62
observed here suggested that the autophagy/lysosomal degradation
pathway was inhibited, and autophagic flow was blocked and
gradually weakened, suggesting that autophagy was gradually
weakened after longer exposure to PA. Consistent with the changes
of p62 protein levels, previous research found that, in H9c2 cells,
the phosphorylation of p70S6K, a direct downstream target of
mTORC1, was reduced from 6 to 18 h, and then increased thereafter,
following PA treatment for 24 h, suggesting that the activity of
mTORC1 was initially suppressed when PA promoted autophagic flux in
the case of short-term exposure (42). However, mTORC1 was reactivated and
the autophagic flux was blocked by prolonged PA treatment, thus
leading to the decrease of autophagy.
Studies have suggested that autophagy could inhibit
apoptosis and facilitate cell survival (20,43).
A recent study suggested that autophagy induction could lead to a
survival response against oxidative stress in BMSCs (44). We found that further increases in
autophagy induced by RA reversed PA-induced apoptosis. Conversely,
the proportion of apoptotic cells was increased when autophagy was
inhibited by 3-MA. This finding was consistent with previous
results showing that autophagy may play a protective role in BMSCs
to prevent PA-induced apoptosis. Taken together, these results
suggest that further induction of autophagy could reverse
PA-induced apoptosis in BMSCs at early stages. However, as
autophagy gradually weakened after 12 h, its protective effect
against PA-induced apoptosis was limited. Since apoptosis is
inevitable under prolonged PA exposure, the number of apoptotic
BMSCs increased significantly.
Under normal physiological conditions, the
generation of ROS is a regular part of cellular metabolism
(45). However, research on the
lipotoxicity of adipocytes has demonstrated that excessive and
long-term treatment with PA induced excessive accumulation of
intracellular ROS in different cell types (46–49).
It is believed that ROS are produced via multiple processes, such
as the mitochondrial electron transport chain, NOX, nitric-oxide
synthase, and xanthine oxidase (46). Among these possibilities, NOX may
be the predominant source of ROS in different cell types (46,47,49).
Previous studies have shown that PA exposure causes Src activation
and increased NOX activity through Src signaling, leading to
overproduction of ROS (46). It is
well known that excessive levels of ROS not only directly damage
cells by oxidizing DNA, protein, and lipid, but also indirectly
damage cells by activating a variety of stress-sensitive
intracellular signaling pathways (50,51).
Meanwhile, accumulation of ROS and reactive nitrogen species cause
oxidative stress, which has been proposed to induce autophagy in
different cell types (42,44). In this study, we found that PA
up-regulated ROS production in a dose-dependent manner in BMSCs. In
addition, pretreating cells with NAC, a well-known antioxidant,
strongly inhibited PA-induced autophagy in BMSCs. These results
suggested that PA induced accumulation of intracellular ROS in
BMSCs. Furthermore, excessive ROS accumulation might disturb
cellular homeostasis, resulting in apoptosis (52). Meanwhile, as a protective mechanism
under various environmental stresses, autophagy is also activated
to decrease the rate of ROS-induced apoptosis.
Previous studies have suggested that apoptosis and
autophagy might be triggered by common upstream signals. On a
molecular level, this means that the apoptotic and autophagic
response mechanisms share common pathways (53,54).
ROS production may lead to oxidative stress that mediates increased
autophagy and decreased ROS-induced apoptosis (55). In addition, apoptotic stimuli may
trigger JNK and p38 MAPK activation through ROS production
(18,56). ROS has also been shown to be
closely related to this signal pathway (57,58).
A recent study suggested that autophagy was induced in colon cancer
cells treated with bufalin, an anti-cancer drug. Bufalin relies on
ROS generation, which leads to the activation of the JNK pathway
(59). In our study, we found that
PA promoted the activation of JNK and p38 MAPK in BMSCs in a
dose-dependent manner. We also investigated whether this signal
pathway was involved in PA-induced autophagy. Pretreatment with the
JNK inhibitor SP600125 or the p38 MAPK inhibitor SB203580 markedly
reduced PA-induced autophagy and inhibited PA-triggered activation
of JNK and p38 MAPK. Taken together, these results indicate that
the ROS-JNK/p38 MAPK pathway plays an important role in PA-induced
autophagy.
In conclusion, the present study demonstrated that
PA can induce autophagy, and that prolonged PA treatment may reduce
autophagy in BMSCs. Moreover, we found that autophagy can protect
cells against PA-induced apoptosis in BMSCs. Furthermore, PA might
induce autophagy through the ROS-JNK/p38 MAPK pathway in BMSCs.
Combined with the above conclusions and our findings, we postulated
that apoptosis induced by palmitate in this in vitro assay
may serve as an explanation for osteoporosis in high saturated FA
consumption people. Our findings can improve the general
understanding of the mechanisms through which BMSCs adapt to
PA-induced apoptosis. This study also provides a novel approach for
the prevention and treatment of PA-induced lipotoxicity in bone
cells. However, it is undeniable that the pathogenesis of
osteoporosis is complex, and the relationship between FA and
osteoporosis still requires in vivo experiments for further
studies.
Acknowledgements
The authors would like to thank the Molecular
Biology Center Laboratory of China Medical University (Shenyang,
China).
Funding
The present study was supported by the Natural
Science Foundation of Liaoning Province (grant no. 201102282) and
the Education Science Foundation of Liaoning Province (grant no.
L2014417).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YYL and QWL conceived and designed the experiments.
YYL, SKZ and NW performed the experiments. YYL, NW and SKZ
conducted the data analysis, and YYL produced the manuscript. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Brown SA and Rosen CJ: Osteoporosis. Med
Clin North Am. 87:1039–1063. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gimble JM, Zvonic S, Floyd ZE, Kassem M
and Nuttall ME: Playing with bone and fat. J Cell Biochem.
98:251–266. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rosen CJ and Bouxsein ML: Mechanisms of
disease: Is osteoporosis the obesity of bone? Nat Clin Pract
Rheumatol. 2:35–43. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pei L and Tontonoz P: Fat's loss is bone's
gain. J Clin Invest. 113:805–806. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Elbaz A, Wu X, Rivas D, Gimble JM and
Duque G: Inhibition of fatty acid biosynthesis prevents adipocyte
lipotoxicity on human osteoblasts in vitro. J Cell Mol Med.
14:982–991. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Orchard TS, Cauley JA, Frank GC, Neuhouser
ML, Robinson JG, Snetselaar L, Tylavsky F, Wactawski-Wende J, Young
AM, Lu B and Jackson RD: Fatty acid consumption and risk of
fracture in the Women's Health Initiative. Am J Clin Nutr.
92:1452–1460. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Turpin SM, Lancaster GI, Darby I, Febbraio
MA and Watt MJ: Apoptosis in skeletal muscle myotubes is induced by
ceramides and is positively related to insulin resistance. Am J
Physiol Endocrinol Metab. 291:E1341–E1350. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Malhi H, Bronk SF, Werneburg NW and Gores
GJ: Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis.
J Biol Chem. 281:12093–12101. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim JE, Ahn MW, Baek SH, Lee IK, Kim YW,
Kim JY, Dan JM and Park SY: AMPK activator, AICAR, inhibits
palmitate-induced apoptosis in osteoblast. Bone. 43:394–404. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
de Bruin EC and Medema JP: Apoptosis and
non-apoptotic deaths in cancer development and treatment response.
Cancer Treat Rev. 34:737–749. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hocking LJ, Whitehouse C and Helfrich MH:
Autophagy: A new player in skeletal maintenance? J Bone Miner Res.
27:1439–1447. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Caramés B, Taniguchi N, Otsuki S, Blanco
FJ and Lotz M: Autophagy is a protective mechanism in normal
cartilage, and its aging-related loss is linked with cell death and
osteoarthritis. Arthritis Rheum. 62:791–801. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lu J, Wang Q, Huang L, Dong H, Lin L, Lin
N, Zheng F and Tan J: Palmitate causes endoplasmic reticulum stress
and apoptosis in human mesenchymal stem cells: Prevention by AMPK
activator. Endocrinology. 153:5275–5284. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gillet C, Spruyt D, Rigutto S, Dalla Valle
A, Berlier J, Louis C, Debier C, Gaspard N, Malaisse WJ, Gangji V
and Rasschaert J: Oleate abrogates palmitate-induced lipotoxicity
and proinflammatory response in human bone marrow-derived
mesenchymal stem cells and osteoblastic cells. Endocrinology.
156:4081–4093. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dong X, Bi L, He S, Meng G, Wei B, Jia S
and Liu J: FFAs-ROS-ERK/P38 pathway plays a key role in adipocyte
lipotoxicity on osteoblasts in co-culture. Biochimie. 101:123–131.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Y, Luo Q, Yuan L, Miao C, Mu X, Xiao W,
Li J, Sun T and Ma E: JNK-dependent Atg4 upregulation mediates
asperphenamate derivative BBP-induced autophagy in MCF-7 cells.
Toxicol Appl Pharmacol. 263:21–31. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Choi SE, Lee SM, Lee YJ, Li LJ, Lee SJ,
Lee JH, Kim Y, Jun HS, Lee KW and Kang Y: Protective role of
autophagy in palmitate-induced INS-1 beta-cell death.
Endocrinology. 150:126–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Martino L, Masini M, Novelli M, Beffy P,
Bugliani M, Marselli L, Masiello P, Marchetti P and De Tata V:
Palmitate activates autophagy in INS-1E β-cells and in isolated rat
and human pancreatic islets. PLoS One. 7:e361882012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xie W, Zhai Z, Yang Y, Kuang T and Wang C:
Free fatty acids inhibit TM-EPCR expression through JNK pathway: An
implication for the development of the prothrombotic state in
metabolic syndrome. J Thromb Thrombolysis. 34:468–474. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Griffith JF, Yeung DK, Ahuja AT, Choy CW,
Mei WY, Lam SS, Lam TP, Chen ZY and Leung PC: A study of bone
marrow and subcutaneous fatty acid composition in subjects of
varying bone mineral density. Bone. 44:1092–1096. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Justesen J, Stenderup K, Ebbesen EN,
Mosekilde L, Steiniche T and Kassem M: Adipocyte tissue volume in
bone marrow is increased with aging and in patients with
osteoporosis. Biogerontology. 2:165–171. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hardouin P, Pansini V and Cortet B: Bone
marrow fat. Joint Bone Spine. 81:313–319. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wren TA, Chung SA, Dorey FJ, Bluml S,
Adams GB and Gilsanz V: Bone marrow fat is inversely related to
cortical bone in young and old subjects. J Clin Endocrinol Metab.
96:782–786. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shen W, Chen J, Gantz M, Punyanitya M,
Heymsfield SB, Gallagher D, Albu J, Engelson E, Kotler D, Pi-Sunyer
X and Gilsanz V: MRI-measured pelvic bone marrow adipose tissue is
inversely related to DXA-measured bone mineral in younger and older
adults. Eur J Clin Nutr. 66:983–988. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fazeli PK, Horowitz MC, MacDougald OA,
Scheller EL, Rodeheffer MS, Rosen CJ and Klibanski A: Marrow fat
and bone-new perspectives. J Clin Endocrinol Metab. 98:935–945.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ahima RS and Flier JS: Adipose tissue as
an endocrine organ. Trends Endocrinol Metab. 11:327–332. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Frühbeck G, Gómez-Ambrosi J, Muruzábal FJ
and Burrell MA: The adipocyte: A model for integration of endocrine
and metabolic signaling in energy metabolism regulation. Am J
Physiol Endocrinol Metab. 280:E827–E847. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lecka-Czernik B: Marrow fat metabolism is
linked to the systemic energy metabolism. Bone. 50:534–539. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rosen CJ, Ackert-Bicknell C, Rodriguez JP
and Pino AM: Marrow fat and the bone microenvironment:
Developmental, functional, and pathological implications. Crit Rev
Eukaryot Gene Expr. 19:109–124. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kawai M, de Paula FJ and Rosen CJ: New
insights into osteoporosis: The bone-fat connection. J Intern Med.
272:317–329. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Maurin AC, Chavassieux PM, Frappart L,
Delmas PD, Serre CM and Meunier PJ: Influence of mature adipocytes
on osteoblast proliferation in human primary cocultures. Bone.
26:485–489. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Clabaut A, Delplace S, Chauveau C,
Hardouin P and Broux O: Human osteoblasts derived from mesenchymal
stem cells express adipogenic markers upon coculture with bone
marrow adipocytes. Differentiation. 80:40–45. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang D, Haile A and Jones LC:
Dexamethasone-induced lipolysis increases the adverse effect of
adipocytes on osteoblasts using cells derived from human
mesenchymal stem cells. Bone. 53:520–530. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Corwin RL, Hartman TJ, Maczuga SA and
Graubard BI: Dietary saturated fat intake is inversely associated
with bone density in humans: Analysis of NHANES III. J Nutr.
136:159–165. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fibbe WE and Noort WA: Mesenchymal stem
cells and hematopoietic stem cell transplantation. Ann N Y Acad
Sci. 996:235–244. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Manolagas SC and Parfitt AM: What old
means to bone. Trends Endocrinol Metab. 21:369–374. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Komiya K, Uchida T, Ueno T, Koike M, Abe
H, Hirose T, Kawamori R, Uchiyama Y, Kominami E, Fujitani Y and
Watada H: Free fatty acids stimulate autophagy in pancreatic
β-cells via JNK pathway. Biochem Biophys Res Commun. 401:561–567.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bjørkøy G, Lamark T, Brech A, Outzen H,
Perander M, Overvatn A, Stenmark H and Johansen T: p62/SQSTM1 forms
protein aggregates degraded by autophagy and has a protective
effect on huntingtin-induced cell death. J Cell Biol. 171:603–614.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu J, Chang F, Li F, Fu H, Wang J, Zhang
S, Zhao J and Yin D: Palmitate promotes autophagy and apoptosis
through ROS-dependent JNK and p38 MAPK. Biochem Biophys Res Commun.
463:262–267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cai N, Zhao X, Jing Y, Sun K, Jiao S, Chen
X, Yang H, Zhou Y and Wei L: Autophagy protects against
palmitate-induced apoptosis in hepatocytes. Cell Biosci. 4:282014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Song C, Song C and Tong F: Autophagy
induction is a survival response against oxidative stress in bone
marrow-derived mesenchymal stromal cells. Cytotherapy.
16:1361–1370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kiffin R, Bandyopadhyay U and Cuervo AM:
Oxidative stress and autophagy. Antioxid Redox Signal. 8:152–162.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gao D, Nong S, Huang X, Lu Y, Zhao H, Lin
Y, Man Y, Wang S, Yang J and Li J: The effects of palmitate on
hepatic insulin resistance are mediated by NADPH Oxidase 3-derived
reactive oxygen species through JNK and p38MAPK pathways. J Biol
Chem. 285:29965–29973. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sato Y, Fujimoto S, Mukai E, Sato H,
Tahara Y, Ogura K, Yamano G, Ogura M, Nagashima K and Inagaki N:
Palmitate induces reactive oxygen species production and β-cell
dysfunction by activating nicotinamide adenine dinucleotide
phosphate oxidase through Src signaling. J Diabetes Investig.
5:19–26. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wei CD, Li Y, Zheng HY, Tong YQ and Dai W:
Palmitate induces H9c2 cell apoptosis by increasing reactive oxygen
species generation and activation of the ERK1/2 signaling pathway.
Mol Med Rep. 7:855–861. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang M, Wang CM, Li J, Meng ZJ, Wei SN,
Li J, Bucala R, Li YL and Chen L: Berberine protects against
palmitate-induced endothelial dysfunction: Involvements of
upregulation of AMPK and eNOS and downregulation of NOX4. Mediators
Inflamm. 2013:2604642013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Klaunig JE, Kamendulis LM and Hocevar BA:
Oxidative stress and oxidative damage in carcinogenesis. Toxicol
Pathol. 38:96–109. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ghosh J, Das J, Manna P and Sil PC:
Taurine prevents arsenic-induced cardiac oxidative stress and
apoptotic damage: Role of NF-kappa B, p38 and JNK MAPK pathway.
Toxicol Appl Pharmacol. 240:73–87. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Manolagas SC: From estrogen-centric to
aging and oxidative stress: A revised perspective of the
pathogenesis of osteoporosis. Endocr Rev. 31:266–300. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang YH, Wu YL, Tashiro S, Onodera S and
Ikejima T: Reactive oxygen species contribute to oridonin-induced
apoptosis and autophagy in human cervical carcinoma HeLa cells.
Acta Pharmacol Sin. 32:1266–1275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lv XC and Zhou HY: Resveratrol protects
H9c2 embryonic rat heart derived cells from oxidative stress by
inducing autophagy: role of p38 mitogen-activated protein kinase.
Can J Physiol Pharm. 90:655–662. 2012. View Article : Google Scholar
|
|
56
|
Dolado I, Swat A, Ajenjo N, De Vita G,
Cuadrado A and Nebreda AR: p38alpha MAP kinase as a sensor of
reactive oxygen species in tumorigenesis. Cancer Cell. 11:191–205.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
West RJ and Sweeney ST: Oxidative stress
and autophagy: Mediators of synapse growth? Autophagy. 8:284–285.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Haberzettl P and Hill BG: Oxidized lipids
activate autophagy in a JNK-dependent manner by stimulating the
endoplasmic reticulum stress response. Redox Biol. 1:56–64. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Xie CM, Chan WY, Yu S, Zhao J and Cheng
CH: Bufalin induces autophagy-mediated cell death in human colon
cancer cells through reactive oxygen species generation and JNK
activation. Free Radic Biol Med. 51:1365–1375. 2011. View Article : Google Scholar : PubMed/NCBI
|