Introduction
With manifestations of liver failure, end-stage
liver disease can be the termination of acute or chronic liver
diseases. Hepatocyte transplantation is currently considered as a
promising replacement resource for these diseases. However,
transplantation is severely limited due to the serious shortage of
liver donors, high expense, immunological rejection of the
transplanted cells and requirement of long-term immunosuppression
(1,2). Therefore, it is necessary to find an
alternative treatment to treat these serious liver injuries.
Mesenchymal stem cells (MSCs) as therapeutic tools as they can be
obtained with relative ease and expanded in culture, along with
features of self-renewal and multidirectional differentiation have
attracted considerable attention. Several studies have reported the
isolation of MSCs from various sources, such as placenta, amniotic
fluid, adipose tissue, bone marrow, and umbilical cord blood
(3,4). Bone-marrow mesenchymal stem cell
(BM-MSC) which can be induced into hepatocyte is once the major
source for MSC isolation. However, collecting bone marrow is an
extremely invasive and painful procedure, and the proliferative
ability, maximal cell lifespan and differentiation potential of
BM-MSCs decrease with aging (5–8).
Umbilical cord (UC)-MSCs, an alternative source for
MSC isolation, can be acquired by a non-invasive procedure and can
be easily cultured, making them potentially superior candidates for
cell transplantation compared with MSCs from other sources
(9). Allogeneic transplantation of
UC-MSCs can be applicable for cell therapy without immunological
cross-reactivity (10). Also, it
has been shown to express a low level of many liver-specific
markers such as albumin (ALB), cytokeratins (CK) 18 and 19,
α-fetoprotein (AFP) (11).
Therefore, UC-MSCs represent a prospective alternative cell source
for hepatic disease therapies. To compare with their hepatic
differentiation potential, both UC-MSCs and BM-MSCs were induced to
differentiate into hepatocytes in this study.
Materials and methods
Isolation and culture of UC-MSCs
With the written informed consent of the donors and
permission of the Institution Review Board and Human Ethics
Committee of Huai'an First People's Hospital, Nanjing Medical
University, fresh human umbilical cords were collected and stored
in 0.9% normal saline containing 100 U/ml penicillin and 100 mg/ml
streptomycin at 4°C after the delivery of the baby. There were ten
donors involved in our experiment for the isolation of UC-MSCs and
their age ranged from 22 to 36 years. The umbilical cord vessels
were removed in 0.9% normal saline following disinfection in 75%
ethanol for 1 min. The cord was cut into cubes of about 1
cm3. After removal of the supernatant fraction, the
precipitate was rinsed with DMEM (Hyclone, Logan, UT, USA) and then
centrifuged at 250 × g for 5 min. The tissue was digested with
collagenase II (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) at 37°C for 1 h and further treated with 0.25% trypsin
(Hyclone) at 37°C for 0.5 h. To neutralize the excess trypsin,
fetal bovine serum (FBS) was added to the mesenchymal tissue. The
dissociated mesenchymal cells were further dispersed with DMEM and
counted using a hemocytometer. The live cells were then plated in a
6-well culture plate at a density of 1×106 cells per
well (Cornings) and the medium was changed twice a week.
Isolation and culture of BM-MSCs
With the written informed consent of donors and
permission of the Institution Review Board and Human Ethics
Committee of Huai'an First People's Hospital, Nanjing Medical
University, bone marrow samples were obtained and isolated as
previously described (12). There
were ten donors involved in our experiment for the isolation of
BM-MSCs and their age ranged from 24 to 47 years. Six of them were
males. A lymphoprep gradient was used to layer the bone marrow and
then it was centrifuged at 2,000 rpm for 15 min. Mononuclear cells
were collected and resuspended in the growth medium. Cells were
cultured in a 6-well tissue culture plate at a density of
1×106 cells per well and the medium was replaced after 3
days. The growth medium was changed twice a week. Cells were
passaged with 0.25% trypsin when the cells reached 80–90%
confluence.
Proliferative ability of UC-MSCs
compared to BM-MSCs
UC-MSCs and BM-MSCs were digested with trypsin and
counted after trypan blue staining when cells reached about 80%
confluence during passages. Mean values of cell counts were
calculated, and the mean population doubling of each passage was
obtained according to the following formula: PD=(logNt-logN0)/log
2, where Nt is the harvested cell number and N0 is the initial cell
number for each passage (13).
Flow cytometry analysis
The phenotype of MSCs was evaluated by flow
cytometry using a flow cytometer (FACScan; BD Sciences, Shanghai,
China). Native third passage UC-MSCs or BM-MSCs were trypsinized
and suspended in PBS at a concentration of 1×107
cells/ml. Antibodies against human antigens CD13, CD105, CD34 and
HLA-DR were purchased from BD Sciences. PE-as well as FITC-labeled
mouse IgG were used as a negative control. The cells and antibodies
were incubated at 4°C for 30 min and washed three times with PBS.
Labeled cells were analyzed with the CELLQUEST Pro software (BD
Sciences).
Osteogenic differentiation
After cells reached ~80% confluence, the growth
medium was changed to the osteogenic differentiation medium,
consisting of DMEM-LG (Invitrogen; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS, 100 U/ml penicillin (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany), 100 nM dexamethasone
(Sigma-Aldrich; Merck KGaA), 100 µg/ml streptomycin (Sigma-Aldrich;
Merck KGaA), 10 nM β-glycerophosphate (Sigma-Aldrich; Merck KGaA),
2 nM L-glutamine (Sigma-Aldrich; Merck KGaA) and 0.2 mM L-ascorbate
(Sigma-Aldrich; Merck KGaA). Cells were cultured in the osteogenic
differentiation medium for 21 days and the medium changed every 3
days. Differentiated cells were analyzed by alizarin red
staining.
Adipogenic differentiation
Cells at passage 3 at a density of 1×104
cells/cm2 were treated with adipogenic medium with
medium changes twice weekly. Briefly, after cells reached 70%
confluence, the medium was replaced with expansion medium
consisting of L-DMEM supplemented with 10% FBS, 2 mM IBMX
(Sigma-Aldrich; Merck KGaA) and 5 µg/ml insulin solution
(Sigma-Aldrich; Merck KGaA). After 3 weeks, the generation of lipid
vacuoles were revealed by Oil Red O staining (Sigma-Aldrich; Merck
KGaA).
Chondrogenic differentiation
Chondrogenic differentiation was carried out
according to a previous method, the 4th passage cells were treated
with chondrogenic medium for 3 weeks (A100701 StemPro Chondro DIFF
kit; Gibco; Thermo Fisher Scientific, Inc.) (14). Medium changes were performed every
3 days, and chondrogenesis was assessed by immonohistochemical
staining for type II collagen (KeyGen Biotech Co., Ltd., Nanjing,
China).
Hepatic differentiation
According to the previous protocol (12), cells at passage 3 were deprived for
2 days in IMDM (Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10 ng/ml bFGF (PeproTech, Inc., Rocky Hill, NJ, USA) and 20
ng/ml EGF (Peprotech) prior to induction using a two-step protocol.
Differentiation was induced by treating MSCs with step-1
differentiation medium, consisting of IMDM supplemented with 20
ng/ml HGF (Peprotech), 10 ng/ml bFGF and 0.61 g/ml nicotinamide
(Sigma-Aldrich; Merck KGaA) for 7 days, followed by treatment with
step-2 maturation medium, containing 20 ng/ml oncostatin M (OSM;
Peprotech), 1 µmol/l dexamethasone (DEX; Sigma-Aldrich; Merck KGaA)
and 50 mg/ml ITS. Medium was changed every 3 days.
Immunofluorescence analysis
Cells were washed twice with cold PBS and fixed in
4% paraformaldehyde (KeyGen Biotech Co., Ltd.) in PBS for 30 min
and permeabilized with PBS containing 0.1% Triton X-100
(Sigma-Aldrich; Merck KGaA) for 20 min. The samples were incubated
with anti-human serum AFP antibody (Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA), anti-human serum ALB antibody (Santa Cruz
Biotechnology, Inc.), and anti-human serum cytochrome P450 3A4
(CYP3A4) antibody (Santa Cruz Biotechnology, Inc.), followed by
incubation with second antibody conjugated with fluorescent
phycobilioroteins, Dylight 594 and Alexa 488 goat anti-mouse
immunoglobulin G (1:2,000; Sigma-Aldrich; Merck KGaA).
Subsequently, the cells were stained with diamidinopheny-lindole
(DAPI; Sigma-Aldrich; Merck KGaA) and observed under a fluorescence
microscope (Olympus, Tokyo, Japan).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacture's protocol. The cDNA templates were synthesized by
oligo(dT) primer and PrimeScript RTase reverse transcriptase
(Takara Biotechnology Co., Ltd., Dalian, China). The products were
then subjected to RT-qPCR analysis using SYBR Green Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.) with the specific
primer pairs and conditions listed in Table I. The details of the thermocycling
conditions were as follows: 95°C for 30 sec (initial denaturation),
followed by 40 cycles at 95°C for 5 sec (exact denaturation) and
60°C for 30 sec (primer annealing and PCR product elongation). The
relative expression levels were determined using the comparative
quantification cycle method, 2−∆∆Cq (15). The mRNA expression levels were
normalized with GAPDH.
 | Table I.Sequences of primers used for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Sequences of primers used for reverse
transcription-quantitative polymerase chain reaction.
| Primer | Sequences
(5′-3′) | Fragment length
(bp) | Annealing temperature
(°C) |
|---|
| ALB | F:
TGCTTGAATGTGCTGATGACAGGG | 162 | 60 |
|
| R:
AAGGCAAGTCAGCAGGCATCTCATC |
|
|
| AFP | F:
GAAACCCACTGGAGATGAACAGTC | 190 | 60 |
|
| R:
AAGTGGGATCGATGCAGGA |
|
|
| TAT | F:
TGAGCAGTCTGTCCACTGCCT | 359 | 60 |
|
| R:
ATGTGAATGAGGAGGATCTGAG |
|
|
| G-6P | F:
GCTGGAGTCCTGTCAGGCATTGC | 349 | 60 |
|
| R:
TAGAGCTGAGGCGGAATGGGAG |
|
|
| CYP3A4 | F:
TGTGCCTGAGAACACCAGAG | 202 | 60 |
|
| R:
GCAGAGGAGCCAAATCTACC |
|
|
| α1AT | F:
CTGGGACAGTGAATCGACAATGC | 560 | 54 |
|
| R:
TCTGTTTCTTGGCCTCTTGGTG |
|
|
| GAPDH | F:
AGAAGGCTGGGGCTCATTTG | 258 | 52 |
|
| R:
AGGGCCATCCACAGTCTTC |
|
|
ELISA
After 1, 2, 3 and 4 weeks of differentiation, cells
were washed twice with PBS and incubated for 2 h in DMEM-LG (5.5 mM
glucose; Gibco; Thermo Fisher Scientific, Inc.). The medium was
collected and stored at −20°C until assayed. ALB and blood urea
nitrogen (BUN) contents were measured using ELISA kit (Human
Albumin ELISA kit ab108788 and Bmassay, Human Blood Ureas Nitrogen
ELISA kit 27013; Abcam, Cambridge, UK) according to the
manufacturer's instructions. TMB substrate was used with absorbance
read at 450 nm.
Western blot analysis
Total cellular protein was extracted using a cell
lysis buffer. Protein were separated by electrophoresis and
transferred to membranes. The membranes were blocked in blocking
solution and incubated with mouse monoclonal Ab against AFP, ALB,
glucose-6phosphate (G-6P), tryosine-aminotransferase (TAT), α1
antitrypsin (α1AT) and CYP3A4 (1:200; Santa Cruz Biotechnology,
Inc., AFP antibody sc-51506, ALB antibody sc-51515, G-6P antibody
sc-373886, TAT antibody sc-365512, α1AT antibody sc-73431, CYP3A4
antibody sc-53850) for 1 h at room temperature. After washing, the
membranes were incubated for 2 h with horseradish peroxidase
(HRP)-linked goat anti-mouse IgG (1:1,000; Santa Cruz
Biotechnology, Inc., goat anti-mouse IgG-HRP, sc-2005). The
membranes were rinsed for 10 sec in substrate buffer to remove
residual detergent. The protein bands were visualized by enhance
chemiluminescence and the images were captured in X-ray film. Mouse
monoclonal Ab against GAPDH (1:5,000; Santa Cruz Biotechnology,
Inc., GAPDH antibody, sc-47724) was used as a housekeeping gene
control. The protein quantities were determined relative to the
internal optical densities of GAPDH reference standards using
ImageJ software.
Statistical analysis
The results obtained from a typical experiment were
expressed as the mean ± standard deviation (SD). Group comparisons
were made by Student's t-test and one-way analysis of variance.
Multiple comparison between the groups was performed using the
Student-Newman-Keuls method. Statistical analysis was carried out
using SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA). A value of
P<0.05 was considered to indicate a statistically significant
difference.
Results
Proliferative ability of UC-MSCs and
BM-MSCs
UC-MSCs and BM-MSCs were respectively isolated from
human umbilical cord and bone marrow. Both UC-MSCs and BM-MSCs were
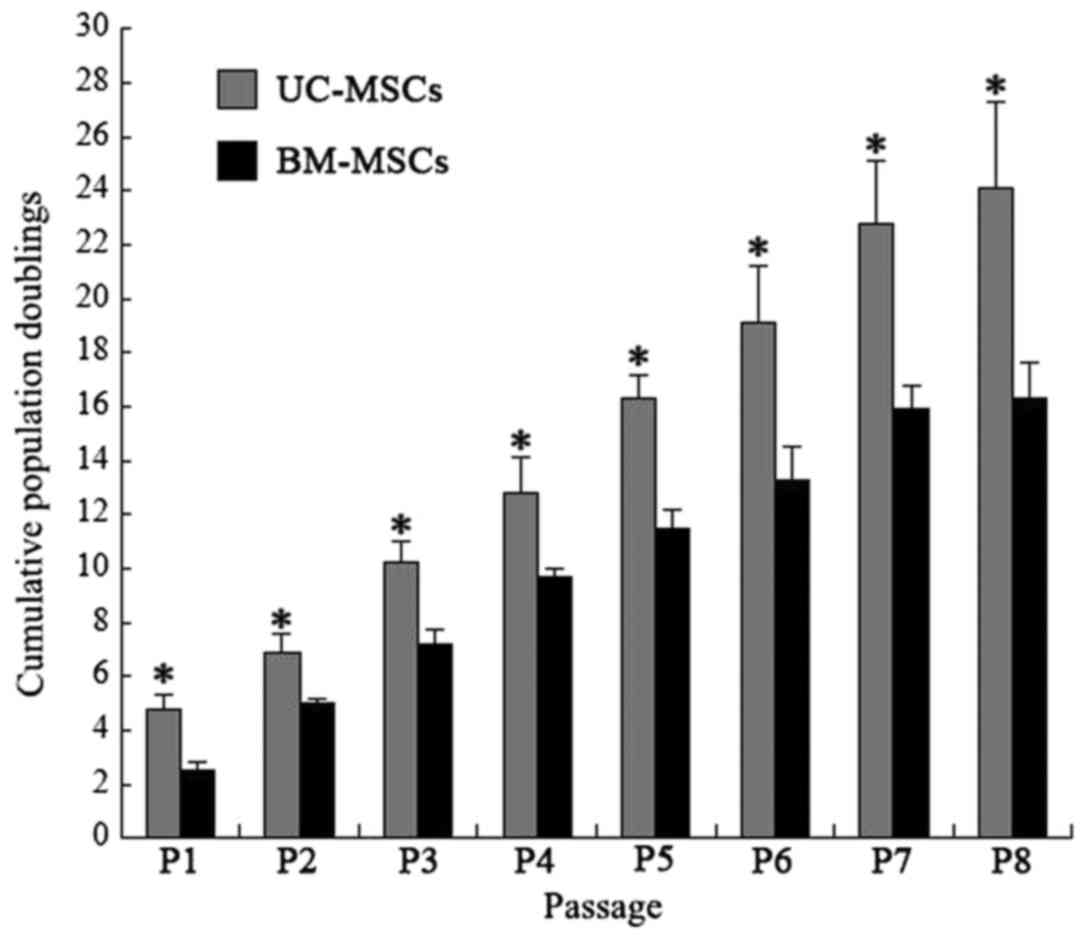
adherent, elongated and spindle-shaped. Cumulative population
doublings of UC-MSCs and BM-MSCs were calculated from passage 1 to
passage 8. The cumulative population doublings of UC-MSCs at
passage 8 was 23.8 while BM-MSCs was 16.5 (Fig. 1), indicating that UC-MSCs had
greater proliferative ability than BM-MSCs.
Characterization of UC-MSCs and
BM-MSCs
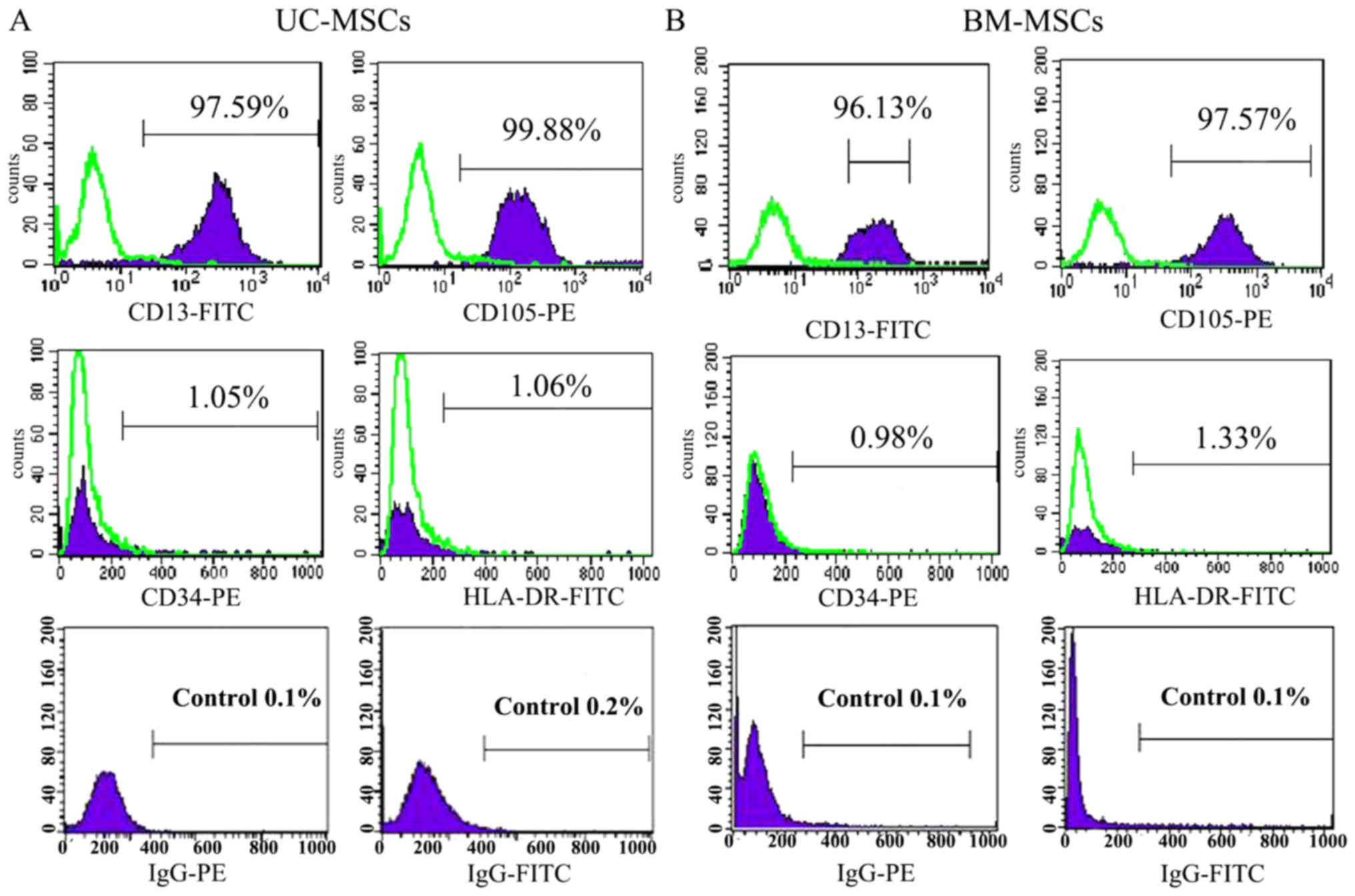
The cultured UC-MSCs expressed high levels of the
MSC marker CD13, CD105 and did not express the hematopoietic marker
CD34 and HLA-DR as a negative control (Fig. 2A). BM-MSCs also showed the similar
expression of CD13, CD105, CD34 and HLA-DR (Fig. 2B). Under certain conditions, MSCs
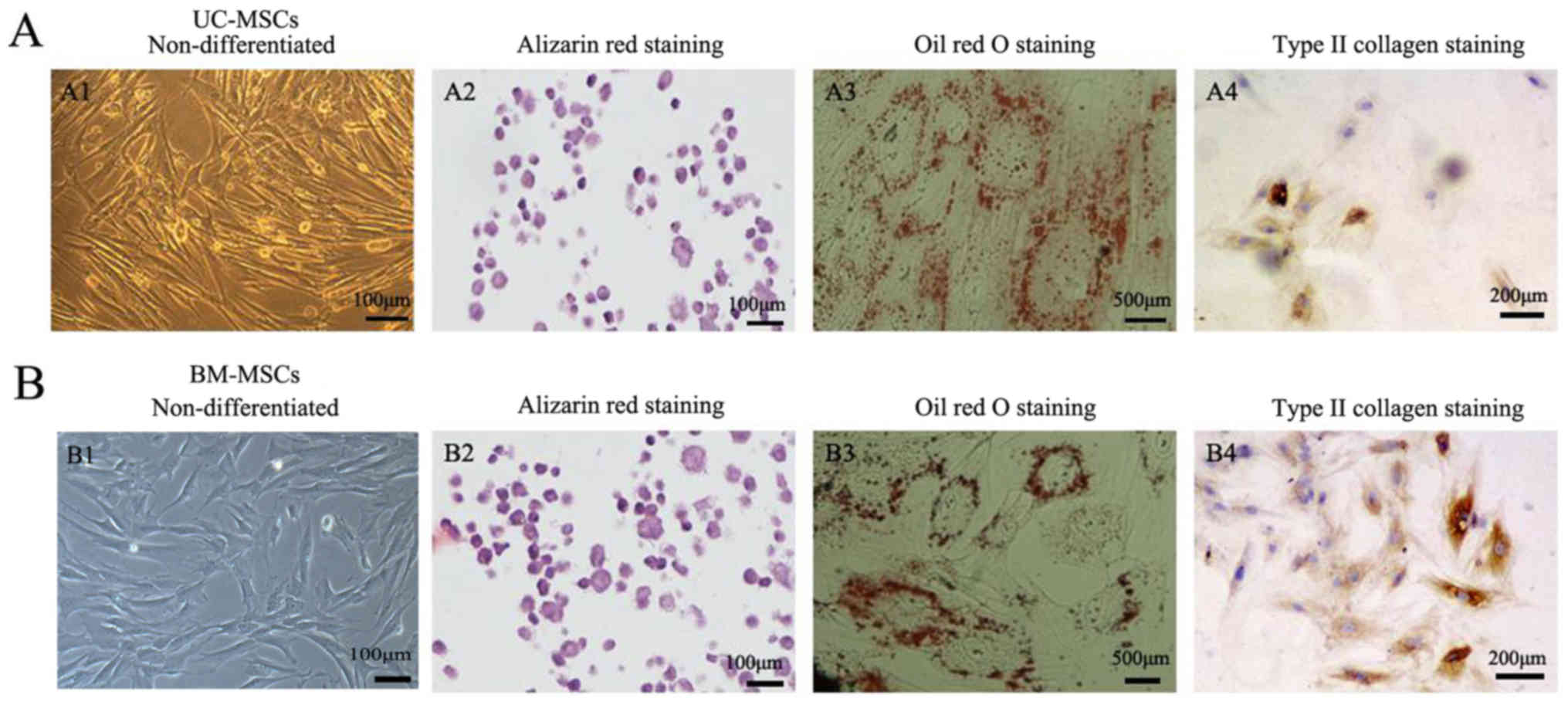
which are characterized as multipotent cells can differentiate into
different cells. Positive staining of alizarin red indicated that
UC-MSCs and BM-MSCs could differentiate into osteogenic cells
(Fig. 3A2 and B2). Oil Red O
staining showed that both UC-MSCs and BM-MSCs were positive for
staining lipid droplets in the cytoplasm after adipogenic
differentiation (Fig. 3A3 and B3).
Both UC-MSCs and BM-MSCs cultured in the chondrogenic medium after
differentiation did show immunohistochemical positive for type II
collagen staining (Fig. 3A4 and
B4).
Hepatogenic differentiation of UC-MSCs
and BM-MSCs
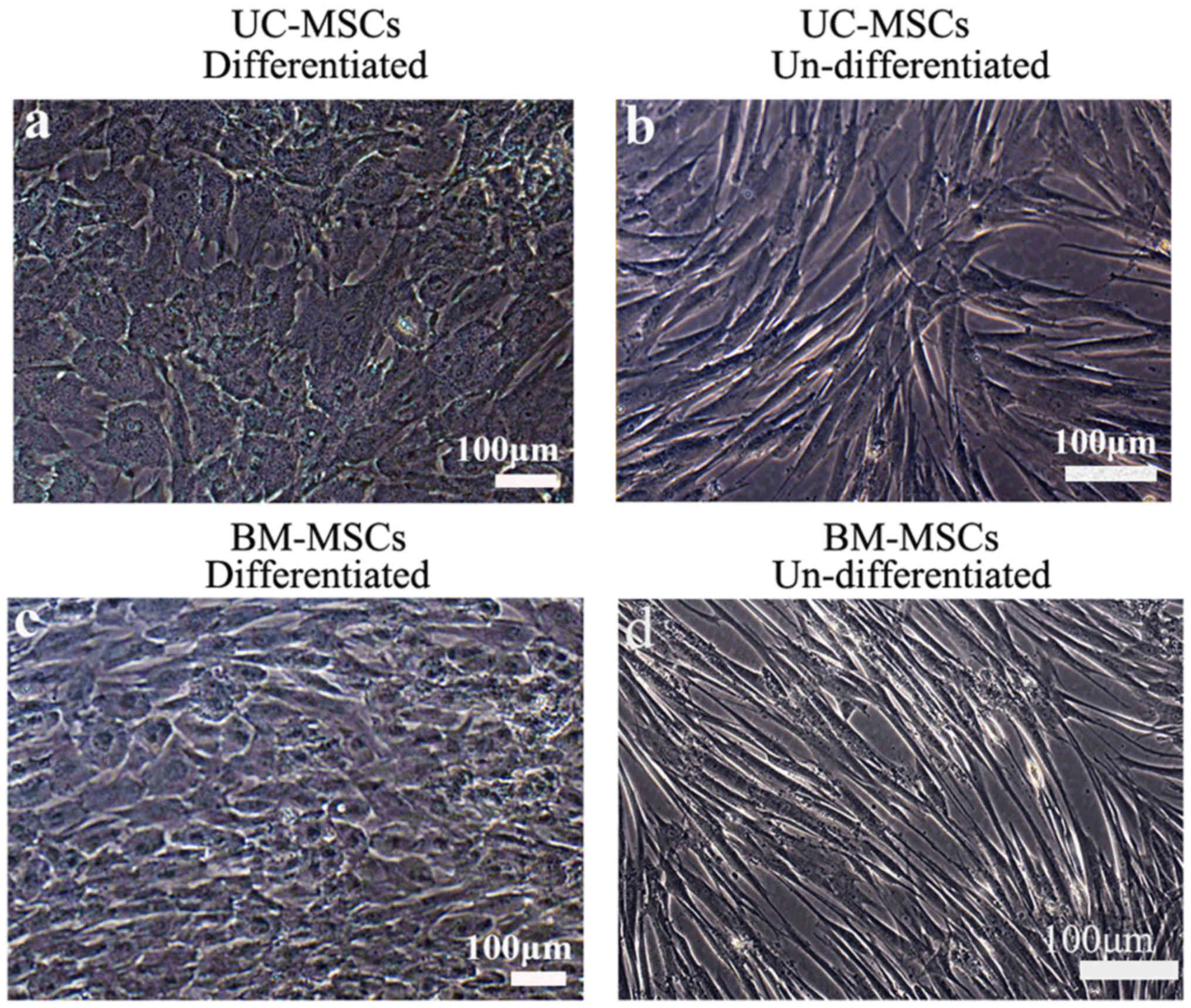
To induce hepatic differentiation, cells were
cultured in the hepatic differentiation medium. Both UC-MSCs and
BM-MSCs gradually formed the polygonal shape of hepatocytes with
the appearance of aboundant granules in the cytoplasm (Fig. 4). We also detected
hepatocyte-specific marker expression by immunofluorescene. UC-MSCs
and BM-MSCs became positive for ALB, CYP3A4 and AFP after they were
incubated in hepatic differentiation medium for 4 weeks. UC-MSCs
and BM-MSCs cultured in growth medium were as negative controls and
they did not show positive signals for these markers (Fig. 5).
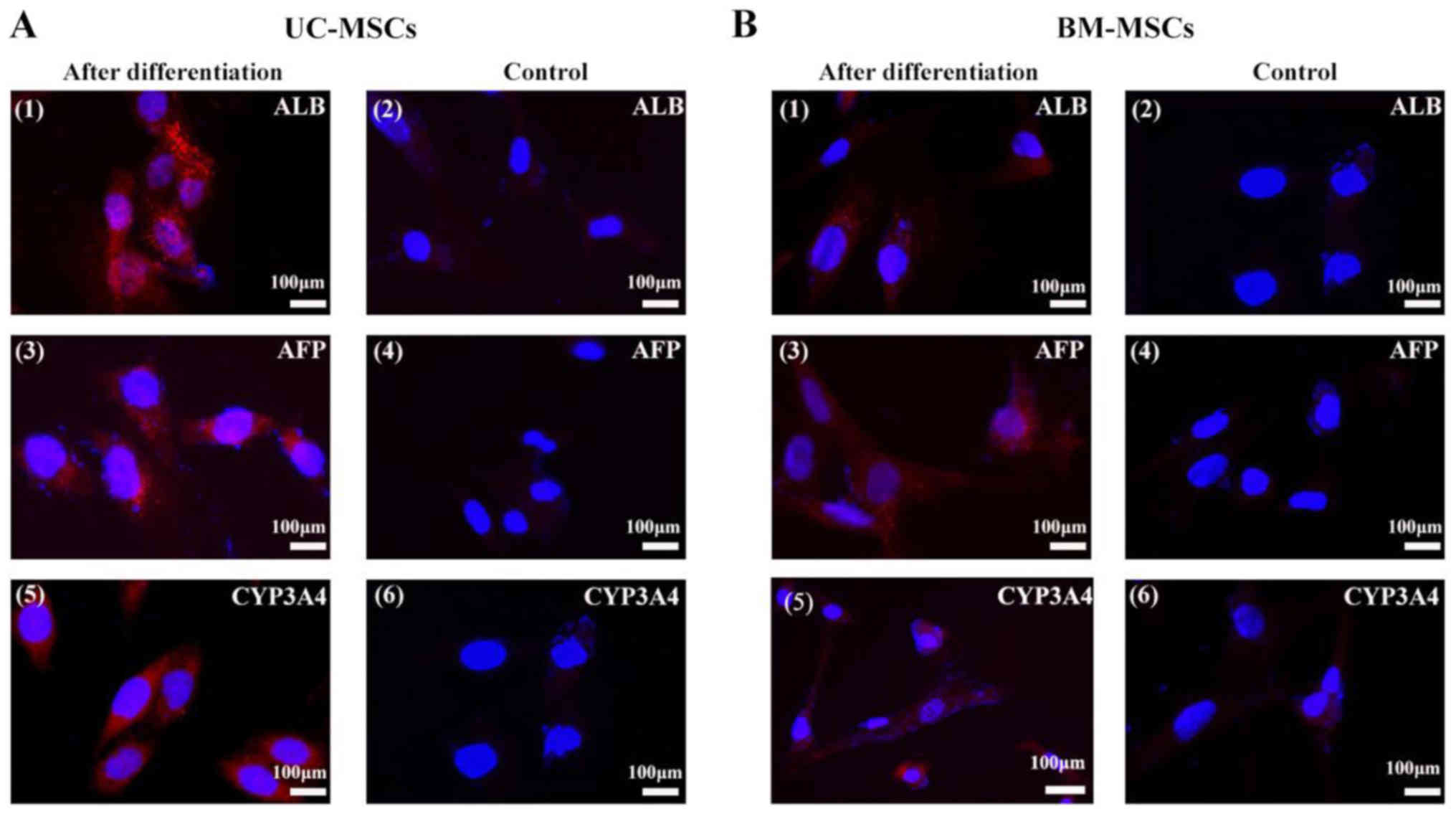 | Figure 5.Immunofluorescent analysis of
hepatocyte-specific proteins. (A) UC-MSCs and (B) BM-MSCs were
examined for their expression of (A1 and B1) ALB, (A3 and B3) AFP,
and (A5 and B5) CYP3A4 following hepatic differentiation for 4
weeks. (A2, 4 and 6, and B2, 4 and 6) Cells cultured in the growth
medium were as negative controls. Scale bars, 100 µm. UC-MSCs,
umbilical cord mesenchymal stem cells; BM-MSCs, bone marrow derived
mesenchymal stem cells; ALB, albumin; AFP, α-fetoprotein; CYP3A4,
cytochrome P450 3A4. |
Hepatocytes-specific gene marker
expression
After 4 weeks of induction, we examined the hepatic
gene expression by RT-qPCR. The gene expression analysis of ALB,
CYP3A4, TAT, G-6P, α1AT from UC-MSCs group showed higher levels
compared with BM-MSCs group while AFP showed lower expression.
Hepatocyes expressed the six markers as a positive control
(Fig. 6).
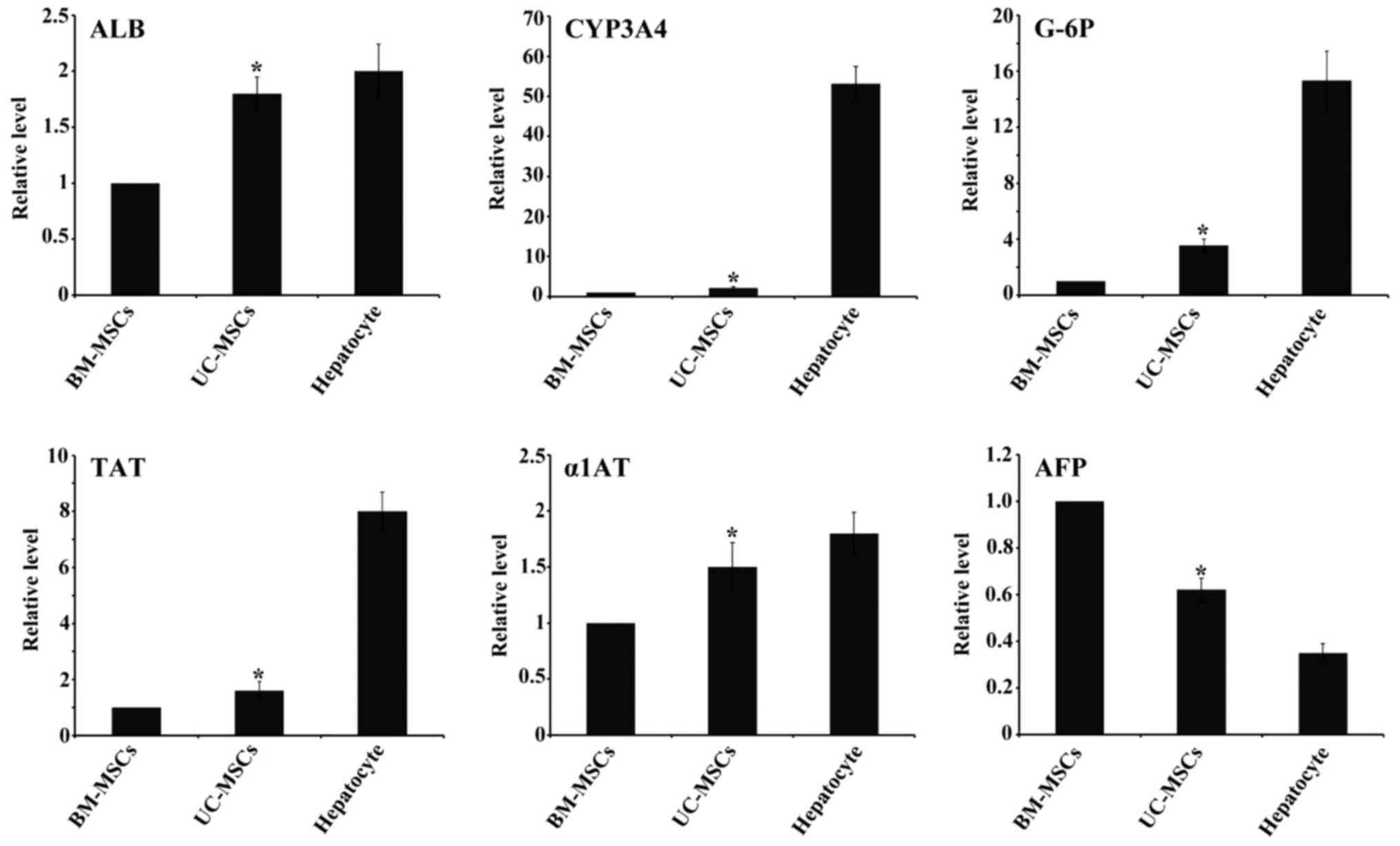 | Figure 6.Expression levels of
hepatocyte-specific genes ALB, TAT, CYP3A4, G-6P, α1AT and AFP by
reverse transcription-quantitative polymerase chain reaction. All
of the data are presented as the mean ± standard deviation (n=3),
and when compared with BM-MSCs, the fold induction for each gene
induced by UC-MSCs was significant. *P<0.05 vs. BM-MSCs.
UC-MSCs, umbilical cord mesenchymal stem cells; BM-MSCs, bone
marrow derived mesenchymal stem cells; ALB, albumin; CYP3A4,
cytochrome P450 3A4; TAT, tyrosine-aminotransferase; G-6P,
glucose-6phosphate; α1AT, α1 antitrypsin; AFP, α-fetoprotein. |
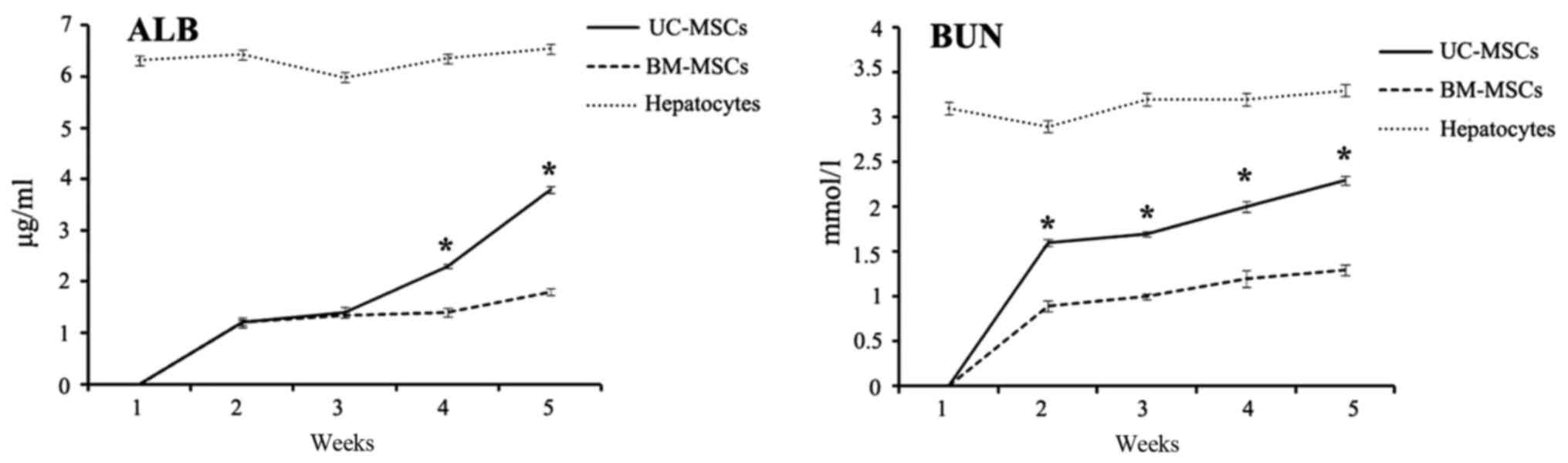
Measurement of secreted ALB and
BUN
Both ALB and BUN are important indications of
functional hepatocytes. From week 1 to 5 after hepatic
differentiation, cell culture supernatants of UC-MSCs and BM-MSCs
were collected every week and examined for the level of secreted
ALB and BUN using ELISA kit. Both ALB and BUN levels were not
detected before 1 week of induction. The ALB and BUN concentration
increased from week 2 to week 5 after hepatic differentiation.
Compared with differentiated BM-MSCs, differentiated UC-MSCs
secreted significantly more ALB and BUN after 4 weeks of induction
(P<0.05). Hepatocytes secreted ALB and BUN as a positive control
(Fig. 7).
Western blot analysis of specific
protein expression
We also examined the protein ALB, CYP3A4, TAT, G-6P,
α1AT and AFP after 4 weeks of induction by western blotting. Also,
the result was in accordance with RT-qPCR (Fig. 8).
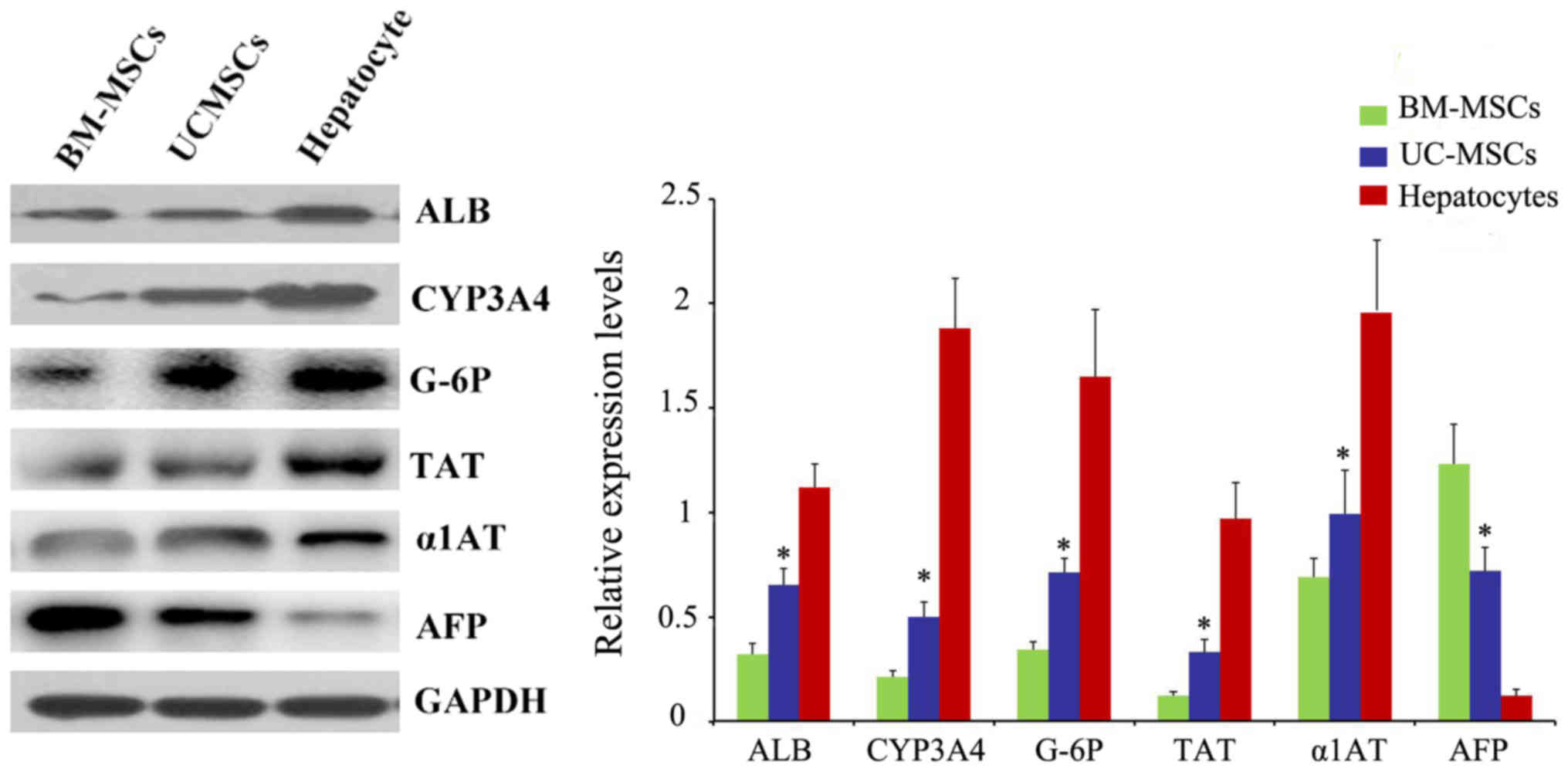 | Figure 8.Protein levels of ALB, TAT, CYP3A4,
G-6P, α1AT and AFP were analyzed by western blot analysis.
*P<0.05 vs. BM-MSCs. UC-MSCs, umbilical cord mesenchymal stem
cells; BM-MSCs, bone marrow derived mesenchymal stem cells; ALB,
albumin; CYP3A4, cytochrome P450 3A4; TAT,
tyrosine-aminotransferase; G-6P, glucose-6phosphate; α1AT, α1
antitrypsin; AFP, α-fetoprotein. |
Discussion
MSCs are present in various tissues and are
excellent candidates for cell therapy because of their capacity for
self-renewal with a high proliferative capacity, multipotency, low
immunogenicity. Our study showed that both UC-MSCs and BM-MSCs
expressed high levels of the MSC markers CD13 and CD105, did not
express the hematopoietic cell marker CD34 and HLA-DR, which were
consistent with previous studies (16). We have also examined other CD
antigens of UC-MSCs and BM-MSCs through flow cytometry analysis. We
found that both BM-MSCs and UC-MSCs expressed CD90, CD44, CD73 and
CD59 (data not shown). Also, they did not express CD45, CD14, CD19
(data not shown). To illustrate their multipotent differentiation
potential, both UC-MSCs and BM-MSCs were examined for their ability
to undergo adipogenic, osteogenic and chondrogenic differentiation.
However, UC-MSCs showed higher proliferation ability than
BM-MSCs.
Previous studies showed that under certain
conditions UC-MSCs and BM-MSCs could differentiate into hepatocyte
(12,17). In this study, UC-MSCs and BM-MSCs
were cultured in the hepatic differentiation medium according to
the protocol described by Lee et al (12). We found that, the two cells
gradually began to form clusters and progressed toward the
polygonal morphology of mature hepatocytes upon exposure to the
differentiation medium. Also, they expressed hepatic protein
markers, such as ALB, CYP3A4 and AFP.
However, which cell has a higher hepatic
differentiation efficiency remains unclear. This study first
focused on comparing the function of hepatocytes differentiated
from UC-MSCs and BM-MSCs. After 4 weeks of induction, we examined
the hepatic gene expression by RT-qPCR. Compared to the gene
expression of ALB, CYP3A4, TAT, G-6P and α1AT in in the BM-MSCs
group, the UC-MSCs group revealed higher level of these genes,
whereas AFP exhibited lower expression. It indicated that the
hepatocytes differentiated from UC-MSCs had a higher degree of
maturity than BM-MSCs. ALB and BUN secretion were the important
indication of functional hepatocytes. Both differentiated UC-MSCs
and BM-MSCs began to secrete ALB and BUN on the 2 week of
induction. Differentiated UC-MSCs secreted both ALB and BUN more
than differentiated BM-MSCs after 4 weeks of induction. The protein
levels of ALB, CYP3A4, TAT, G-6P, α1AT and AFP were also examined
by western blotting, and the results were in accordance with the
RT-qPCR findings.
In conclusion, both UC-MSCs and BM-MSCs could be
induced into hepatocytes under some conditions. Also, UC-MSCs had
higher hepatic differentiation potential than BM-MSCs. Furthermore,
without causing pain to donors, UC-MSCs can be obtained more easily
compared with BM-MSCs, and the procedure avoids technical and
ethical issues. UC-MSCs have a higher proliferation rate and are
more primitive than BM-MSCs. Therefore, UC-MSCs has advantages over
BM-MSCs for the treatment of end-stage liver disease.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
FZ and FZQ conceived and designed the experiments.
YBY performed the experiments and wrote the manuscript. YS and YC
performed the experiments and analyzed the data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The present study was performed with the permission
of the Institution Review Board and Human Ethics Committee of
Huai'an First People's Hospital, Nanjing Medical University
(Jiangsu, China); written informed consent was obtained from all
donors.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dhawan A, Puppi J, Hughes RD and Mitry RR:
Human hepatocyte transplantation: Current experience and future
challenges. Nat Rev Gastroenterol Hepatol. 7:288–298. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou WL, Medine CN, Zhu L and Hay DC: Stem
cell differentiation and human liver disease. World J
Gastroenterol. 18:2018–2025. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
El-Tantawy WH and Haleem EN: Therapeutic
effects of stem cell on hyperglycemia, hyperlipidemia, and
oxidative stress in alloxan-treated rats. Mol Cell Biochem.
391:193–200. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Anker In 't PS, Scherjon SA, Kleijburg-van
der Keur C, de Groot-Swings GM, Claas FH, Fibbe WE and Kanhai HH:
Isolation of mesenchymal stem cells of fetal or maternal origin
from human placenta. Stem Cells. 22:1338–1345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stenderup K, Justesen J, Clausen C and
Kassem M: Aging is associated with decreased maximal life span and
accelerated senescence of bone marrow stromal cells. Bone.
33:919–926. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nishida S, Endo N, Yamagiwa H, Tanizawa T
and Takahashi HE: Number of osteoprogenitor cells in human bone
marrow markedly decreases after skeletal maturation. J Bone Miner
Metab. 17:171–177. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mueller SM and Glowacki J: Age-related
decline in the osteogenic potential of human bone marrow cells
cultured in three-dimensional collagen sponges. J Cell Biochem.
82:583–590. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang H, Fazel S, Tian H, Mickle DA,
Weisel RD, Fujii T and Li RK: Increasing donor age adversely
impacts beneficial effects of bone marrow but not smooth muscle
myocardial cell therapy. Am J Physiol Heart Circ Physiol.
289:H2089–H2096. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fan CG, Zhang QJ and Zhou JR: Therapeutic
potentials of mesenchymal stem cells derived from human umbilical
cord. Stem Cell Rev. 7:195–207. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Anzalone R, Lo Iacono M, Loria T, Di
Stefano A, Giannuzzi P, Farina F and La Rocca G: Wharton's jelly
mesenchymal stem cells as candidates for beta cells regeneration:
Extending the differentiative and immunomodulatory benefits of
adult mesenchymal stem cells for the treatment of type 1 diabetes.
Stem Cell Rev. 7:342–363. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Prasajak P and Leeanansaksiri W:
Developing a new two-step protocol to generate functional
hepatocytes from Wharton's Jelly-derived mesenchymal stem cells
under hypoxic condition. Stem Cells Int. 2013:7621962013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee KD, Kuo TK, Whang-Peng J, Chung YF,
Lin CT, Chou SH, Chen JR, Chen YP and Lee OK: In vitro hepatic
differentiation of human mesenchymal stem cells. Hepatology.
40:1275–1284. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kern S, Eichler H, Stoeve J, Klüter H and
Bieback K: Comparative analysis of mesenchymal stem cells from bone
marrow, umbilical cord blood, or adipose tissue. Stem Cells.
24:1294–1301. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang HS, Shyu JF, Shen WS, Hsu HC, Chi TC,
Chen CP, Huang SW, Shyr YM, Tang KT and Chen TH: Transplantation of
insulin-producing cells derived from umbilical cord stromal
mesenchymal stem cells to treat NOD mice. Cell Transplant.
20:455–466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dominici M, Le Blanc K, Mueller I,
Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A,
Prockop Dj and Horwitz E: Minimal criteria for defining multipotent
mesenchymal stromal cells. The International society for cellular
therapy position statement. Cytotherapy. 8:315–317. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Campard D, Lysy PA, Najimi M and Sokal EM:
Native umbilical cord matrix stem cells express hepatic markers and
differentiate into hepatocyte-like cells. Gastroenterology.
134:833–848. 2008. View Article : Google Scholar : PubMed/NCBI
|