Introduction
Chronic obstructive pulmonary disease (COPD), a
disabling and life-threatening disease, is associated with chronic
inflammatory responses. By 2020, COPD is predicted to be the third
leading cause of morbidity and mortality in the world (1,2).
Long-term exposure to cigarette smoke (CS) is a major risk factor
for COPD and accounts for 80~90% of COPD cases (3). CS exposure results in severe lung
inflammation characterized by influx of inflammatory cells and
secretion of cytokines in susceptible individuals (4). One type of these inflammatory cells
are macrophages which play a crucial role in the progression of
COPD as they secrete pro-inflammatory cytokines such as TNF-α, IL-1
and IL-8 (5,6). Although the mechanisms for the
pathogenesis of COPD remain elusive, it is clear that CS-induced
airway inflammatory response contributes to COPD. At present,
standard COPD treatment includes inhaled corticosteroids in
combination with long-acting β2-agonists. However, these treatments
are often ineffective due to reduced responsiveness to
corticosteroids (7,8). Therefore, new therapeutic strategies
targeted to airway inflammatory response need to be devised.
The nuclear factor-κB (NF-κB) transcription factor
exists mainly as a heterodimer contained subunits of the Rel family
p50 and p65 and is trapped in the cytoplasm of unstimulated cells.
The inhibitor proteins of NF-κB (IκB) masks the nuclear
localization sequence which is contained in the Rel homology domain
and retain NF-κB in the cytoplasm (9). When stimilated, the IκB kinase (IKK)
complex is activated and triggers phosphorylation and degradation
of IκBα (10). The resulting free
NF-κB is translocated to the nucleus and induces the transcription
of pro-inflammatory mediators including cytokines in macrophages in
COPD (5,6). Increased nuclear localization of p65
was observed in sputum macrophages and in bronchial biopsies of
COPD patients (11,12). Additionally, the increased NF-κB
levels in serum were found in COPD patients compared to health
controls (13). More and more
studies showed that the NF-κB pathway has been implicated in COPD
and indicated that modulating NF-κB activity may act as a
therapeutic strategy for COPD treatment (6,14).
Club cell secretory protein (CC16) is a secretory
protein with anti-inflammatory and immunomodulatory effects
(15). CC16 is expressed primarily
by non-ciliated bronchiolar epithelial cells, known as club cells
(16). CC16 is also synthesized by
the epithelial lining of the nose (17). Exaggerated airway inflammation has
been reported in CC16 knockout mice compared with wild-type mice
(18). Reduced levels of CC16 in
the bronchoalveolar lavage fluid (BALF) have been reported to be
associated with various lung disorders, including asthma and COPD
(19,20). Our previous study revealed that
recombinant rat CC16 protein (rCC16) exerted an anti-inflammatory
effect by inhibiting the expression of MMP-9 and pro-inflammatory
cytokines in epithelial cells and macrophages (21,22).
Given the anti-inflammatory function of rCC16, the aim of the
present study was to investigate the effects of rCC16 on CS-induced
lung inflammation in mice and the possible mechanism through which
rCC16 suppresses CS-induced lung inflammation.
Materials and methods
Animals
Adult male C57/BL6 mice (6–8 weeks old; weight,
18–20 g) were purchased from the Laboratory Animal Center of Shanxi
Medical University (Shanxi, China) and were maintained under
specific pathogen-free conditions in a 12-h light-dark cycle with
food and water provided ad libitum. All experiments were
approved by the Animal Ethics Committee of Shanxi Medical
University (Permit no. SXMU-2014-16) and carried out according to
the guidelines of the National Institutes of Health Guide for the
Care and Use of Laboratory Animals (NIH publication no. 85–23,
revised 1996) (23).
Generation of COPD mice and rCC16
treatment
The COPD mouse model was prepared as previously
described (24). Briefly, mice
were divided into three groups (10 mice/group), including a control
group, COPD group and rCC16 treatment group. For CS exposure, the
mice were exposed (whole body) to smoke produced by six
commercially filtered cigarettes, whose filters were removed before
lighting, (Daguang brand, 1.3 mg nicotine, 13 mg tar and 15 mg
carbon monoxide per cigarette; Shanxi Kunming Tobacco Co., Ltd.,
Taiyuan, China) in a 27 l chamber (plexiglass, self-made, 3×3×3 m)
for 2 h/day, 5 days/week for 24 weeks. rCC16 was prepared according
to our published method, and the biological activity of rCC16 was
measured by analyzing the activity of phospholipase A2,
the substrate of CC16 (12).
During the last month of the study, rCC16-treated mice were treated
intranasally with 2.5 µg/g/body weight rCC16 [dissolved in 20 µl
sterile phosphate-buffered saline (PBS); 10 µl to each nostril]
each day prior to CS exposure as previously described (25,26).
Control animals were exposed to normal room air for 24 weeks.
CS-exposed mice and control mice were instilled with 20 µl sterile
PBS intranasally (10 µl to each nostril) each day during the last
month. Mice were weighed before they were sacrificed.
Collection of blood and BALF
After anesthetization with sodium pentobarbital (75
mg/kg body weight), blood samples were collected from 5 mice in
group via retro-orbital plexus puncture and stored at −20°C for
analysis. The chest wall was removed and the left bronchus was
ligated with a silk suture (0.5 mm; Bangshan Medtec, Guangzhou,
China). The trachea was cannulated using an intravenous catheter
(20G Intima; Sanxin Medtec, Jiangxi, China), and the right lung was
gently lavaged 1 time via a tracheal cannula with 1 ml ice-cold
PBS, followed by 4 times with 1 ml PBS. BALF was collected and
centrifuged at 290 × g for 10 min at 4°C. Supernatants of the first
fraction were stored at −80°C for subsequent analysis of
inflammatory cytokines using ELISA. BALF cells from the five
different fractions were combined and resuspended in 500 µl PBS.
The total number of cells was counted using a hemocytometer. A
differential cell count was performed using Wright-Giemsa
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) staining for
cytological determination. Finally, tissues of the left lung were
removed and stored at −70°C for mRNA analysis.
Lung histological analysis
After anesthetization, the chest wall was removed
from another five mice in each group. BALF cells from the right
lung of each animal were collected as described above for further
analysis. The right bronchus was ligated and the left lung was
perfused with 4% paraformaldehyde through the main left bronchus
before it was immersed and maintained in 4% paraformaldehyde for
another 24 h. The tissue was embedded in paraffin, and 5-µm thick
sections were prepared. The mean linear intercept (MLI) and the
mean alveolar number (MAN) were visualized using hematoxylin and
eosin (H&E) staining, and this was assessed in five randomly
selected fields on each slice by Image-Pro Plus 6.0 (Media
Cybernetics, Inc., Rockville, MD, USA). One investigator was
responsible for inflating the lung with paraformaldehyde and two
blinded investigators analyzed the MLI and MAN in order to avoid
observer bias.
Immunohistochemistry
Immunohistochemical analysis based on the
streptavidin-biotin complex was performed as previously described
(27). The primary antibodies were
CC16 (1:250) and NF-κB/p65 (1:100; both from Santa Cruz
Biotechnology, Inc., Dallas, TX, USA). Horseradish
peroxidase-conjugated secondary antibodies were obtained from
Beijing Zhongshan Jinqiao Biotechnology Co., Ltd. (Beijing, China).
The appearance of a tan color indicated positive staining for the
target protein, and staining was analyzed using Leica QWin image
processing software (Leica Microsystems GmbH, Wetzlar, Germany).
Data are presented as the gray value mean ± standard deviation
(SD), and a higher gray value corresponds to lower protein
expression.
Gene expression analysis by
RT-qPCR
RNA was isolated from mouse lung tissues (whole
lung) using the RNApure Tissue & Cell kit (CWBIO, Beijing,
China) according to the manufacturer's protocols and was reverse
transcribed using a Reverse Transcription kit (CWBIO). For gene
expression analysis, RT-qPCR was performed using UltraSYBR mixture
(CWBIO) and each reaction was performed in duplicate. The averages
of the obtained Ct values were used for further calculations. Gene
expression levels were normalized to the expression of the
reference gene, β-actin. Gene expression levels were calculated
using the comparative Ct method (2−ΔΔCt) (28). The primer sequences are the same as
in a previous study (22).
Determining the levels of
cytokines
Serum and BALF levels of tumor necrosis factor
(TNF)-α, interleukin (IL)-6 and IL-8 proteins were measured using
an ELISA kit (Shanghai Westang Bio-Tech. Co., Ltd., Shanghai,
China) according to the manufacturer's instructions. The BALF CC16
level was measured using ELISA (Qiaodu Biotechnology, Shanghai,
China) according to the manufacturer's protocols.
Immunoblotting
An NE-PER Nuclear and Cytoplasmic Extraction kit
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) was used to
extract nuclear and cytoplasmic proteins from BALF cells and
proteins were quantified using the BCA protein assay reagent. The
total protein was prepared using SDS lysis buffer containing 50 mM
Tris-HCl (pH 6.8), 10% glycerol and 2% SDS. Proteins (20 µg) were
loaded onto a 12% SDS-PAGE gel, separated and transferred onto a
polyvinylidene fluoride (PVDF) membrane. The membrane was blocked
with 5% skimmed milk, incubated overnight with the indicated
primary antibodies at 4°C and then incubated with an anti-rabbit
HRP-conjugated IgG antibody for 1 h at room temperature. Protein
bands were visualized using the ECL blot detection system (CWBIO).
Lamin B and β-actin prime antibodies (Cell Signaling Technology,
Inc., Danvers, MA, USA) served as references to detect nuclear
proteins, total protein and cytosolic proteins.
Electrophoretic mobility shift assay
(EMSA)
Nuclear proteins were extracted from lung tissues
and incubated with a biotin-labelled double-stranded
oligonucleotide containing the consensus sequence of NF-κB-DNA
binding site (5′-AGTTGAGGGGACTTTCCCAGG-3′) as previously described
(29). Binding reaction mixtures
(20 µl), containing 8 µg nuclear extract protein, 1 µg poly(dI-Dc)
(Thermo Fisher Scientific, Inc.) and 20 ficomoles biotin-labeled
probe in binding buffer (10 mM Tris, 50 mM potassium chloride, 1 mM
dithiothreitol, 0.1 mM EDTA, 2.5% glycerol and 5 mM magnesium
chloride) were incubated for 20 min at room temperature.
DNA-protein complexes were resolved on 6% non-denaturing
polyacrylamide gels and blotted onto a Biodyne B (pore size, 0.45
µm) positively charged nylon membrane (Thermo Fisher Scientific,
Inc.). Bands were detected by chemiluminescence using the
LightShift Chemiluminescent EMSA kit according to the
manufacturer's instructions (Thermo Fisher Scientific, Inc.).
Statistical analysis
Data are presented as the mean ± standard deviation.
One-way ANOVA followed by the S-N-K method was used for two-group
comparisons. All statistical analyses were conducted using GraphPad
Prism software (GraphPad Software, Inc., La Jolla, CA, USA).
Differences with P<0.05 were considered to be statistically
significant.
Results
rCC16 attenuates pathological changes
in lung tissues
The amino acid sequences for mouse and rat CC16 were
analyzed using DNAMAN software. The alignment showed that mouse and
rat CC16 shared a 89.58% homology (Fig. 1A).
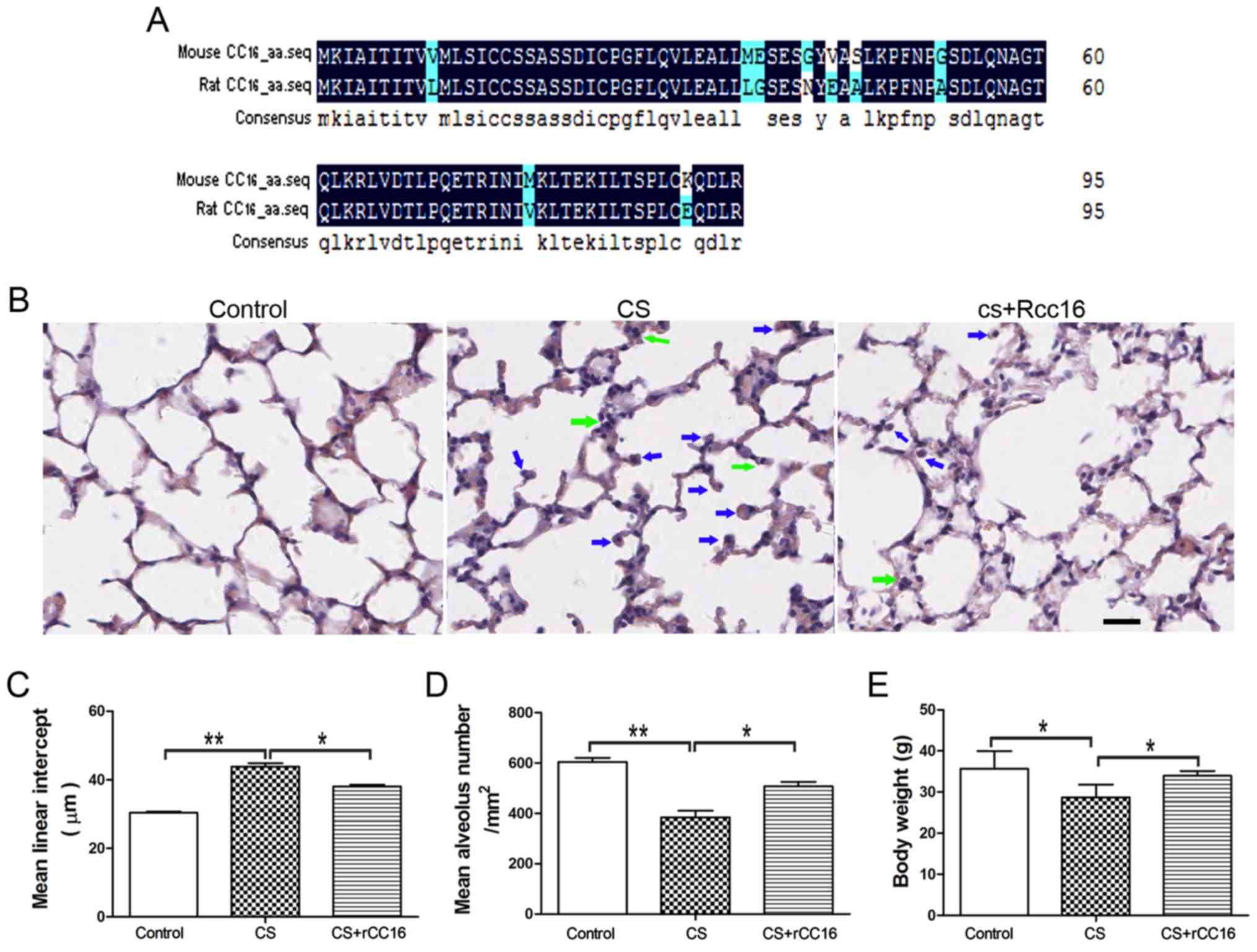 | Figure 1.rCC16 ameliorates CS-induced
pathological changes in the lungs of COPD mice. (A) Amino acid
sequences of mouse CC16 (NP_035811.1) and rat CC16 (NP_037183.1)
were aligned using DNAMAN (Lynnon, Quebec, Canada). The degree of
homology is indicated by different colors. Black, 100%; blue, 50%.
(B) H&E-stained lung sections from control mice, CS-exposed
mice and rCC16-treated mice. Exposure to CS induced the
infiltration of inflammatory cells into the lungs, as indicated by
arrows (neutrophils, green; macrophages, blue). Treatment with
rCC16 reduced the degree of pulmonary inflammation, as indicated by
arrows. (C) rCC16 reduced the mean linear intercept, which was
increased by CS exposure. (D) rCC16 augmented the mean alveolar
number, which was decreased by CS exposure. (E) rCC16 treatment led
to an increase in body weight, which was significantly reduced in
CS-exposed mice. Data are presented as the mean ± SD (n=5/group).
*P<0.05 and **P<0.01 as indicated. Scale bars represent 50
µm. COPD, chronic obstructive pulmonary disease; CS, cigarette
smoke; rCC16, recombinant club cell secretory protein; SD, standard
deviation. |
CS exposure for 24 weeks resulted in the typical
pathological features of COPD, with major enlargement of alveolar
spaces distributed throughout the parenchyma. rCC16 treatment could
reduce the degree of alveolar enlargement, and lung structures in
the control group were normal. H&E staining also revealed that,
compared with exposure to CS alone, rCC16 administration alleviated
the influx of inflammatory cells, which was characterized by a
large number of neutrophils and macrophages in the alveolar spaces
(Fig. 1B). Quantitative
morphological analysis showed that MLI was much higher in
CS-exposed mice (45.62±1.036 µm) compared with normal mice
(31.49±0.2619 µm), and that rCC16 could decrease the MLI
(39.37±0.5287 µm; Fig. 1C).
Additionally, quantitative morphological analysis showed that MAN
was significantly decreased in CS mice (384.3±26.35/mm2)
compared with the normal group (604.4±15.63/mm2), and
rCC16 treatment increased MAN (506.7±18.36/mm2; Fig. 1D). rCC16 treatment also led to
increased body weight compared with CS mice (Fig. 1E). These data demonstrate that a
CS-induced mouse COPD model was successfully established and that
rCC16 was able to attenuate pathological changes in the lungs
induced by CS exposure.
rCC16 attenuates the expression of
TNF-α, IL-6 and IL-8 in CS-exposed mice
CC16 is known to possess anti-inflammatory
properties (30). In order to
determine the potential effect of rCC16 on the expression of
pro-inflammatory cytokines, a mouse model of COPD was established
and mice were treated with rCC16. Cytokine mRNA expression in the
lungs and cytokine protein levels in the serum and BALF were
analyzed using qPCR and ELISA assays, respectively. Compared with
control mice, exposure to CS markedly increased the production of
TNF-α, IL-6 and IL-8 (Fig. 2), and
CS-stimulated overexpression of cytokines was inhibited by rCC16 at
the mRNA (Fig. 2A-C) and protein
levels (Fig. 2D-F). These results
suggest that rCC16 effectively suppressed the production of
pro-inflammatory cytokines in CS-exposed mice.
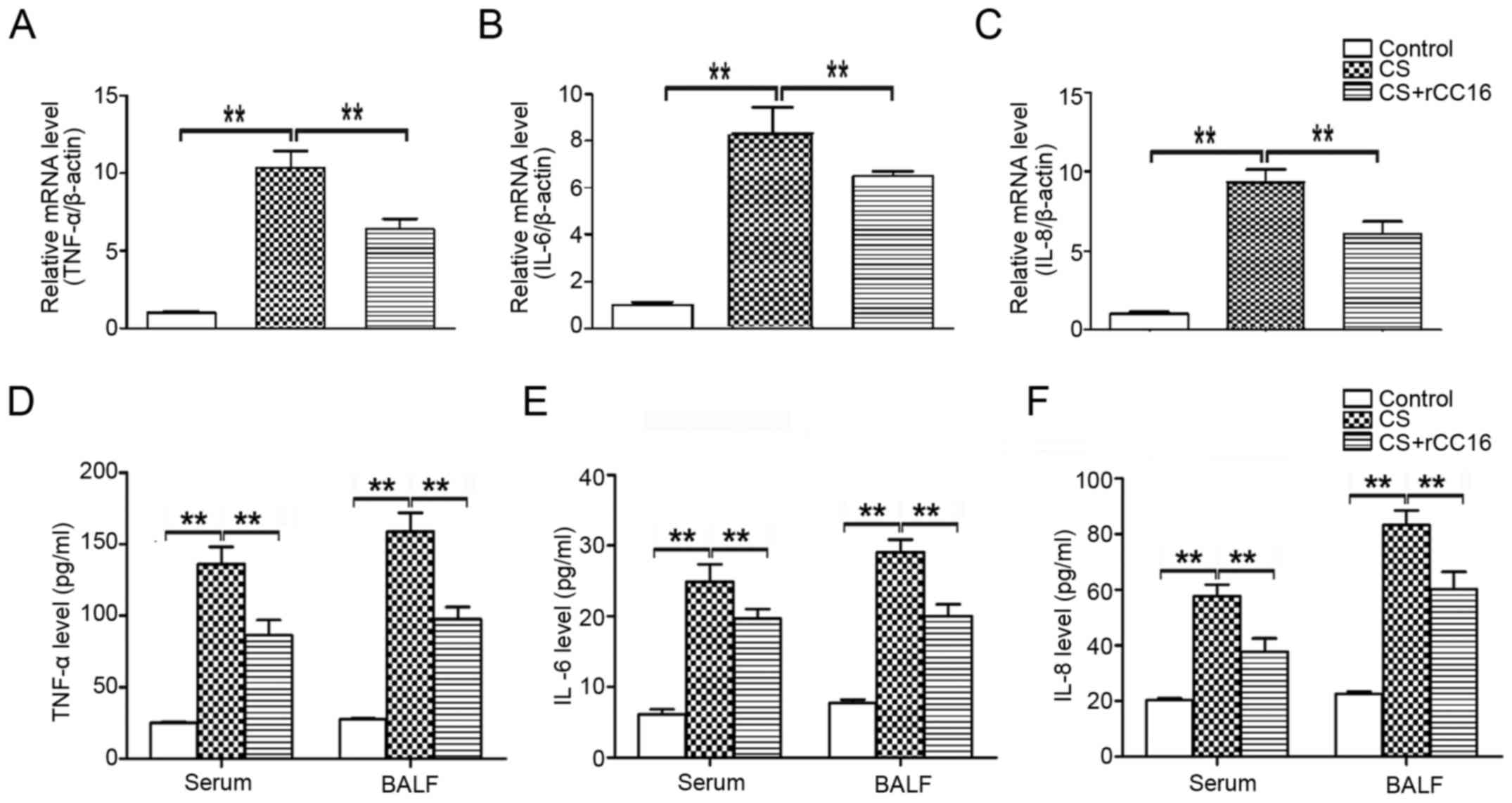 | Figure 2.Effects of rCC16 on TNF-α, IL-6 and
IL-8 expression in CS-exposed mice. The expression of TNF-α, IL-6
and IL-8 (A-C) mRNA and (D-F) protein in CS-exposed mice treated
with rCC16. mice were exposed to CS for 24 weeks and treated
intranasally with rCC16 for the last 4 weeks. For qPCR
measurements, the results are expressed as ratios of
2−∆∆Ct values for TNF-α, IL-6 and IL-8 mRNA/β-actin. For
ELISA measurements, results are expressed as the absolute protein
values of TNF-α, IL-6 and IL-8. Data are presented as the mean ± SD
(n=5/group). *P<0.05 and **P<0.01 as indicated. BALF,
bronchoalveolar lavage fluid; CS, cigarette smoke; rCC16,
recombinant club cell secretory protein; SD, standard
deviation. |
rCC16 inhibits the nuclear
translocation and DNA binding of NF-κB/p65
Pro-inflammatory cytokines are secreted from the
endothelium and immunocytes in response to various stimuli
(31,32). The total number of leukocytes,
neutrophils, macrophages and lymphocytes in BALF was assessed. As
shown in Table I, the CS-exposed
group exhibited a significant increase in the number of
neutrophils, lymphocytes, macrophages and total cells compared with
the control group. rCC16 treatment reduced the total number cells
and macrophages in BALF but had no effect on neutrophils and
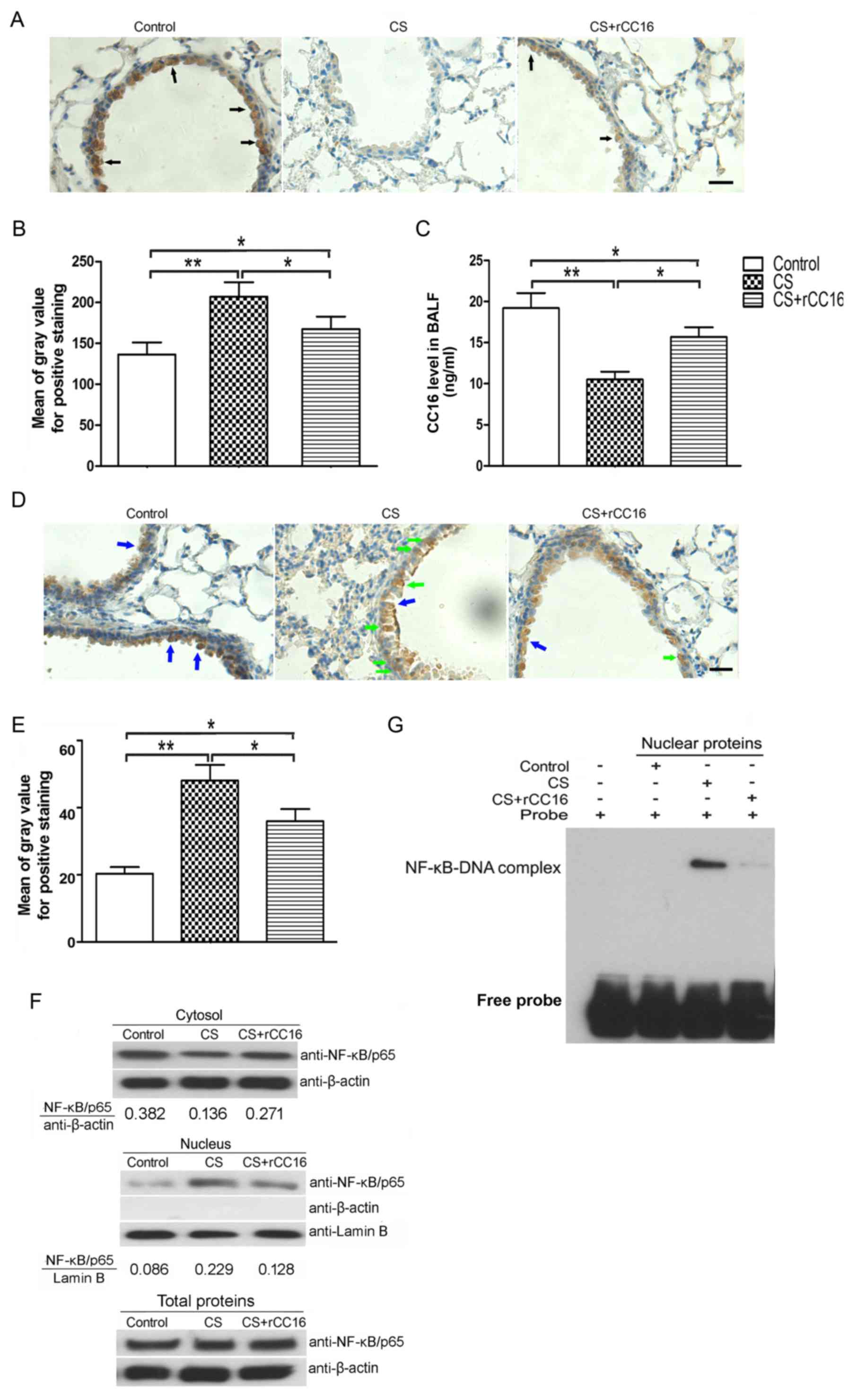
lymphocytes. Endogenous CC16 expression in club cells distributed
throughout the respiratory tract was measured using
immunohistochemical analysis, and levels of secreted CC16 in BALF
were analyzed using ELISA. Immunohistochemistry showed that CC16
was mainly expressed in bronchial epithelial cells and that CS
exposure led to a decrease in CC16 expression, while rCC16
treatment ameliorated this effect. However, rCC16 treatment did not
completely rescue the level of endogenous CC16 compared with the
control group (Fig. 3A and B).
ELISA analysis showed that CS exposure decreased CC16 expression,
while rCC16 treatment increased the level of CC16 in BALF (Fig. 3C).
 | Table I.BALF cells analyzed by Wright-Giemsa
staining. |
Table I.
BALF cells analyzed by Wright-Giemsa
staining.
| Cells | Control | CS | CS+rCC16 |
|---|
| Total cells
(×105/ml) | 3.60±1.18 |
14.93±4.79a |
10.57±3.89b |
| Macrophages
(×105/ml) | 3.57±1.09 |
14.53±4.04a |
10.91±4.31b |
| Neutrophils
(×105/ml) | 0 |
0.38±0.03a |
0.33±0.02 |
| Lymphocytes
(×105/ml) | 0.02±0.01 |
0.31±0.06a |
0.30±0.01 |
Our previous studies showed that rCC16 inhibits an
inflammatory mediator via the NF-κB pathway (21,22).
To determine whether the NF-κB pathway is also involved in the
regulation of TNF-α, IL-6 and IL-8 expression by rCC16 in
CS-exposed mice, we investigated the distribution of NF-κB/p65 in
bronchiolar epithelial and BALF cells. As shown in Fig. 3D and E, NF-κB/p65 in the nuclei of
bronchial airway epithelial cells was increased in CS-exposed mice,
and this was significantly reduced by rCC16 treatment.
Additionally, NF-κB/p65 was upregulated in the nuclei of BALF cells
and this was significantly ameliorated by rCC16 treatment.
NF-κB/p65 levels in the cytosol were decreased in CS-exposed BALF
cells. Treatment with rCC16 ameliorated this decrease without
affecting the total cellular levels of NF-κB/p65 (Fig. 3F). Moreover, to further verify the
inhibitory effect of rCC16 on NF-κB activity, EMSA was employed to
determine whether rCC16 inhibited the DNA binding activity of
NF-κB. As shown in Fig. 3G, CS
exposure markedly augmented the DNA binding activity of NF-κB, and
rCC16 suppressed CS-induced NF-κB-DNA binding. These findings
indicate that rCC16 suppresses the activity of NF-κB by inhibiting
nuclear translocation and blocking binding to DNA.
Discussion
The primary characteristic of COPD is restricted
pulmonary airflow. This is typically progressive and is associated
with an abnormal inflammatory response to the inhalation of noxious
particles and gases. CS is a complex mixture of oxidant radicals
and different chemical compounds, including reactive aldehydes and
semiquinones. CS is widely recognized as a primary risk factor that
is associated with the progression of COPD (33).
CC16 is a secretory protein release by club cells,
which are present throughout the respiratory tract epithelium from
the nose to the respiratory bronchioles. CC16 possesses
anti-inflammatory and immunomodulatory properties (34). Bernard et al (35) first reported that CC16 levels in
the serum and BALF were significantly reduced in COPD patients
compared with non-smoker controls. In addition, markedly decreased
serum levels of CC16 were detected in healthy smokers and COPD
patients compared with non-smoker controls (36). The expression of CC16 in the airway
was inversely correlated with the severity of airflow obstruction
in COPD patients (37), and
patients with COPD GOLD stages III–IV have a much lower expression
of CC16 in the airway epithelium compared with patients with COPD
GOLD I–II (38). Notably,
increased CC16 levels in the BALF and plasma samples were found to
be associated with improved bronchial dysplasia following smoking
cessation (39). In mouse models
of chronic CS exposure, decreased CC16 expression was detected in
the airway epithelium (38). Using
the CC16-deficient (CC16−/−) mouse model, Zhu
et al (40) reported the
enlargement of alveolar airspaces and increased lung inflammatory
cell counts in CC16−/− mice exposed to CS
compared with wild-type mice that were exposed to CS. Together,
these studies suggest that CC16 serves a role in the progression of
COPD. However, the underlying mechanism of CC16 remains
unclear.
As CC16 has anti-inflammatory properties (22,38),
several groups have attempted to use CC16 augmentation approaches
to treat inflammatory diseases, including COPD. It has been
reported that adenoviral-mediated CC16 overexpression in airway
epithelial cells in mice that were acutely exposed to CS resulted
in reduced CS-induced lung inflammation, and that this effect is
associated with reduced activation of NF-κB in the lung (38). rCC16 protein was delivered to
infants via the intra-tracheal route, and markers of acute lung
inflammation and injury were reduced (26). The recombinant human CC16 protein
had also been applied to respiratory distress syndrome and acute
lung injury models; promising anti-inflammatory effects were
reported (25,41). In our previous study, recombinant
rat CC16 protein was demonstrated to have biological activity
(21). There is an 89.58%
similarity in the amino acid sequences of mouse and rat CC16
proteins. Importantly, rCC16 has been used as an anti-inflammatory
mediator in lipopolysaccharide-activated mouse macrophages
(RAW264.7) in vitro (22).
In the present study, a mouse model of COPD was prepared by
chronically exposing mice to CS. Moreover, rCC16 was delivered via
intranasal instillation. The anti-inflammatory effects of CC16 were
evaluated and its underlying mechanism was investigated.
The administration of anti-inflammatory reagents via
a nasal route to treat respiratory inflammation has been reported
to be beneficial (42,43). In the present study, a murine COPD
model was established by exposure to CS for 24 weeks, and it was
found that COPD mice displayed the hallmarks of COPD, including
enlarged alveolar spaces, increased MLI and decreased MAN. When
rCC16 was administered intranasally, pathological impairment in
CS-exposed mice was alleviated (Fig.
1). However, the present study is not without limitations; the
pulmonary function test was not performed as the equipment was
unavailable. Since lung structure can partially represent the
change of pulmonary function, we evaluated the structure of the
lungs by analyzing pathological alterations (MLI and MAN) after CS
exposure in the presence and absence of rCC16 treatment.
Pro-inflammatory cytokines serve an important role
in the development of COPD. TNF-α can trigger the activation of
other pro-inflammatory cytokines, such as IL-6 and IL-8 (44). Both IL-6 and IL-8 are involved in
regulating the recruitment and activation of neutrophils to
regulate neutrophilic inflammation (45). Along with damage to the lung
parenchyma, increased TNF-α, IL-6 and IL-8 were observed in the
BALF and serum of CS-exposed mice. In contrast, rCC16 treatment
downregulated pro-inflammatory cytokines in both the serum and BALF
(Fig. 2).
Pro-inflammatory cytokines are produced by both
epithelial cells and immune cells. The accumulation of inflammatory
cells, such as macrophages, neutrophils and lymphocytes, is seen in
BALF from CS-exposed mice. Among these cells, the number of
macrophages is markedly increased in CS-exposed mice. It is known
that an increased number of macrophages in the airway lumen are
involved in the inflammatory responses of COPD, and
anti-inflammation treatment is a primary strategy for COPD
(46,47). In the present study, we found that
rCC16 administration decreased the number of macrophages. Previous
studies (21,22,38)
have revealed that CC16 functions as an anti-inflammation mediator
via the NF-κB pathway. First, the levels of endogenous CC16 in BALF
and club cells were analyzed using ELISA and immunohistochemistry.
Our data showed that rCC16 treatment upregulated endogenous CC16 in
both BALF and club cells, which were reduced in CS-exposed mice. In
our previous studies, ultrastructural damage was observed in club
cells and a reduced number of club cells were detected in the
CS-exposed rats. Moreover, the levels of CC16 protein and mRNA were
also decreased in lung tissues from CS-exposed rats (48,49).
Therese results suggest that CS exposure damages club cells and
affected the synthesis of CC16. Notably, exogenous delivery of
rCC16 limited injury to club cells and increased CC16 expression in
the airway (48,49).
Nuclear translocation of NF-κB/p65, which is known
to activate the NF-κB signaling pathway (48), was assessed. Immunohistochemistry
showed that CS exposure increased the nuclear distribution of
NF-κB/p65 in bronchial epithelial cells, and the administration of
rCC16 reversed this effect. Immunoblotting also showed that CS
exposure promoted nuclear translocation of NF-κB/p65 in BALF cells,
and rCC16 treatment reversed this translocation. Additionally, EMSA
further demonstrated that rCC16 suppressed CS-induced NF-κB-DNA
binding. All these findings indicate that rCC16 suppresses the
activity of NF-κB, which was activated by CS exposure, by
inhibiting its nuclear translocation and blocking its binding with
DNA. Other studies showed that NF-κB was expressed and activated in
club cells in rats with induced respiratory inflammation that were
exposed to hexavalent chromium (49). Nuclear translocation of the NF-κB
p65 subunit and activation of NF-κB were observed in respiratory
syncytial virus-infected human club-like lung (H441) cells
(50). However, it was reported
that CC16 does not interact with NF-κB p65 subunits directly
(19). Hence, we speculated that
CC16 may be directly or indirectly involved in regulating IκB-α
phosphorylation, which is normally present in the cytosol and forms
complexes with NF-κB dimers, thereby preventing the nuclear
localization of NF-κB and ensuring a low basal transcriptional
activity (51). This hypothesis
needs to be investigated further.
Our present findings are in agreement with those of
previous studies (30,38) which showed that airway CC16
expression was decreased in CS-induced COPD murine model, CC16
deficiency promoted CS-induced airspace enlargement and pulmonary
inflammation, and that these changes were associated with increased
NF-κB activation. Moreover, all of these studies revealed that
CS-induced pulmonary inflammation and injury were reversed by CC16
augmentation. Here we further revealed that CC16 augmentation
reduced NF-κB activation not only in lung tissue, but also in
bronchiolar epithelial and BALF cells. These findings may better
explain the possible mechanism of how CC16 acts in the development
of COPD.
In the present study, CS-exposure led to a decrease
in body weight in COPD mice, and rCC16 treatment partially reversed
the effect. Here, we speculate that this may be because CS exposure
leads to dysfunction of respiratory muscles and causes malnutrition
in COPD mice. By contrast, rCC16 treatnent inhibits inflammaton in
the lungs and improves the strength and endurance of respiratory
muscles. The recovery of lung fuction may ameliorate nutritional
imbalance in COPD mice (52). In
summary, our results suggest that the administration of rCC16
alleviates pathological lung injuries and decreases the production
of pro-inflammatory cytokines in CS-exposed mice by inactivating
the NF-κB pathway. The anti-inflammatory effects exerted by rCC16
suggest that it may have potential as a treatment for COPD.
However, further studies should be conducted to determine the
underlying mechanisms of rCC16 to contribute to the discovery of
new therapeutic agents for COPD treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Research
Project Supported by Shanxi Scholarship Council of China (grant no.
2015-101), the Fund Program for the Scientific Activities of
Selected Returned Overseas Professionals in Shanxi Province (grant
no. 2016-097) and the Startup Foundation for Doctors of Shanxi
Medical University (grant no. 03201539).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
MP conceived and designed the experiments. TL, DW,
HYL and XYH performed the experiments. BFY and XRZ analyzed and
interpreted the data. RG and HLW contributed in designing the
present study and drafted the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the Animal
Ethics Committee of Shanxi Medical University (Taiyuan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Khan MA, Kianpour S, Stämpfli MR and
Janssen LJ: Kinetics of in vitro bronchoconstriction in an
elastolytic mouse model of emphysema. Eur Respir J. 30:691–700.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Van Dijk EM, Culha S, Menzen MH, Bidan CM
and Gosens R: Elastase-induced parenchymal disruption and airway
hyper responsiveness in mouse precision cut lung slices: Toward an
ex vivo COPD model. Front Physiol. 7:6572016.PubMed/NCBI
|
|
3
|
Rabe KF, Hurd S, Anzueto A, Barnes PJ,
Buist SA, Calverley P, Fukuchi Y, Jenkins C, Rodriguez-Roisin R,
van Weel C, et al: Global strategy for the diagnosis, management
and prevention of chronic obstructive pulmonary disease: GOLD
executive summary. Am J Respir Crit Care Med. 176:532–555. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hogg JC, Chu F, Utokaparch S, Woods R,
Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson
HO and Paré PD: The nature of small-airway obstruction in chronic
obstructive pulmonary disease. N Engl J Med. 350:2645–2653. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barnes PJ: Alveolar macrophages as
orchestrators of COPD. Copd. 1:59–70. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Barnes PJ: Inflammatory mechanisms in
patients with chronic obstructive pulmonary disease. J Allergy Clin
Immunol. 138:16–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Di Marco F, Santus P, Scichilone N,
Solidoro P, Contoli M, Braido F and Corsico AG: Symptom variability
and control in COPD: Advantages of dual bronchodilation therapy.
Respir Med. 125:49–56. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barnes PJ: Corticosteroid resistance in
patients with asthma and chronic obstructive pulmonary disease. J
Allergy Clin Immunol. 131:636–645. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ,
Lee K, Hong YS and Lee JJ: Inhibition of NF-kappa B activation
through targeting I kappa B kinase by celastrol, a quinone methide
triterpenoid. Biochem Pharmacol. 72:1311–1321. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Park HJ, Kim IT, Won JH, Jeong SH, Park
EY, Nam JH, Choi J and Lee KT: Anti-inflammatory activities of
ent-16alphaH,17-hydroxy-kauran-19-oic acid isolated from the roots
of Siegesbeckia pubescens are due to the inhibition of iNOS and
COX-2 expression in RAW 264.7 macrophages via NF-kappaB
inactivation. Eur J Pharmacol. 558:185–193. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Caramori G, Romagnoli M, Casolari P,
Bellettato C, Casoni G, Boschetto P, Chung KF, Barnes PJ, Adcock
IM, Ciaccia A, et al: Nuclear localisation of p65 in sputum
macrophages but not in sputum neutrophils during COPD
exacerbations. Thorax. 58:348–351. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Di Stefano A, Caramori G, Oates T, Capelli
A, Lusuardi M, Gnemmi I, Ioli F, Chung KF, Donner CF, Barnes PJ and
Adcock IM: Increased expression of nuclear factor-kappaB in
bronchial biopsies from smokers and patients with COPD. Eur Respir
J. 20:556–563. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhuan B, Yu Y, Yang Z, Zhao X and Li P:
Mechanisms of oxidative stress effects of the NADPH
oxidase-ROS-NF-κB transduction pathway and VPO1 on patients with
chronic obstructive pulmonary disease combined with pulmonary
hypertension. Eur Rev Med Pharmacol Sci. 21:3459–3464.
2017.PubMed/NCBI
|
|
14
|
Edwards MR, Bartlett NW, Clarke D, Birrell
M, Belvisi M and Johnston SL: Targeting the NF-kappaB pathway in
asthma and chronic obstructive pulmonary disease. Pharmacol Ther.
121:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nogaki T, Asano K, Furuta A, Kanai K,
Suzaki I, Kanei A and Suzaki H: Enhancement of clara cell 10-kD
protein (CC10) production from nasal epithelial cells by
fexofenadine hydrochloride. Asian Pac J Allergy Immunol.
30:139–145. 2012.PubMed/NCBI
|
|
16
|
Mukherjee AB, Kundu GC, Mantile-Selvaggi
G, Yuan CJ, Mandal AK, Chattopadhyay S, Zheng F, Pattabiraman N and
Zhang Z: Uteroglobin: A novel cytokine? Cell Mol Life Sci.
55:771–787. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Johansson S, Keen C, Ståhl A, Wennergren G
and Benson M: Low levels of CC16 in nasal fluid of children with
birch pollen-induced rhinitis. Allergy. 60:638–642. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang H, Long XB, Cao PP, Wang N, Liu Y,
Cui YH, Huang SK and Liu Z: Clara cell 10-kD protein suppresses
chitinase 3-like 1 expression associated with eosinophilic chronic
rhinosinusitis. Am J Respir Crit Care Med. 181:908–916. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Long XB, Hu S, Wang N, Zhen HT, Cui YH and
Liu Z: Clara cell 10-kDa protein gene transfection inhibits NF-κB
activity in airway epithelial cells. PLoS one. 7:e359602012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Van Vyve T, Chanez P, Bernard A, Bousquet
J, Godard P, Lauwerijs R and Sibille Y: Protein content in
bronchoalveolar lavage fluid of patients with asthma and control
subjects. J Allergy Clin Immunol. 95:60–68. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pang M, Wang H, Bai JZ, Cao D, Jiang Y,
Zhang C, Liu Z, Zhang X, Hu X, Xu J and Du Y: Recombinant rat CC16
protein inhibits LPS-induced MMP-9 expression via NF-kappaB pathway
in rat tracheal epithelial cells. Exp Biol Med. 240:1266–1278.
2015. View Article : Google Scholar
|
|
22
|
Pang M, Yuan Y, Wang D, Li T, Wang D, Shi
X, Guo M, Wang C, Zhang X, Zheng G, et al: Recombinant CC16 protein
inhibits the production of pro-inflammatory cytokines via NF-kappaB
and p38 MAPK pathways in LPS-activated RAW264.7 macrophages. Acta
biochimica et biophysica Sinica. 1–9. 2017.
|
|
23
|
Bayne K: Revised Guide for the Care and
Use of Laboratory Animals available. American Physiological
Society. Physiologist. 39(199): 208–111. 1996.
|
|
24
|
Ma R, Gong X, Jiang H, Lin C, Chen Y, Xu
X, Zhang C, Wang J, Lu W and Zhong N: Reduced nuclear translocation
of serum response factor is associated with skeletal muscle atrophy
in a cigarette smoke-induced mouse model of COPD. Int J Chron
Obstruct Pulmon Dis. 12:581–587. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wolfson MR, Funanage VL, Kirwin SM, Pilon
AL, Shashikant BN, Miller TL and Shaffer TH: Recombinant human
Clara cell secretory protein treatment increases lung mRNA
expression of surfactant proteins and vascular endothelial growth
factor in a premature lamb model of respiratory distress syndrome.
Am J Perinatol. 25:637–645. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Levine CR, Gewolb IH, Allen K, Welch RW,
Melby JM, Pollack S, Shaffer T, Pilon AL and Davis JM: The safety,
pharmacokinetics and anti-inflammatory effects of intratracheal
recombinant human Clara cell protein in premature infants with
respiratory distress syndrome. Pediatr Res. 58:15–21. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chilkoti A, Tan PH and Stayton PS:
Site-directed mutagenesis studies of the high-affinity
streptavidin-biotin complex: Contributions of tryptophan residues
79, 108 and 120. Proc Natl Acad Sci USA. 92:1754–1758. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yan C, Wang H, Aggarwal B and Boyd DD: A
novel homologous recombination system to study 92 kDa type IV
collagenase transcription demonstrates that the NF-kappaB motif
drives the transition from a repressed to an activated state of
gene expression. FASEB J. 18:540–541. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Laucho-Contreras ME, Polverino F,
Tesfaigzi Y, Pilon A, Celli BR and Owen CA: Club cell protein 16
(CC16) augmentation: A potential disease-modifying approach for
chronic obstructive pulmonary disease (COPD). Expert Opin Ther
Targets. 20:869–883. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nicholas A, Jeon H, Selasi GN, Na SH, Kwon
HI, Kim YJ, Choi CW, Kim SI and Lee JC: Clostridium
difficile-derived membrane vesicles induce the expression of
pro-inflammatory cytokine genes and cytotoxicity in colonic
epithelial cells in vitro. Microb Pathog. 107:6–11. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nishino R, Fukuyama T, Watanabe Y,
Kurosawa Y, Kosaka T and Harada T: Significant upregulation of
cytokine secretion from T helper type 9 and 17 cells in a NC/Nga
mouse model of ambient chemical exposure-induced respiratory
allergy. J Pharmacol Toxicol Methods. 80:35–42. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shen LL, Liu YN, Shen HJ, Wen C, Jia YL,
Dong XW, Jin F, Chen XP, Sun Y and Xie QM: Inhalation of
glycopyrronium inhibits cigarette smoke-induced acute lung
inflammation in a murine model of COPD. Int Immunopharmacol.
18:358–364. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Irander K, Palm JP, Borres MP and Ghafouri
B: Clara cell protein in nasal lavage fluid and nasal nitric
oxide-biomarkers with anti-inflammatory properties in allergic
rhinitis. Clin Mol Allergy. 10:42012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bernard A, Marchandise FX, Depelchin S,
Lauwerys R and Sibille Y: Clara cell protein in serum and
bronchoalveolar lavage. Eur Respir J. 5:1231–1238. 1992.PubMed/NCBI
|
|
36
|
Lomas DA, Silverman EK, Edwards LD,
Locantore NW, Miller BE, Horstman DH and Tal-Singer R: Evaluation
of COPD Longitudinally to Identify Predictive Surrogate Endpoints
study investigators: Serum surfactant protein D is steroid
sensitive and associated with exacerbations of COPD. Eur Respir J.
34:95–102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pilette C, Godding V, Kiss R, Delos M,
Verbeken E, Decaestecker C, De Paepe K, Vaerman JP, Decramer M and
Sibille Y: Reduced epithelial expression of secretory component in
small airways correlates with airflow obstruction in chronic
obstructive pulmonary disease. Am J Respir Crit Care Med.
163:185–194. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Laucho-Contreras ME, Polverino F, Gupta K,
Taylor KL, Kelly E, Pinto-Plata V, Divo M, Ashfaq N, Petersen H,
Stripp B, et al: Protective role for club cell secretory protein-16
(CC16) in the development of COPD. Eur Respir J. 45:1544–1556.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen J, Lam S, Pilon A, McWilliams A,
Macaulay C and Szabo E: Higher levels of the anti-inflammatory
protein CC10 are associated with improvement in bronchial dysplasia
and sputum cytometric assessment in individuals at high risk for
lung cancer. Clin Cancer Res. 14:1590–1597. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhu L, Di PY, Wu R, Pinkerton KE and Chen
Y: Repression of CC16 by cigarette smoke (CS) exposure. PLoS One.
10:e01161592015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Miller TL, Shashikant BN, Melby JM, Pilon
AL, Shaffer TH and Wolfson MR: Recombinant human Clara cell
secretory protein in acute lung injury of the rabbit: effect of
route of administration. Pediatr Crit Care Med. 6:698–706. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ma H, Wang H, Luo Y, Guo S and Song C:
Mir-20b-induced increase in myeloid-derived suppressor cells in the
lungs of mice with chronic asthma. Ann Clin Lab Sci. 47:76–82.
2017.PubMed/NCBI
|
|
43
|
Reno FE, Normand P, McInally K, Silo S,
Stotland P, Triest M, Carballo D and Piché C: A novel nasal powder
formulation of glucagon: Toxicology studies in animal models. BMC
Pharmacol Toxicol. 16:292015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Drost EM and MacNee W: Potential role of
IL-8, platelet-activating factor and TNF-alpha in the sequestration
of neutrophils in the lung: Effects on neutrophil deformability,
adhesion receptor expression and chemotaxis. Eur J Immunol.
32:393–403. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gibson PG, Simpson JL and Saltos N:
Heterogeneity of airway inflammation in persistent asthma: Evidence
of neutrophilic inflammation and increased sputum interleukin-8.
Chest. 119:1329–1336. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kaku Y, Imaoka H, Morimatsu Y, Komohara Y,
Ohnishi K, Oda H, Takenaka S, Matsuoka M, Kawayama T, Takeya M, et
al: Overexpression of CD163, CD204 and CD206 on alveolar
macrophages in the lungs of patients with severe chronic
obstructive pulmonary disease. PLoS One. 9:e874002014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shapiro SD: The macrophage in chronic
obstructive pulmonary disease. Am J Respir Crit Care Med.
160:S29–S32. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Oeckinghaus A, Hayden MS and Ghosh S:
Crosstalk in NF-κB signaling pathways. Nat Immunol. 12:695–708.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhao L, Song Y, Pu J, Guo J, Wang Y, Chen
Z, Chen T, Gu Y and Jia G: Effects of repeated Cr (VI)
intratracheal instillation on club (Clara) cells and activation of
nuclear factor-kappa B pathway via oxidative stress. Toxicol Lett.
231:72–81. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Song W, Liu G, Bosworth CA, Walker JR,
Megaw GA, Lazrak A, Abraham E, Sullender WM and Matalon S:
Respiratory syncytial virus inhibits lung epithelial Na+
channels by up-regulating inducible nitric-oxide synthase. J Biol
Chem. 284:7294–7306. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Baeuerle PA and Henkel T: Function and
activation of NF-kappa B in the immune system. Annu Rev Immunol.
12:141–179. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sahebjami H and Sathianpitayakul E:
Influence of body weight on the severity of dyspnea in chronic
obstructive pulmonary disease. Am J Respir Crit Care Med.
161:886–890. 2000. View Article : Google Scholar : PubMed/NCBI
|