Introduction
Diabetes mellitus is one of the most chronic and
severe non-communicable diseases. Hyperglycemia-induced cardiac
injury is one of the serious complications of diabetes, which can
lead to myocardial oxidative stress, inflammation, calcium
overload, cell death and ultimately myocardial failure.
There are different forms of cell death, including
necrosis, apoptosis, autophagy, necroptosis and mitoptosis.
Recently, necroptosis as a form of regulated necrosis has attracted
widespread attention (1).
Apoptosis is a caspase-dependent process, as is widely established.
During apoptosis, caspase activation is mediated by extrinsic
signals that activate cell surface receptors or intrinsic signals
that induce cytochrome c release from mitochondria. Similar to
apoptosis, necroptosis is a form of regulated cell death, though it
is not mediated by caspases. Necroptosis has been reported in the
pathogenesis of various diseases, including myocardial ischemia,
brain ischemia and inflammation (2).
Necroptosis is mediated by the receptor-interacting
protein kinase (RIP)1 and RIP3-dependent pathway. The association
between high glucose (HG)-induced injury and necroptosis has been
investigated by Liang et al (3), who reported that RIP3 and reactive
oxygen species (ROS) both participate in high glucose-induced
inflammation in H9c2 cardiac cells (4); however, the detailed mechanism was
unclear. Since ROS overload and necroptosis both participate in
HG-induced cardiac cell injury, an interesting research question is
whether interventions can be selected to inhibit the release of ROS
and/or inhibit necroptosis in order to mediate a protective effect
on the cardiac cells.
It was widely reported that the activation of
aldehyde dehydrogenase (ALDH)2 has a number of implications on the
brain, heart, lung, liver injury and cancer (5,6). The
previous studies identified that the activation of ALDH2 can
attenuate diabetes-induced oxidative stress and myocardial injury
as well as inhibiting apoptosis (7–9).
However, it is unclear whether the activation of ALDH2 represses
necroptosis as a mechanism for protecting cardiac cells. In the
present study, the alterations in necroptosis in HG-induced injury
of H9c2 cardiac cells were observed and the association between
ALDH2 and necroptosis was investigated.
Materials and methods
Cell culture
The myoblast H9c2 rat cardiac cell line from the
heart was purchased from Shanghai GeneChem Co., Ltd., (Shanghai,
China). The cells were cultured in Dulbecco's modified Eagle's
medium (DMEM; HyClone; GE Healthcare Life Sciences Logan, UT, USA)
supplemented with 10% fetal bovine serum (HyClone GE Healthcare
Life Sciences) and 1% penicillin-streptomycin solution (Beyotime
Institute of Biotechnology, Shanghai, China) at 37°C in a
humidified 5% CO2 atmosphere. The cells were grown until
70–85% confluence was reached for subsequent experiments.
Chemicals and antibodies
Alda-1, the specific activator of ALDH2,
necrostatin-1 (Nec-1), the specific inhibitor of necroptosis and
the dihydroethidium (DHE) fluorescent probe were obtained from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). The mitochondrial
ALDH2 activity assay kit was obtained from Abcam (Cambridge, UK).
The ALDH2 antibody was obtained from Santa Cruz Biotechnology,
Inc., (Dallas, TX, USA). The RIP1 and RIP3 antibodies were obtained
from Abcam. The mixed lineage kinase domain like pseudokinase
(MLKL) antibody was obtained from Affinity Biosciences (Cambridge,
UK). The cleaved caspase-3 antibody was obtained from Cell
Signaling Technology, Inc., (Danvers, MA, USA). The GAPDH antibody
was acquired from Boster Biological Technology Co., Ltd. (Wuhan,
China). A Cell Counting Kit-8 (CCK-8) assay kit was from Bestbio
Life Technology (Shanghai, China; http://www.bestbio.com.cn; cat. no. BB-4202-1).
Primers for ALDH2, RIP1, RIP3, MLKL and GAPDH were acquired from
Sangon Biotech Co., Ltd. (Shanghai, China; sequences are presented
in Table I). A RevertAid RT
Reverse Transcription kit was purchased from Thermo Fisher
Scientific, Inc., (Waltham, MA, USA). The SYBR® Premix
DimerEraser™ (Perfect Real Time) was acquired from Takara Bio, Inc.
(Otsu, Japan).
 | Table I.Primer sequences for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Primer | Sequence (5′-3′) | Product size
(bp) |
|---|
| ALDH2 | Forward |
GTGTTCGGAGACGTCAAAGA | 187 |
|
| Reverse |
GCAGAGCTTGGGACAGGTAA |
|
| RIP1 | Forward |
AGGTACAGGAGTTTGGTATGGGC | 123 |
|
| Reverse |
GGTGGTGCCAAGGAGATGTATG |
|
| RIP3 | Forward |
TAGTTTATGAAATGCTGGACCGC | 145 |
|
| Reverse |
GCCAAGGTGTCAGATGATGTCC |
|
| MLKL | Forward |
GCCACTGGAAAGATCCCGTT | 108 |
|
| Reverse |
CAACAACTCGGGGCAATCCT |
|
| GAPDH | Forward |
ACAGCAACAGGGTGGTGGAC | 255 |
|
| Reverse |
TTTGAGGGTGCAGCGAACTT |
|
Experimental protocols
The cells were incubated at 37°C in a humidified 5%
CO2 atmosphere, the experiments were divided into 5
groups as follows: i) Normal control group, where H9c2 cardiac
cells were cultured in DMEM supplemented with 10% fetal bovine
serum; ii) a hypertonic control group (HPG), where H9c2 cardiac
cells were subjected to 35 mM mannitol as an osmotic control for 24
h; iii) a high glucose group (HG), where H9c2 cardiac cells were
subjected to 35 mM glucose stress for 24 h to induce cell injury;
iv) an HG+Alda-1 group where H9c2 cardiac cells, which were
subjected to HG stress were treated with 20 µM Alda-1 (a specific
activator of ALDH2) (9) for 24 h
and v) an HG+Nec-1 group, where H9c2 cardiac cells that were
subjected to HG stress were treated with 100 µM Nec-1 for 24 h.
The hypertonic control group was used to exclude the
role of the hypertonic effect. The aim of the HG+Alda-1 group was
to investigate whether the activation of ALDH2 can attenuate HG
induced-cardiac cell injury. The purpose for the HG+Nec-1 group was
to observe whether inhibiting necroptosis can attenuate HG-induced
cardiac cell injury.
CCK-8 assay
The CCK-8 assay kit was used to detect cell
viability, according to the manufacturer's protocol. In brief, H9c2
cardiac cells (100 µl/well) were seeded into 96-well plates at a
density of 8×103 cells/plate and incubated at 37°C and
5% CO2 overnight. A total of 10 µl CCK-8 solution was
added to each well following the different treatments described
above. Following 1–4 h incubation in the dark at 37°C, cell
viability was determined by measuring the absorbance at a
wavelength of 450 nm using a microplate reader (BioTek Instruments,
Inc., Winooski, VT, USA).
Detection of alterations in superoxide
production by DHE staining
Superoxide production was evaluated using the DHE
staining method. H9c2 cardiac cells were seeded in 6-well plates at
a density of 2×105 cells/plate. Following treatment with
different interventions, the cells were incubated with 10 µM DHE
solution at 37°C for 30 min in a chamber in the dark at 37°C and
then washed with PBS 2–3 times. The cells were subsequently
incubated with 1 µg/ml DAPI at 37°C for 15 min in the dark. The
fluorescent image and intensity of DHE was measured at 485 nm
(excitation wavelength) and 590 nm (emission wavelength). DAPI was
measured at 358 nm (excitation wavelength) and 461 nm (emission
wavelength) with a fluorescent microscope camera (Olympus IX71;
Olympus Corporation, Tokyo, Japan). Superoxide production was
expressed as a mean fluorescence ratio (fluorescence of exposed
cells/fluorescence of control cells) and the mean fluorescence
intensity was analyzed with Image J software (version 1.48,
National Institutes of Health, Bethesda, MD, USA).
Analysis of ALDH2 activity
ALDH2 activity may be determined by the
Mitochondrial Aldehyde Dehydrogenase (ALDH2) Activity Assay kit
(cat. no. ab115348; Abcam) and following the production of NADH in
the following ALDH2-catalyzed reaction: Acetaldehyde +
NAD+ → acid + NADH. The reaction reduces a colorless
probe to a colored product with strong absorbance at 450 nm. This
experiment was conducted according to the manufacturer's
protocol.
Detection of ALDH2, RIP1, RIP3 and
MLKL levels by reverse transcription-quantitative polymerase chain
reaction (RT-qPCR)
Total RNA in H9c2 cardiac cells was isolated using
TRIzol® reagent following the manufacturer's protocol
(Invitrogen; Thermo Fisher Scientific Inc.). The purity and
concentration of total RNA were detected using a microplate reader
(Biotek™ Epoch™; Biotek Instruments, Inc.). A total of 3 µg total
RNA was used to synthesize cDNA according to the protocol of the
RT-qPCR kit (cat. no. K1691; Thermo Fisher Scientific Inc.). The
total volume of each tube was 20 µl. The mixture was placed on a
PCR machine under conditions of 42°C 60 min and 70°C, 5 min to
obtain the cDNA. The volume of reagents used was determined
according to the protocol of the qPCR kit (cat. no. RR091A; Takara
Bio Inc.): 2 µl cDNA template (50 ng/µl); 0.6 µl upstream and
downstream primers (final concentration, 0.3 µmol/l); 0.4 µl Rox
reference dye; and 10 µl SYBR Premix DimerErase with the addition
of enzyme-free water to a total volume of 20 µl. The thermocycling
conditions were as follows: i) Pre-denaturation at 95°C for 30 sec;
ii) denaturation at 95°C for 5 sec; iii) annealing at 60°C for 30
sec and iv) extension at 72°C for 34 sec. The steps 2–4 were
repeated for 40 cycles. The dissolution curve was automatically
generated by the thermocycler. The internal control GAPDH was used
for correction and the normal group was used as a control. The
2−ΔΔCq value was calculated as the relative gene
expression level of each sample and the data was analyzed (10). The sequences of the primers used
are presented in Table I and the
reference gene was GAPDH.
Detection of ALDH2, RIP1, RIP3, MLKL
and cleaved caspase-3 levels by western blotting
The expression of ALDH2, RIP1, RIP3, MLKL and
cleaved caspase-3 proteins was detected by western blotting. H9c2
cardiac cells in each group were homogenized in the mixture buffer
of radioimmunoprecipitation assay lysate (cat. no. P0013B; Beyotime
Institute of Biotechnology) and PMSF (0.1 mM) for 60 min on ice.
The mixture was centrifuged at 1,0621 × g for 15 min at 4°C to
obtain the total protein, and the concentration of protein was
measured by bicinchoninic acid assay (cat. no. P0010; Beyotime
Institute Biotechnology). Equal quantities of protein lysates (40
µg) were electrophoresed on 10% SDS-PAGE and transferred to a
polyvinylidene difluoride membrane. Following blocking by 5% nonfat
milk for 150 min at room temperature, the membrane was probed
overnight at 4°C with primary antibodies against ALDH2 (1:500; cat.
no. sc-100496), RIP1 (1:500; cat. no. ab106393), RIP3 (1:400; cat.
no. ab62344), MLKL (1:500; cat. no. DF7412) and cleaved caspase-3
(1:300; cat. no. 9664). Following washing, the membrane was probed
with the corresponding secondary antibodies (goat anti-rabbit IgG;
1:8,000; cat. no. AP132P; Merck KGaA) at the water of 37°C for 40
min then agitating (100 rpm for 20 min) at room temperature.
Finally, the membrane was visualized using the enhanced
chemiluminescence method (Immobilon Western Chemiluminescent HRP
Substrate; cat. no. WBKLS0100; Merck KGaA). GAPDH (1:2,000; cat.
no. PB0141) was used as a loading control. The autoradiographs were
scanned using the ChemiDoc XRS Gel Image system and analyzed with
the Image Lab software (version 3.0; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The density value of each band was expressed as
arbitrary units.
Statistical analysis
The data are presented as the mean ± the standard
error of the mean. Software used for statistical analysis was
Graphpad Prism® (v6.0 for Windows; GraphPad Software,
Inc., La Jolla, CA, USA). Statistical significance was determined
using one-way analysis of variance followed by Newman-Keuls for
comparisons between multiple samples. Experiments were repeated
independently more than three times. P<0.05 was considered to
indicate a statistically significant difference.
Results
Alterations in cell viability in the
different treatment groups
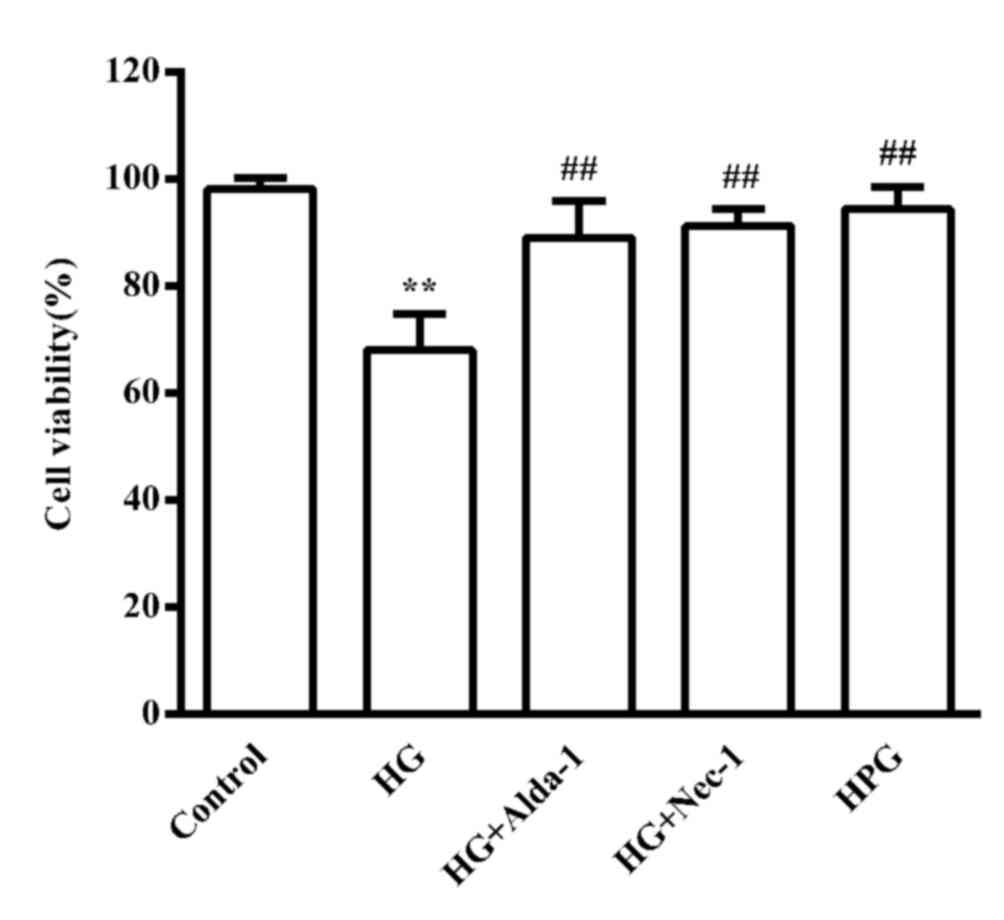
Compared with the control group, HG significantly
decreased the viability of H9c2 cardiac cells (P<0.01).
Treatments with the ALDH2 activator, Alda-1 and the RIP1 inhibitor,
Nec-1, significantly increased cell viability compared with the HG
group (P<0.01). There was no marked difference in cell viability
between the control and the HPG group (Fig. 1). As the hypertonic solution had no
obvious effect on cell viability based on the CCK-8 results,
further experiments were not performed on the HPG group.
Alterations in the activity, mRNA and
protein expression of ALDH2 in different groups
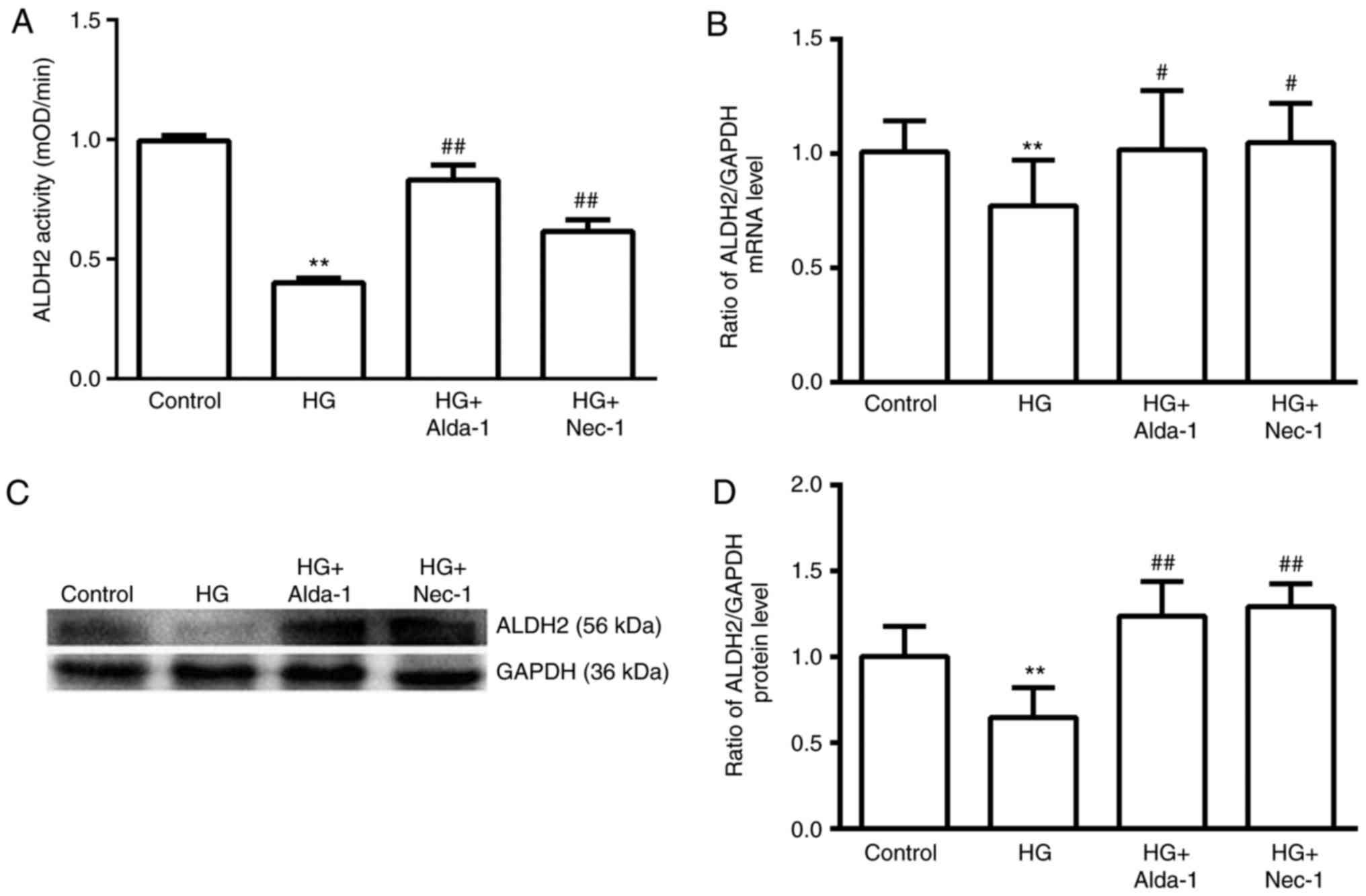
Compared with the control group, HG intervention
significantly decreased ALDH2 activity and ALDH2 expression at the
mRNA and protein levels (P<0.01; Fig. 2). In the Alda-1+HG and Nec-1+HG
groups, the activity and mRNA and protein expression of ALDH2 were
significantly increased compared with in the HG group (P<0.05;
Fig. 2). These results suggested
that inhibiting necroptosis with Nec-1 induced increases in ALDH2
activity and expression, which was similar to the apparent
mechanism of ALDH2 activation by Alda-1.
Alterations in ROS production in
different treatment groups
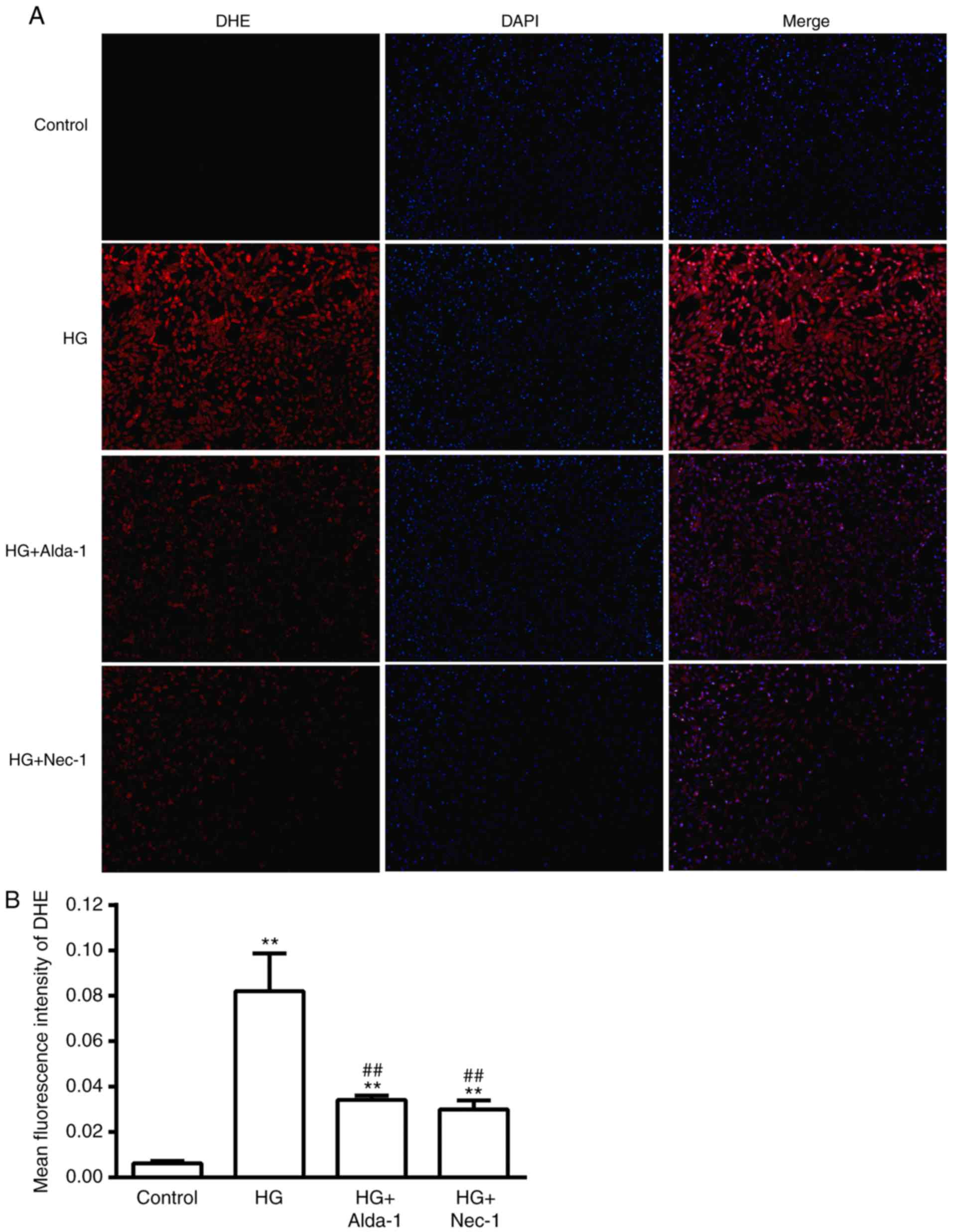
Compared with the control group, DHE fluorescence
intensity was significantly increased in the HG group, indicating
an increase in ROS (P<0.01). DHE fluorescence intensity was
significantly decreased when the cells were treated with Alda-1 and
Nec-1 under HG conditions (P<0.01; Fig. 3), which suggested that the
activation of ALDH2 or the inhibition of necroptosis could reduce
the over-production of ROS in HG-induced cardiac cell injury.
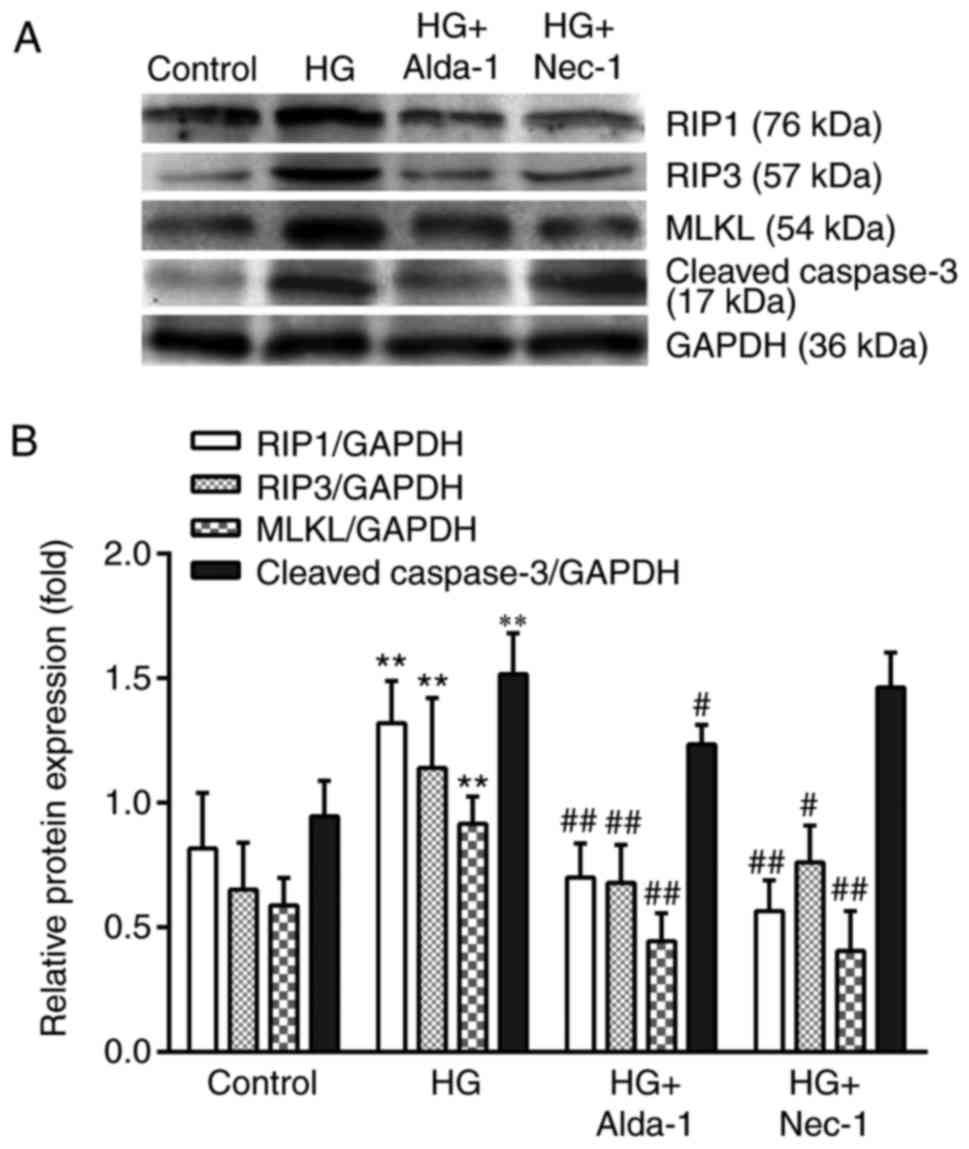
Alterations in RIP1, RIP3 and MLKL
mRNA and protein expression in different treatment groups
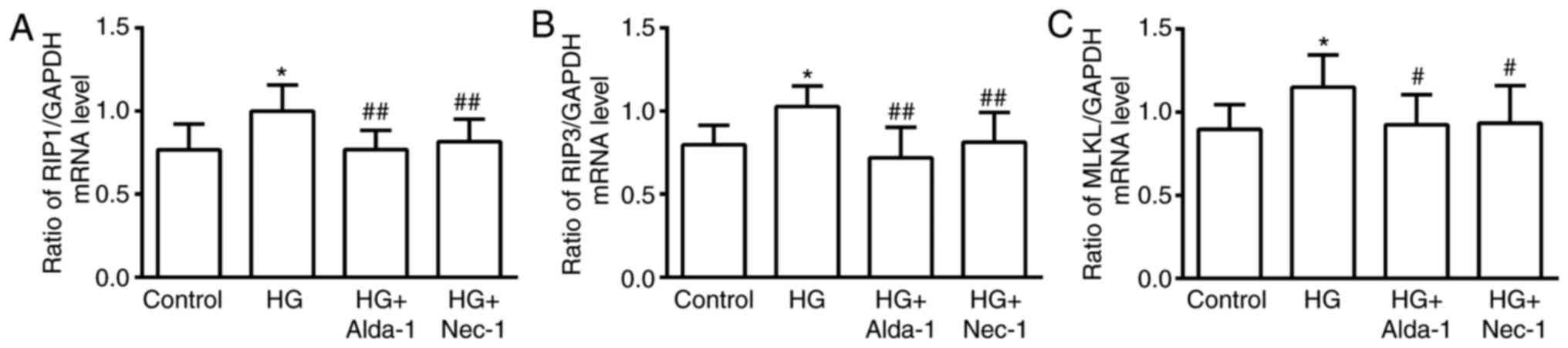
Compared with the control group, the expression of
RIP1, RIP3 and MLKL at the mRNA and protein levels was
significantly increased when the cells were subjected to HG
intervention (P<0.05). In the Alda-1+HG and Nec-1+HG groups, the
levels of RIP1, RIP3 and MLKL mRNA and protein expression were
significantly decreased compared with in the HG group (P<0.05;
Figs. 4 and 5). The results suggested that necroptosis
occurs in HG-induced cardiac cell injury. Furthermore, the
activation of ALDH2 could downregulate necroptosis, which was
similar to the apparent mechanism of inhibiting necroptosis via
Nec-1.
Alterations in the expression of
cleaved caspase-3 protein in different treatment groups
Compared with the control group, the expression of
cleaved caspase-3 protein was increased in the HG group. Compared
with the HG group, the expression of cleaved-caspase 3 protein was
significantly decreased in the Alda-1+HG group (P<0.05).
However, there was no difference between the HG group and the
Nec-1+HG group (Fig. 5). The
results suggested that the activation of ALDH2 could attenuate both
the occurrence of apoptosis and necroptosis, while the inhibition
of necroptosis by Nec-1 only attenuated necroptosis without having
an effect on apoptosis (Fig.
6).
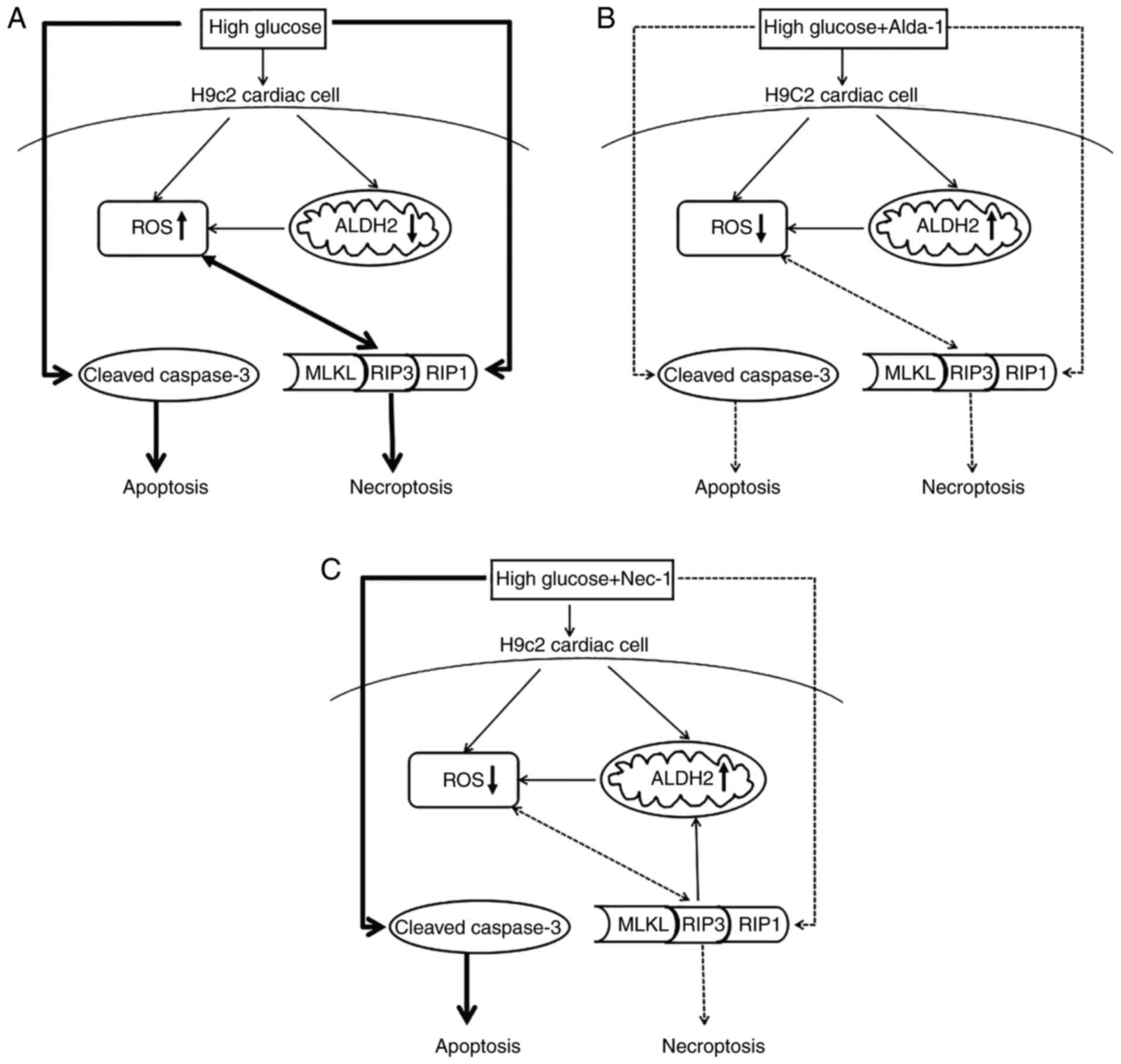 | Figure 6.Possible mechanisms of the protective
effects of activation of ALDH2 and inhibition of necroptosis in
high glucose induced H9c2 cardiac cells injury. (A) Under high
glucose conditions, ROS were over-released, the level of ALDH2 was
downregulated and apoptosis, and necroptosis pathways were both
activated. (B) With the presence of Alda-1 (the activator of
ALDH2), accompanied by the increase of ALDH2, the overproduction of
ROS was inhibited and apoptosis, and necroptosis pathways were
inhibited. (C) When the necroptosis pathway was inhibited by Nec-1,
the inhibitor of RIP1, accompanied by the inhibition of
necroptosis, the overproduction of ROS was blocked, ALDH2 was
increased but the apoptosis pathway was not affected. The solid
black lines and arrowheads indicate activation; the dashed black
lines and arrowheads indicate inhibition. MLKL, mixed lineage
kinase domain like pseudokinase; RIP, receptor-interacting protein
kinase 1; ROS, reactive oxygen species; ALDH2, aldehyde
dehydrogenase 2; Nec-1, necrostatin-1. |
Discussion
In the present study, it was first observed that
when H9c2 cardiac cells were subjected to HG stress, the activity
and expression of ALDH2 were downregulated, which was accompanied
by increases in the expression of key mediators of necroptosis
(RIP1, RIP3 and MLKL). This suggested that hyperglycemia induced
the occurrence of necroptosis in a model of HG-induced H9c2 cardiac
cell injury. Then, it was observed that the necroptosis inhibitor,
Nec-1 and the ALDH2 activator, Alda-1, increased the activity and
expression of ALDH2 and inhibited necroptosis. This result
suggested that the activation of ALDH2 could protect cardiac cells
by inhibiting RIP1/RIP3/MLKL-mediated necroptosis.
Recently, it has been reported that necroptosis
(also named programmed necrosis) is one form of regulated cell
death, which occurs in a number of diseases. Necroptosis is
mediated by RIP1, RIP3 and MLKL, which forms the RIP1-RIP3-MLKL
complex termed, the ‘necrosome’. This complex is considered an
important mediator of the necroptotic pathway (11–13).
ROS are small and highly reactive molecules with
important signaling transduction functions when maintained at
proper cellular concentrations. ROS levels are increased when the
body is in a pathological state and the accumulation of ROS can
elicit necroptosis as well as apoptosis (14–16).
ROS production is considered as an effector of necroptosis in
primary red blood cells, Jurkat T cells and U937 monocytes when
they are subjected to HG stress (17). ROS may also be considered as an
effector of necroptosis (18).
ALDH2 is the mitochondrial isoform of the ALDH superfamily and is
located in the mitochondrial matrix for removal of aldehyde
substrates (19). The
cardioprotective role of ALDH2 has been noted in epidemiological
and experimental studies, ALDH2 mutation reduces ALDH2 activity and
is associated with cardiovascular, neurological, and digestive
complications (19,20). The activation of ALDH2 can
attenuate diabetes-induced myocardial injury (7,8,21).
ALDH2 had been widely recognized as a protective factor during
organ injury triggered by different causes and one of its important
protective mechanisms is inhibition of the overproduction of ROS
(4). However, there are a limited
number of reports regarding ALDH2 and necroptosis. Therefore, the
present study aimed to observe the alterations in ROS, ALDH2 and
necroptosis, and to analyze the likely mechanisms that are involved
in HG-induced cardiac cell injury using H9c2 cells.
In the present study, it was observed that when H9c2
cardiac cells were subjected to HG stress, which was accompanied by
a decrease in cell viability and ROS production, ALDH2 activity was
decreased and the levels of ALDH2 mRNA and protein expression were
downregulated. Additionally, the levels of RIP1, RIP3 and MLKL mRNA
and protein expression were all increased. These results suggested
that hyperglycemia induced necroptosis in H9c2 cardiac cells, which
was accompanied by a decline in ALDH2 activity and expression, and
an increase in ROS production. These results suggest that
necroptosis and ALDH2 may be linked.
The inhibition of necroptosis can exert beneficial
effects both in vivo and ex vivo. Nec-1 is a specific
inhibitor of necroptosis that inhibits the interaction between RIP1
and RIP3, Nec-1 can mediate the protective effects in myocardial
and brain ischemic injury in adult rodent models (22–25).
Cardiac cells under HG stress were treated with Nec-1 in order to
verify that necroptosis occurred under hyperglycemic conditions and
to determine whether inhibiting necroptosis can protect cardiac
cells. As a result, the expression of RIP1, RIP3, MLKL mRNA and
protein were decreased; by contrast, cell viability and the
activity and expression of ALDH2 were increased, and oxidative
stress was also inhibited. These results indicated that under
hyperglycemic conditions, the inhibition of necroptosis via the
activation of ALDH2 and downregulation of ROS production could
mediate a cardiac-protective effect.
As ALDH2 participates in the protective effect of
Nec-1, it is interesting to speculate on a potential association
between ALDH2 and necroptosis. In a study regarding ALDH2 and
necroptosis, Shen et al (26) reported that ALDH2 deficiency may
lead to unexpected cardiac dysfunctions by enhancing myocardial
apoptosis and necroptosis in a mouse model. In the present study,
it was observed that ALDH2 activity and expression were increased
when necroptosis was inhibited. On the basis of the previously
reported studies (7–9,26)
and the present study, whether ALDH2 is a regulatory factor in
necroptosis was further questioned. Therefore, a specific ALDH2
agonist, Alda-1 was selected to treat H9c2 cardiac cells under HG
conditions. The results indicated that with an increase in ALDH2
activity and expression, there was a decrease of the release of
ROS, and the levels of RIP1, RIP3 and MLKL mRNA and protein
expression were all decreased. This result suggested that
inhibiting necroptosis was also involved in the protective
mechanism of ALDH2 activation.
In the present study, another type of programmed
cell death, apoptosis, was observed to be happening in HG induced
cell injury. The results demonstrated Alda-1 but not Nec-1 could
inhibit the protein expression of cleaved caspase-3, a terminal
effector of apoptosis. The results were in accordance with the
study by Liu et al (27),
where they reported that Nec-1 did not alter the expression of
caspase-3 following spinal cord injury in adult mice. However,
there are different viewpoints. Chang et al (28) reported that in a mouse
intracerebral hemorrhage model, Nec-1 not only suppressed
necroptosis but also suppressed apoptosis and autophagy. Therefore,
there may be cross-talk between necroptosis, apoptosis and
autophagy following mouse intracerebral hemorrhage. Liu et
al (29) indicated that the
inhibition of RIP3 may serve as a therapeutic strategy for
traumatic brain injury by suppressing inflammation, oxidative
stress and apoptosis in a mouse model. Whether altering the levels
of other proteins, including inhibiting RIP3 or MLKL, can exhibit
an effect on apoptosis in HG-induced cardiac cell injury requires
further investigation to analyze the connections of different cell
death pathways.
In conclusion, necroptosis occurred in HG-induced
H9c2 cardiac cell injury, suggesting that inhibiting necroptosis
could have a cardiac-protective role. Furthermore, the activation
of ALDH2 could improve cell injury by inhibiting necroptosis and
apoptosis. The present study offers further insights into the role
of ALDH2 in diseases. Inhibiting necroptosis may be a novel
therapeutic tool for the prevention and treatment of
diabetes-induced complications.
Acknowledgements
Not applicable.
Funding
The present study was supported by research grants
from the Natural Science Foundation of China (grant no. 81770297),
the Anhui Province University Top-Notch Talent Project (grant no.
gxbjZD18), the Anhui Province Education Key Project (grant no.
KJ2017A221) and the Bengbu Medical College Graduate Students
Innovation Project (grant nos. Byycx1604 and Byycx1704).
Availability of data and materials
All data used and/or analyzed during this study are
included in this published article.
Authors' contributions
TF, RC and QG designed the study, performed the
experiments including cell culture, RT-qPCR, western blotting and
draft the manuscript. WW and HY performed the experiments including
CCK8 assay, ALDH2 activity measurement and collected the
experimental data. LS performed the DHE staining measurement, ZL
and JH contributed to do the statistical analysis and interpret the
experimental data. All authors revised the manuscript critically
for important intellectual content and given the final approval of
the version to be published. All authors read and approved the
manuscript.
Ethics approval and consent to
participate
All H9c2 cardiac cells experiments were approved by
the Institutional Animal Care and Use Committee of Bengbu Medical
College of China (Bengbu, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Arora D, Sharma PK, Siddiqui MH and Shukla
Y: Necroptosis: Modules and molecular switches with therapeutic
implications. Biochimie. 137:35–45. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weinlich R, Oberst A, Beere HM and Green
DR: Necroptosis in development, inflammation and disease. Nat Rev
Mol Cell Biol. 18:127–136. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liang W, Chen M, Zheng D, He J, Song M, Mo
L, Feng J and Lan J: A novel damage mechanism: Contribution of the
interaction between necroptosis and ROS to high glucose-induced
injury and inflammation in H9c2 cardiac cells. Int J Mol Med.
40:201–208. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liang W, Chen M, Zheng D, Li J, Song M,
Zhang W, Feng J and Lan J: The opening of ATP-sensitive K+ channels
protects H9c2 cardiac cells against the high glucose-induced injury
and inflammation by inhibiting the ROS-TLR4-necroptosis pathway.
Cell Physiol Biochem. 41:1020–1034. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luo XJ, Liu B, Ma QL and Peng J:
Mitochondrial aldehyde dehydrogenase, a potential drug target for
protection of heart and brain from ischemia/reperfusion injury.
Curr Drug Targets. 15:948–955. 2014.PubMed/NCBI
|
|
6
|
Matsumoto A, Thompson DC, Chen Y, Kitagawa
K and Vasiliou V: Roles of defective ALDH2 polymorphism on liver
protection and cancer development. Environ Health Prev Med.
21:395–402. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao Q, Wang HJ, Wang XM, Kang PF, Yu Y, Ye
HW, Zhou H and Li ZH: Activation of ALDH2 with ethanol attenuates
diabetes induced myocardial injury in rats. Food Chem Toxicol.
56:419–424. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kang PF, Wu WJ, Tang Y, Xuan L, Guan SD,
Tang B, Zhang H, Gao Q and Wang HJ: Activation of ALDH2 with low
concentration of ethanol attenuates myocardial ischemia/reperfusion
injury in diabetes rat model. Oxid Med Cell Longev.
2016:61905042016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guo Y, Yu W, Sun D, Wang J, Li C, Zhang R,
Babcock SA, Li Y, Liu M, Ma M, et al: A novel protective mechanism
for mitochondrial aldehyde dehydrogenase (ALDH2) in type i
diabetes-induced cardiac dysfunction: Role of AMPK-regulated
autophagy. Biochim Biophys Acta. 1852:319–331. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fuchs Y and Steller H: Live to die another
way: Modes of programmed cell death and the signals emanating from
dying cells. Nat Rev Mol Cell Biol. 16:329–344. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun L, Wang H, Wang Z, He S, Chen S, Liao
D, Wang L, Yan J, Liu W, Lei X and Wang X: Mixed lineage kinase
domain-like protein mediates necrosis signaling downstream of RIP3
kinase. Cell. 148:213–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu W, Liu P and Li J: Necroptosis: An
emerging form of programmed cell death. Crit Rev Oncol Hematol.
82:249–258. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cano M, Wang L, Wan J, Barnett BP,
Ebrahimi K, Qian J and Handa JT: Oxidative stress induces
mitochondrial dysfunction and a protective unfolded protein
response in RPE cells. Free Radic Biol Med. 69:1–14. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Osipov RM, Bianchi C, Feng J, Clements RT,
Liu Y, Robich MP, Glazer HP, Sodha NR and Sellke FW: Effect of
hypercholesterolemia on myocardial necrosis and apoptosis in the
setting of ischemia-reperfusion. Circulation. 120 11 Suppl:S22–S30.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ,
Lin SC, Dong MQ and Han J: RIP3, an energy metabolism regulator
that switches TNF-induced cell death from apoptosis to necrosis.
Science. 325:332–336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
LaRocca TJ, Sosunov SA, Shakerley NL, Ten
VS and Ratner AJ: Hyperglycemic conditions prime cells for
RIP1-dependent necroptosis. J Biol Chem. 291:13753–13761. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang X, Yousefi S and Simon HU:
Necroptosis and neutrophil-associated disorders. Cell Death Dis.
9:1112018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen CH, Joshi AU and Mochly-Rosen D: The
role of mitochondrial aldehyde dehydrogenase 2 (ALDH2) in
neuropathology and neurodegeneration. Acta Neurol Taiwan.
25:111–123. 2016.PubMed/NCBI
|
|
20
|
Lao-Sirieix P, Caldas C and Fitzgerald RC:
Genetic predisposition to gastro-oesophageal cancer. Curr Opin
Genet Dev. 20:210–217. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang J, Wang H, Hao P, Xue L, Wei S, Zhang
Y and Chen Y: Inhibition of aldehyde dehydrogenase 2 by oxidative
stress is associated with cardiac dysfunction in diabetic rats. Mol
Med. 17:172–179. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Adameova A, Hrdlicka J, Szobi A, Farkasova
V, Kopaskova K, Murarikova M, Neckar J, Kolar F, Ravingerova T and
Dhalla NS: Evidence of necroptosis in hearts subjected to various
forms of ischemic insults. Can J Physiol Pharmacol. 95:1163–1169.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vieira M, Fernandes J, Carreto L,
Anuncibay-Soto B, Santos M, Han J, Fernández-López A, Duarte CB,
Carvalho AL and Santos AE: Ischemic insults induce necroptotic cell
death in hippocampal neurons through the up-regulation of
endogenous RIP3. Neurobiol Dis. 68:26–36. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Oerlemans MI, Koudstaal S, Chamuleau SA,
de Kleijn DP, Doevendans PA and Sluijter JP: Targeting cell death
in the reperfused heart: Pharmacological approaches for
cardioprotection. Int J Cardiol. 165:410–422. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shen C, Wang C, Han S, Wang Z, Dong Z,
Zhao X, Wang P, Zhu H, Sun X, Ma X, et al: Aldehyde dehydrogenase 2
deficiency negates chronic low-to-moderate alcohol
consumption-induced cardioprotecion possibly via ROS-dependent
apoptosis and RIP1/RIP3/MLKL-mediated necroptosis. Biochim Biophys
Acta. 1863:1912–1918. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu M, Wu W, Li H, Li S, Huang LT, Yang
YQ, Sun Q, Wang CX, Yu Z and Hang CH: Necroptosis, a novel type of
programmed cell death, contributes to early neural cells damage
after spinal cord injury in adult mice. J Spinal Cord Med.
38:745–753. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chang P, Dong W, Zhang M, Wang Z, Wang Y,
Wang T, Gao Y, Meng H, Luo B, Luo C, et al: Anti-necroptosis
chemical necrostatin-1 can also suppress apoptotic and autophagic
pathway to exert neuroprotective effect in mice intracerebral
hemorrhage model. J Mol Neurosci. 52:242–249. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu ZM, Chen QX, Chen ZB, Tian DF, Li MC,
Wang JM, Wang L, Liu BH, Zhang SQ, Li F, et al: RIP3 deficiency
protects against traumatic brain injury (TBI) through suppressing
oxidative stress, inflammation and apoptosis: Dependent on AMPK
pathway. Biochem Biophys Res Commun. 499:112–119. 2018. View Article : Google Scholar : PubMed/NCBI
|