Introduction
Cerebral stroke is a leading cause of disability
worldwide, and it is estimated that ischemic stroke accounts for
approximately 85% of the case (1).
Moreover, hospitalizations for ischemic stroke have shown a
year-on-year increase among adolescents and young adults (aged 5–44
years) (2). The pathophysiological
processes of ischemic stroke, which trigger neuronal necrosis and
apoptosis, are complex and extensive. These include a cascade of
bioenergetics failure, loss of cellular ion homeostasis, increased
intracellular calcium-induced excitotoxicity, reactive oxygen
species (ROS)-mediated toxicity, activation of neuronal and glial
cells, cytokine-mediated cytotoxicity and disruption of the
blood-brain barrier (3).
Currently, intravenous recombinant tissue plasminogen activator
(r-TPA) to induce thrombolysis combined with a neuroprotective drug
to rescue dying neurons is the common clinical strategy for acute
ischemic stroke therapy (3).
Ischemic stroke triggers multiple and overlapping
cell signaling pathways that may contribute to cell damage or cell
survival. The mitogen-activated protein kinases (MAPKs), which
control a broad spectrum of cellular processes including apoptosis,
growth, inflammation and stress responses, are important modulators
of a variety of diseases. There are also increasing evidences that
MAPKs are crucial regulators of hemorrhagic and ischemic cerebral
disease, furthermore, raising the possibility that MAPKs may be a
drug discovery target for ischemic stroke (4,5). P38
and JNK are two of the main members of the MAPKs signaling group.
Thus, emerging evidences suggest that activation of p38 and JNK may
play an important role in ischemia-induced neuronal apoptosis. The
apoptosis is triggered by the enhanced pro-apoptotic activity of
p53 and phosphorylation of the c-Jun regulated by p38 and JNK
activities (6).
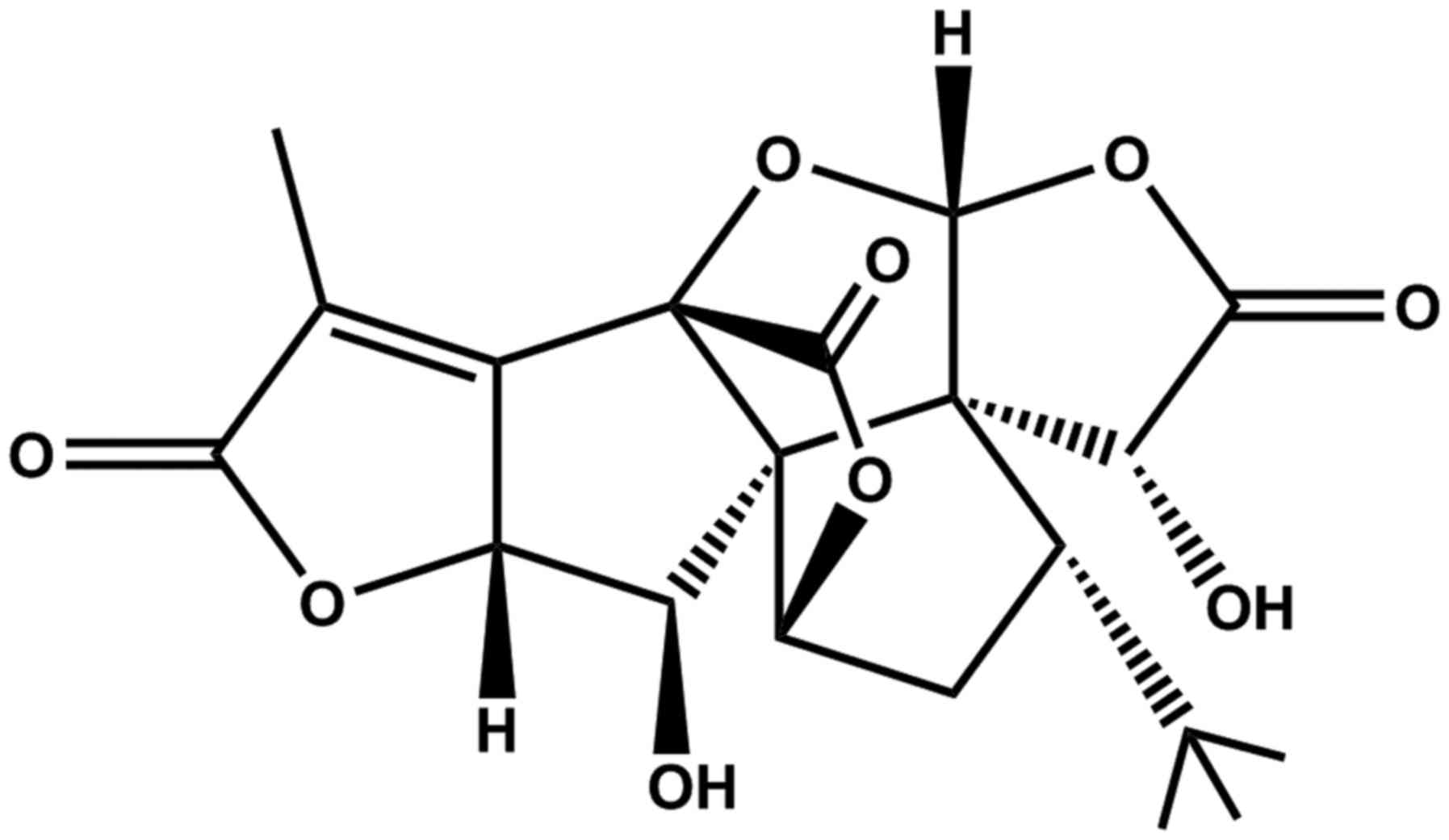
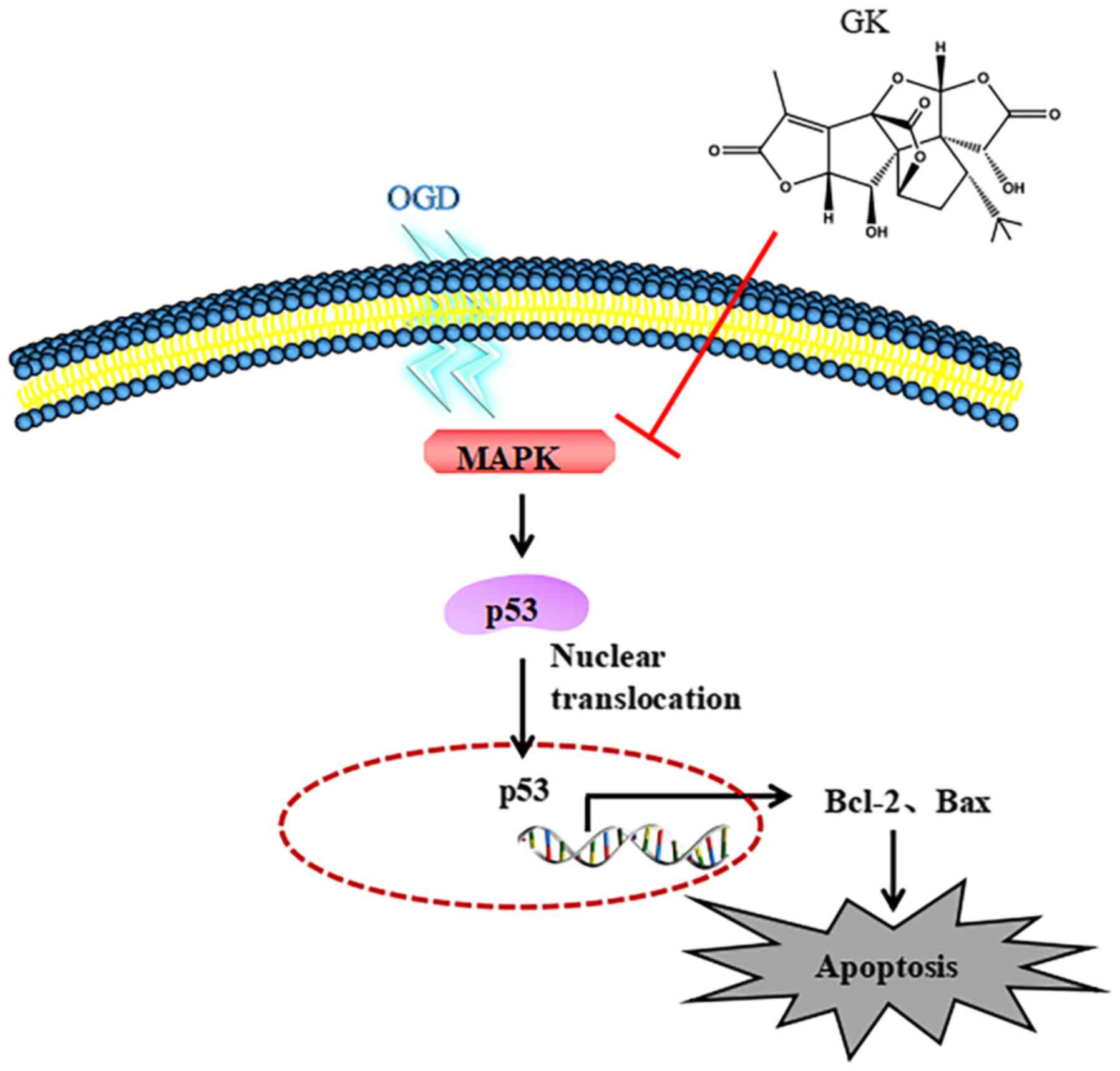
Ginkgolide K (GK:
C20H22O9 as shown in Fig. 1) is a diterpene lactone compound
isolated from the leaves of Ginkgo biloba which has a long
history of therapeutic application as a natural medicine for
cardiovascular diseases in humans (7). Recently, GK has been reported to
protect the heart against ER stress injury by activating the
IRE1α/XBP1 pathway (8), and also
markedly protect PC12 cells against
H2O2-induced cytotoxicity by ameliorating
oxidative stress and mitochondrial dysfunction (9). Oxygen-glucose deprivation (OGD) is
widely used as an in vitro model for stroke due to its
similarities with the in vivo models of brain ischemia, and
it is a simple and highly useful technique, not only for the
elucidation of the role of key cellular and molecular mechanisms,
but also for the development of novel neuroprotective strategies.
SH-SY5Y cells exposed to OGD constitute a classical model used to
mimic cerebral ischemic injury. In the present study, the
neuroprotective effect and functional mechanism of GK on cerebral
ischemia were further confirmed by OGD-stimulated SH-SY5Y cells
in vitro.
Materials and methods
GK was extracted and separated by Jiangsu Kanion
Modern Traditional Chinese Medicine Research Institute with 98%
purity. SH-SY5Y cells were purchased from Cell Bank of the Chinese
Academy of Sciences (no. CRL-2266) which is imported from the ATCC
(Shanghai, China). Fetal bovine serum (FBS) and RPMI-1640 medium
were obtained from Gibco; Thermo Fisher Scientific, Inc. (Waltham,
MA, USA). The Cell Counting Kit-8 (CCK-8) was obtained from Bestbio
Biotechnology (Shanghai, China). The ROS assay kit was purchased
from Beyotime Institute of Biotechnology (Shanghai, China). Rabbit
antibodies against p38, p-p38 (Thr180/Tyr182), JNK, p-JNK
(Thr183/Tyr185), p53, p-p53 (Ser15), c-Jun, p-c-Jun (Ser73), Bcl-2,
cleaved caspase-3, caspase-3, tubulin, actin and the secondary
antibody were obtained from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Rabbit antibodies against Bax and cleaved
caspase-9 were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). PVDF membrane and ECL western detection reagent
were obtained from Bio-Rad Laboratory (Hercules, CA, USA). All
other reagents were purchased from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany) unless otherwise stated.
Cell viability assay
SH-SY5Y cells were cultured in RPMI-1640 medium
supplemented with 10% FBS in a 5% CO2, 37°C incubator.
The SH-SY5Y cells of logarithmic growth were seeded in 96-well
plates (2×104 cells/well) and cultured overnight. For
OGD and reoxygenation model, the culture medium of SH-SY5Y cells
was first replaced with RPMI-1640 medium containing no glucose, and
then the plates were placed in a hypoxia chamber aerated with 95%
N2 and 5% CO2 for 4 h in a 37°C incubator.
Afterwards, the plates were transferred to the 5% CO2,
37°C incubator with reoxygenation for 1 h.
After OGD 4 h, SH-SY5Y cells treated with GK at a
dose of 25 µg/ml were cultured with reoxygenation for different
times (1, 2, 4 and 6 h). The CCK-8 assay, a sensitive colorimetric
assay for determination of the number of viable cells, was used in
the cell proliferation and cytotoxicity analysis. WST-8 (10 µl) was
added to each well, and then the cells were cultured for an
additional 2 h to allow for the reaction of WST-8. Furthermore,
WST-8 is reduced by dehydrogenases in cells to give a
yellow-colored product (formazan), which is soluble in the tissue
culture medium. The amount of the formazan dye generated by the
activity of dehydrogenases in cells is directly proportional to the
number of living cells. Finally, the absorbance at 450 nm was
measured using a microplate reader. OD450 nm values were converted
to a percentage and all groups were compared to the control group
(100%).
In addition, SH-SY5Y cells were treated with
different concentrations of GK (12.5, 25 and 50 µg/ml) followed by
reoxygenation for 1 and 24 h respectively. Relative cell viability
was also measured by CCK-8 assay.
Nuclear staining by Hoechst 33258
To observe the nuclear changes occurring during OGD,
the chromatin-specific dye, Hoechst 33258, was used to stain the
nuclei. After treatment with different concentrations of GK (12.5,
25 and 50 µg/ml) and reoxygenation for 1 h, cells were washed with
PBS and fixed in 4% paraformaldehyde at 4°C overnight, and then
cells were permeabilized with 0.3% Triton X-100 at room temperature
for 30 min. The cells were subsequently incubated with 10 ng/ml
Hoechst 33258 in the dark for 10 min. After two additional rinses
with PBS, cells were photographed under a fluorescent microscope at
350 and 460 nm (Leica Microsystems GmbH, Wetzlar, Germany) with
×200 magnification.
Detection of ROS and mitochondrial
membrane potential
After exposure to OGD for 4 h, followed by treatment
with different concentrations of GK (12.5, 25 and 50 µg/ml) and
reoxygenation for 1 h, cells were incubated with 10 mM DCFHDA in
the dark at 37°C for 20 min, and washed twice with PBS. Then the
fluorescence intensity of DCF was measured with a microplate
reader.
Additionally, GK-treated cells were incubated with 1
µM rhodamine-123 in the dark at 37°C for 20 min. After two
additional rinses with PBS, rhodamine-123 intensity was determined
by flow cytometry. Cells with reduced fluorescence (less
rhodamine-123) were counted as the collapse of mitochondrial
membrane potential.
Western blot analysis
After exposure to OGD for 4 h, SH-SY5Y cells
(5×106/dish of 100-mm2 size) treated with GK
and reoxygenation for 1 h were collected on ice, and optimal cell
lysis solution was added to completely release the proteins for 2
h. The supernatants were collected after centrifuging at 14,000 × g
at 4°C for 10 min, and then protein concentrations were assayed
with a BCA kit. After SDS-PAGE electrophoresis, proteins were
transferred to a PVDF membrane. The transferred membranes were
blocked with 5% nonfat milk for 2 h at room temperature, and
incubated with primary antibodies at 4°C overnight. After three
rinses with TBST, the membranes were incubated with secondary
antibodies for 1 h at room temperature. Finally, after three
additional rinses with TBST, the immune complexes were detected
using ECL western detection reagents and photographed with
ChemiDoc™ XRS+ software to calculate gray value statistics.
Statistical analysis
All data are presented as the mean ± SD. Data were
analyzed with one-way ANOVA analysis followed by Tukey's post hoc
test using GraphPad Prism software version 5.0 (GraphPad Software,
Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
GK increases OGD-damaged SH-SY5Y cells
viability
We first examined the effect of GK on the
proliferation of SH-SY5Y cells. The CCK-8 assay showed that GK did
not affect the viability of SH-SY5Y cells at concentrations of
12.5, 25 and 50 µg/ml (Fig.
2A).
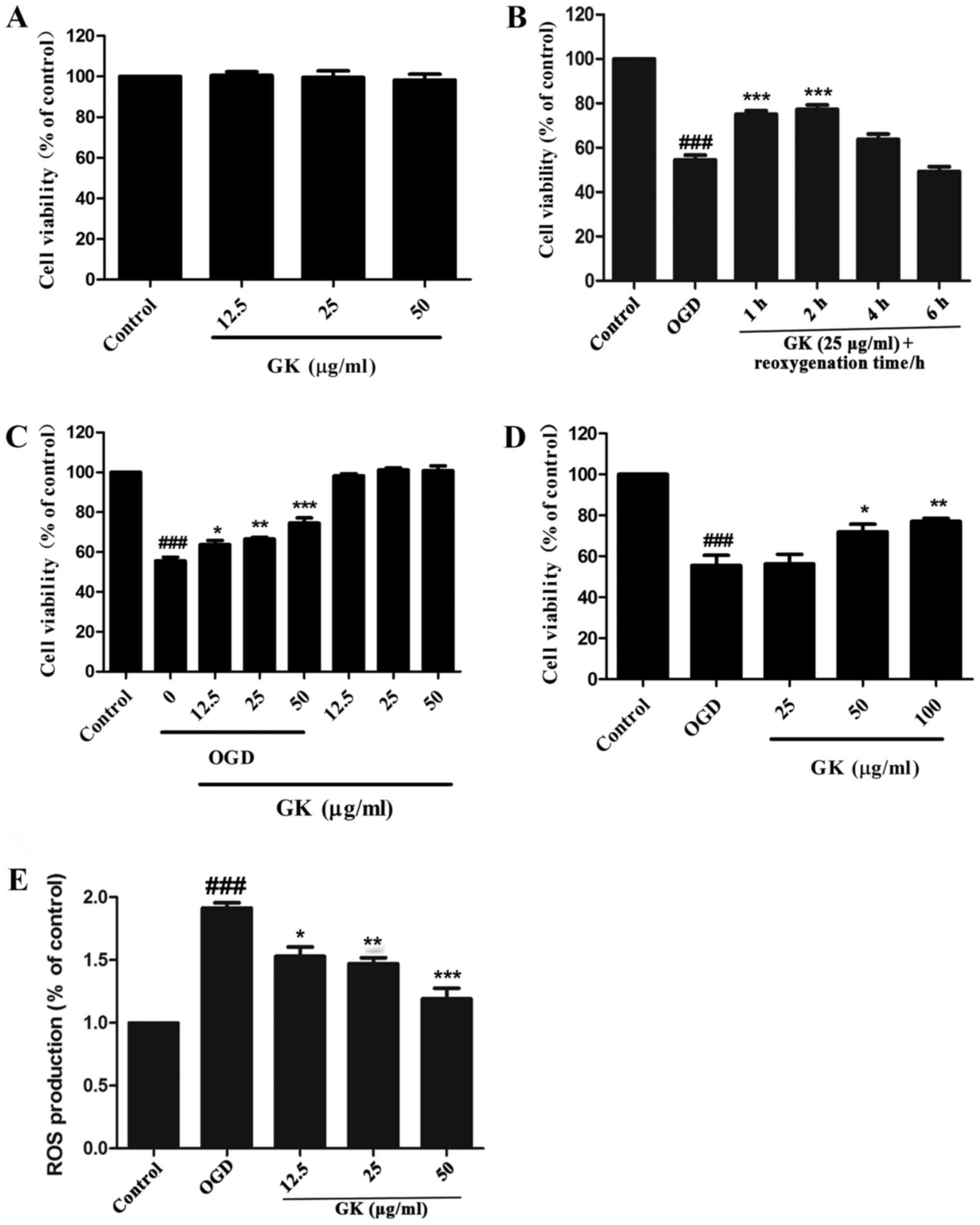 | Figure 2.GK increased cell viability and ROS
generation of SH-SY5Y cells damaged by OGD. (A) Cells were treated
with 12.5, 25 and 50 µg/ml of GK for 24 h and cell viability was
measured with the CCK-8 assay. (B) After 4 h OGD, the SH-SY5Y cells
were treated with GK and reoxygenation with various times (1, 2, 4
and 6 h). Cell viability was assessed by CCK-8 assay. (C) After 4 h
OGD, the SH-SY5Y cells were treated with GK at different
concentrations (12.5, 25 and 50 µg/ml) and reoxygenation for 1 h.
Cell viability was assessed by CCK-8 assay. (D) After 4 h OGD, the
SH-SY5Y cells were treated with GK at different concentrations (25,
50 and 100 µg/ml) and reoxygenation for 24 h. Cell viability was
assessed by CCK-8 assay. (E) ROS generation was quantified by
DCFHDA assay at excitation 488 nm and emission 525 nm. Data are
represented as mean ± SD from six experiments.
###P<0.001 vs. control group. *P<0.05, **P<0.01
and ***P<0.001 vs. OGD group. GK, ginkgolide K; ROS, reactive
oxygen species; CCK-8, Cell Counting Kit-8; OGD, oxygen-glucose
deprivation. |
After exposure to OGD for 4 h, SH-SY5Y cells were
treated with a moderate dose of GK (25 µg/ml) and reoxygenation for
different durations (1, 2, 4 and 6 h). To evaluate the effect of
GK, cells viability was measured using the CCK-8 method. The
results showed that cell viability significantly decreased after
the exposure to OGD. However, GK treatment at a dose of 25 µg/ml
for 1 and 2 h significantly increased the cells viability
respectively (Fig. 2B). While the
cell viability decreased after reoxygenation and GK treatment for 4
and 6 h, this was considered to be caused by increased damage due
to prolonged glucose deprivation. Considering the time-dependent
nature of this effect, we selected reoxygenation and GK treatment
for 1 h as the optimal duration in the subsequent experiments.
Next, we examined the effects of GK treatment at
different concentrations (12.5, 25 and 50 µg/ml). The assay
demonstrated that GK significantly increased the cell viability in
a dose-dependent manner (Fig. 2C).
In general, GK plays a neuroprotective role in OGD-damaged SH-SY5Y
cells, in a dose-dependent manner. We also assayed reoxygenation
after OGD stimulation and GK treatment at different concentrations
(25, 50 and 100 µg/ml) for 24 h to verify whether GK has protective
effect on cell damage or not. As predicted, GK significantly
suppressed cell death following OGD for 4 h and reoxygenation for
24 h, although 25 µg/ml of GK had no marked effect (Fig. 2D).
GK decreases the intracellular ROS
content
The intracellular ROS levels in the presence of
oxidative stress induced by OGD were measured by a DCFHDA assay.
The results demonstrated that treatment with GK at concentrations
of (12.5, 25 and 50 µg/ml) for 1 h had significantly decreased the
ROS levels by 1.56±0.07, 1.45±0.07 and 1.24±0.05% compared with
untreated SH-SY5Y cells, respectively (Fig. 2E).
GK protects OGD-induced SH-SY5Y cells
from apoptosis
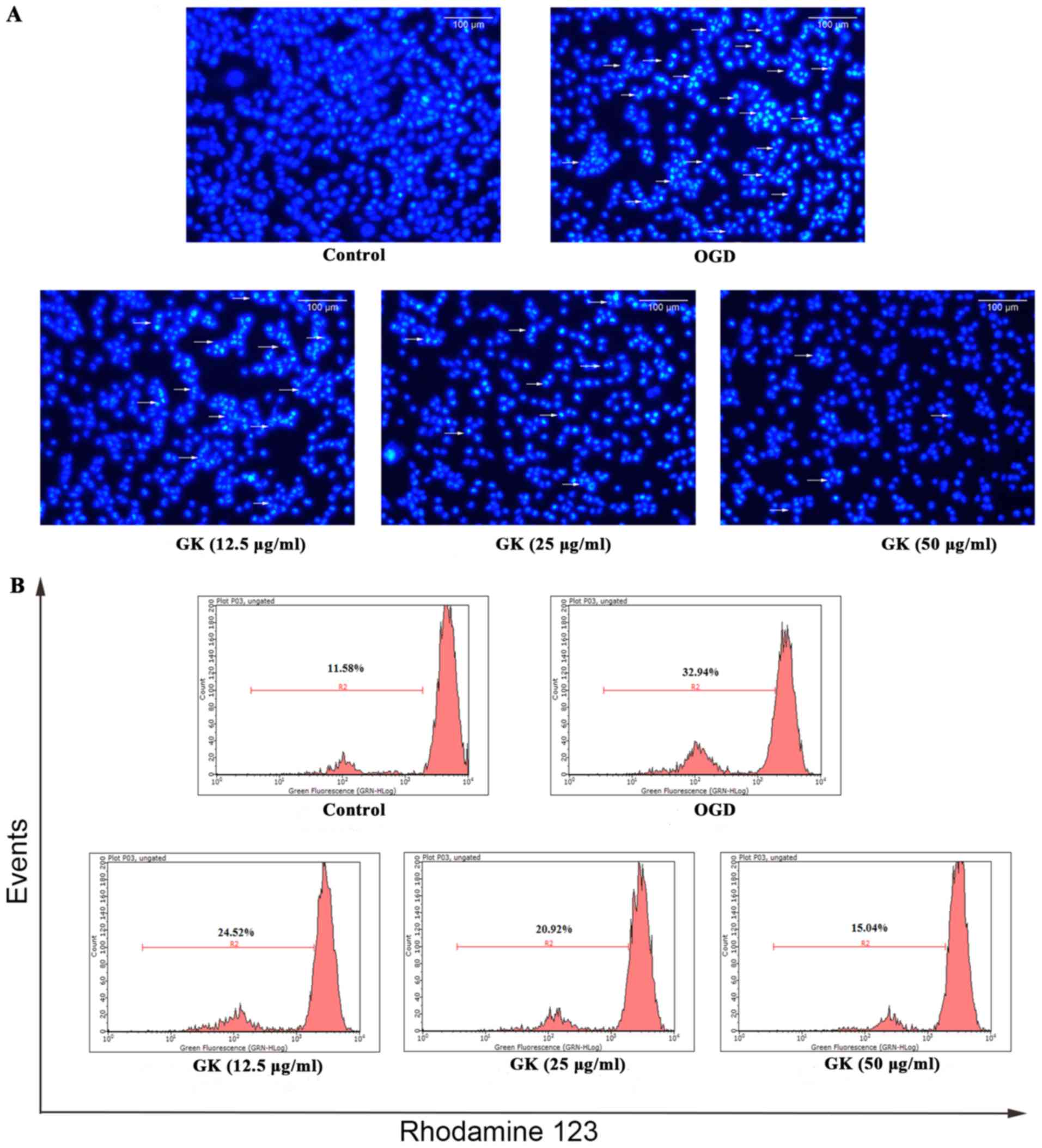
Hoechst-33258 staining (blue) was used to observe
the morphology of the nuclei, in order to demonstrate apoptosis in
OGD-induced SH-SY5Y cells. By comparison with the control group,
cell nuclear pyknosis, chromatin condensation, chromosome
fragmentation, the formation of apoptotic bodies, and other
apoptotic changes were observed in OGD-induced SH-SY5Y cells. By
contrast, we observed that GK treatment decreased appearance of
these morphological features, indicating the attenuation of
apoptosis by GK treatment (Fig.
3A).
In addition, we also observed the collapse of
mitochondrial membrane potential in OGD-induced SH-SY5Y cells with
rhodamine-123 staining. The value was measured by flow cytometry.
As shown in Fig. 3B, after
exposure to OGD for 4 h, the quantity of SH-SY5Y cells with
dissipation of mitochondrial membrane potential was increased from
11.58 to 32.94%. However, mitochondrial membrane potential was
dose-dependently decreased in SH-SY5Y cells treated with GK at
12.5, 25 and 50 µg/ml concentrations. Taken together, these data
suggest that GK inhibited the apoptosis of SH-SY5Y cells induced by
sustained OGD damage.
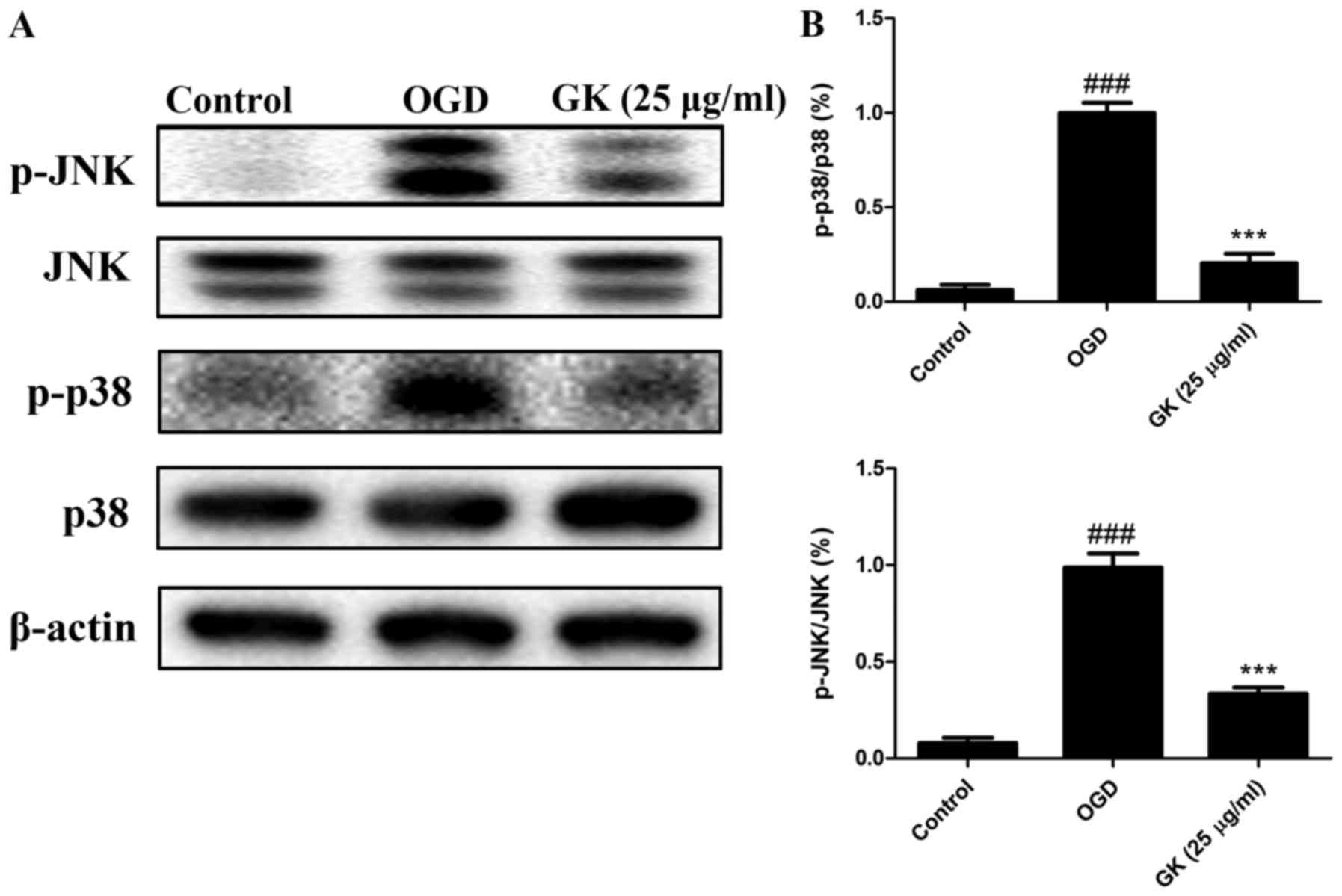
GK suppresses p38 and JNK activation
in OGD-induced SH-SY5Y cells
To investigate the mechanism through which GK
prevents cellular apoptosis in response to OGD, we next examined
the effects of GK on the p38 and JNK signaling via western blot.
The results showed that p-p38 (Thr180/Tyr182) and p-JNK
(Thr183/Tyr185) expressions were notably increased after OGD for 4
h (Fig. 4A). However, after
treatment with GK, p-p38 (Thr180/Tyr182) and p-JNK (Thr183/Tyr185)
proteins were significantly down-regulated compared with the
non-treated control (Fig. 4B),
suggesting that GK could suppress the p38 and JNK pro-apoptotic
signaling pathways to protect OGD-damaged SH-SY5Y cells.
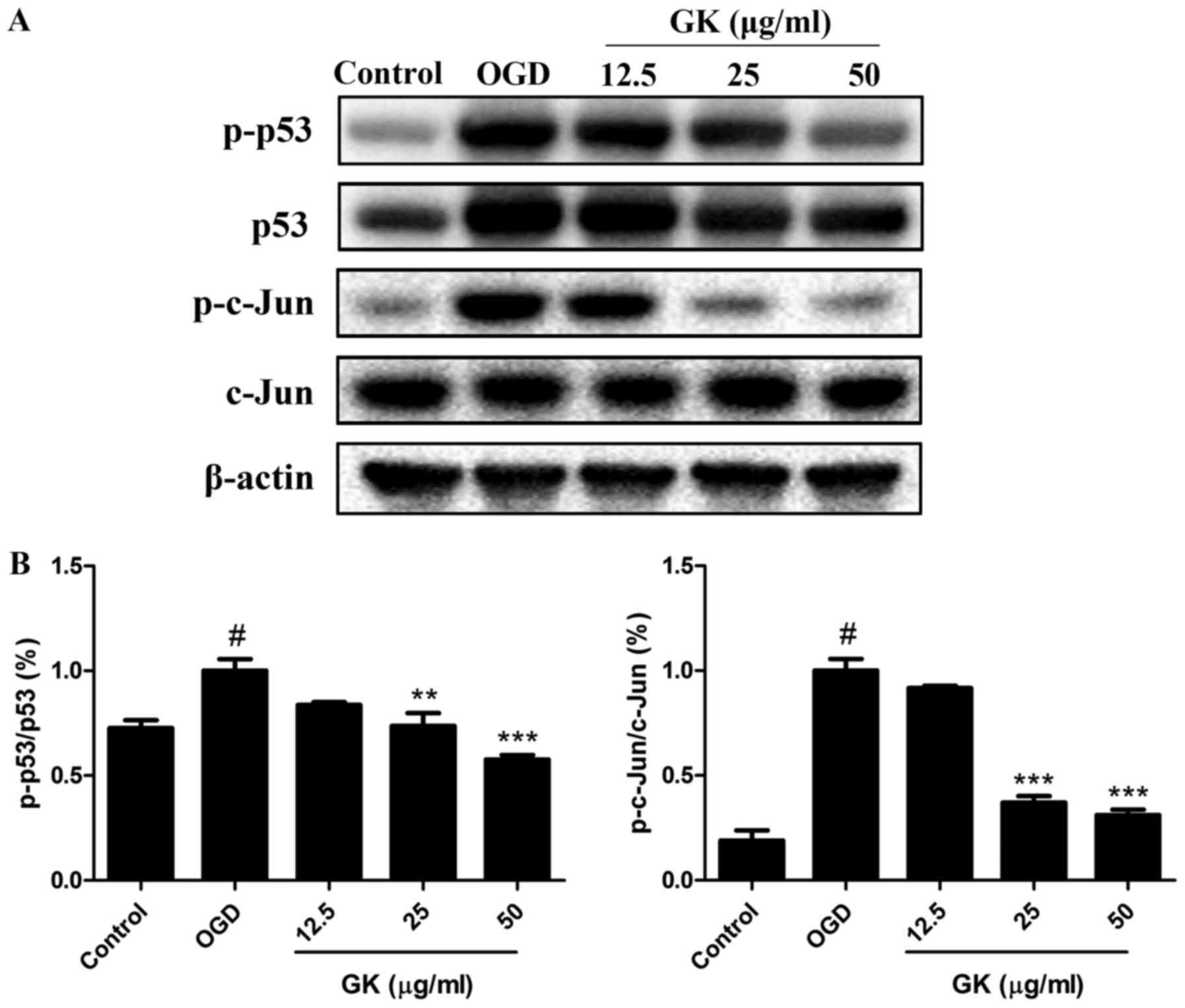
GK reduces p53 and c-Jun transcription
factor activation in OGD-induced SH-SY5Y cells
After activation by intracellular and extracellular
stimuli, JNK and p38 can directly enhance the pro-apoptotic
activity of p53 and the phosphorylation of the c-Jun to induce
apoptosis (10,11). Thus, the activities of p53 and
c-Jun were analyzed by western blotting. As shown in Fig. 5A, B, OGD treatment increased the
phosphorylation levels of p53 (ser15) and c-Jun (ser73) compared
with the control. However, by comparison with the OGD group, GK
treatment significantly decreased the levels of p-p53 (ser15) and
p-c-Jun (ser73) in a dose-dependent manner, indicating the
inhibition of p53 and c-Jun activities by GK in OGD-induced SH-SY5Y
cells.
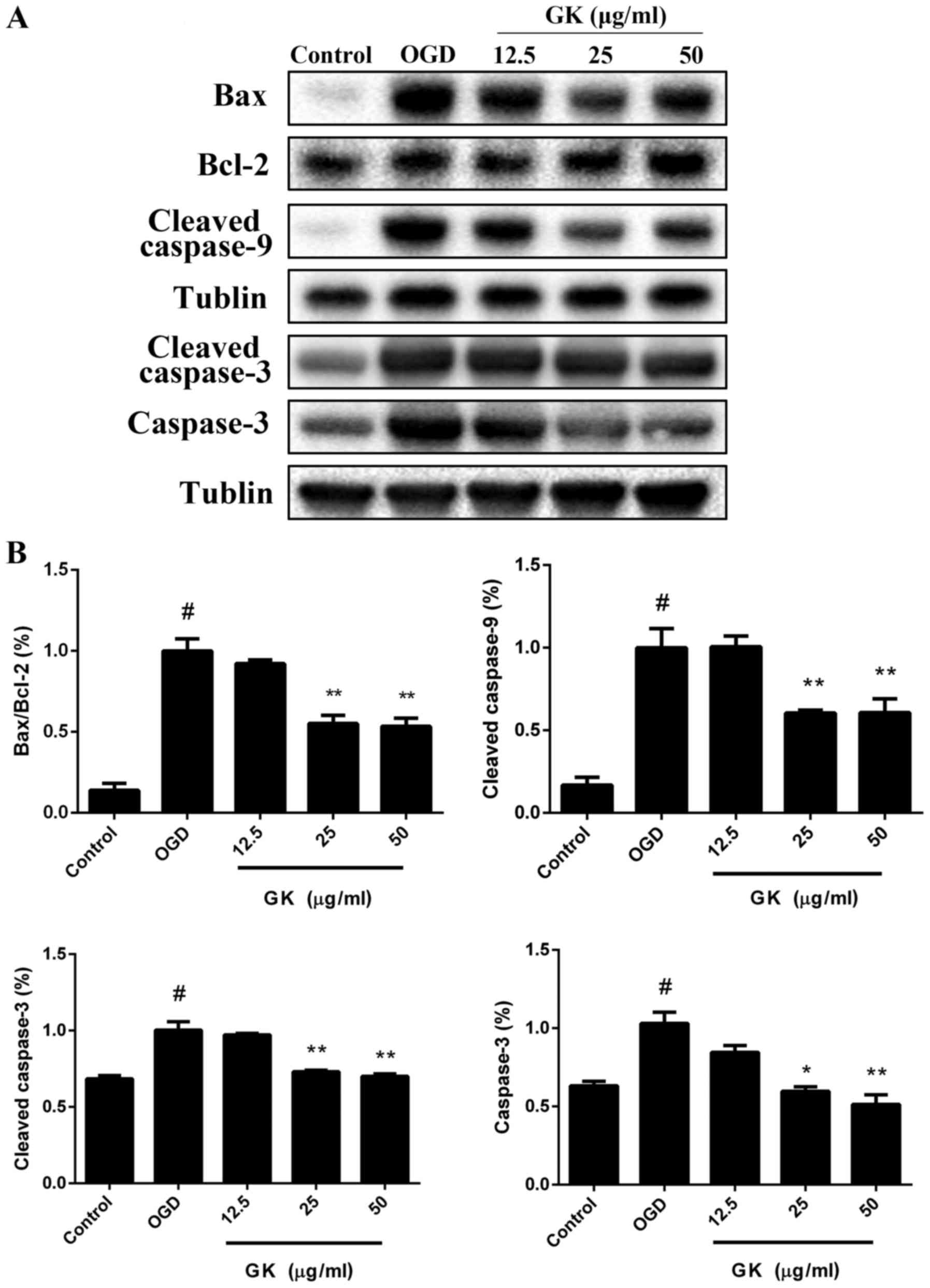
GK decreases the mitochondrial-related
Bax/Bcl-2 ratio to rescue caspase-dependent apoptosis in
OGD-induced SH-SY5Y cells
Activations of p53 and c-Jun immediately trigger the
expression of a number of apoptosis regulatory proteins, such as
Bax and Bad, but reverse the anti-apoptotic function of Bcl-2
(12). The Bcl-2 family proteins
are localized on the mitochondrial outer membrane and to initiate
mitochondria-mediated apoptosis. Therefore, we next examined the
effects of GK on the protein levels of the caspase and Bcl-2
families. Western blot analysis showed that treatment with GK
reduced the protein level of Bax and increased the level of Bcl-2,
thus decreasing the ratio of Bax/Bcl-2 following OGD-induced
apoptosis (Fig. 6A, B). Moreover,
cleaved caspase-9 and cleaved caspase-3 were reduced after
treatment with 12.5, 25 and 50 µg/ml GK compared with OGD group
(Fig. 6A and B). In addition, GK
also decreased the expression of total caspase-3 in a
dose-dependent manner. Collectively, these data demonstrate that GK
significantly repressed Bcl-2 family protein-regulated caspase
activity in OGD-induced SH-SY5Y cells.
Discussion
Ginkgo biloba extracts, especially
ginkgolides mainly including ginkgolide A, B and C have been
reported to possess potent protective properties by antagonizing
platelet activating factor (PAF), thereby inhibiting platelet
aggregation to protect against ischemic stroke (1,13,14).
In this study, we established that GK, a newly isolated compound in
ginkgolide family, protected SH-SY5Y cells against OGD-induced
apoptosis. The selective inhibition of the p38 and JNK pathways
play a crucial role in the neuroprotective effect of GK on cerebral
ischemia. These results indicated that GK conferred profound
neuroprotection in response to ischemic stroke.
The mitochondrial apoptotic pathway may play an
important role in neuronal cell death after cerebral ischemia. When
neuronal ischemic injury occurs, there are at least three factors
that induce mitochondrial pore channels: the overload of calcium
ions in the mitochondria, the oxidative damage to the mitochondrial
membrane and the decline of energy levels (6). After death stimuli, the permeability
of the mitochondria may increase, which causes the release of
Apaf-1, cytochrome c and procaspase-9 from the mitochondria to
cytosol. Subsequently, cytochrome c binds to Apaf-1 and leads to
the formation of cytochrome c/Apaf-1 multimeric complex.
Procaspase-9 gets recruited to the multimeric complex in a 1:1
ratio through the interaction between Apaf-1 and caspase-9. Thus,
the procaspase-9 molecules are activated by auto cleavage.
Moreover, capase-3 is activated by caspase-9 to trigger the further
downstream apoptotic processes (15–18).
In addition, the Bcl-2 family proteins play a crucial role in
regulating the mitochondrial permeability after cerebral ischemia
(19). The protein levels of Bax
and translocation from the cytosolic to the mitochondria have been
observed to increase after ischemic injury. Furthermore, Bax
promotes the release of procaspase-9 and the cytochrome c from the
mitochondria coincides to cytosolic through interacting with the
voltage-dependent anion channel and the mitochondrial adenine
nucleotide translocator (12). On
the other hand, the protein levels of Bcl-2 have been reported to
decrease in ischemic rats (20).
It was previously demonstrated that the anti-apoptotic effects of
Bcl-2 were accompanied by decreased cytochrome c release and
reduced activation of caspase-3 (21). In the present study, our results
demonstrated that GK exerted a dose-dependent inhibitory on Bcl-2
down-regulation, Bax up-regulation and decreased the caspase-9 and
caspase-3 activities in OGD-induced SH-SY5Y cells. These results
suggested that GK conferred a neuroprotective effect in the
simulated cerebral ischemia in vitro by inhibiting the
mitochondria-mediated death pathway.
P38 and JNK are two of the main members of the MAPKs
signaling group, which are crucial regulators of hemorrhagic and
ischemic cerebral disease. The activation of p38 can promote p53
phosphorylation at Ser15 residues to inhibit the ubiquitination and
degradation of the p53 (22,23).
Similarly, JNK phosphorylates c-Jun at Ser63 and Ser73 regions to
activate the pro-apoptotic effects of c-Jun (24,25).
Both activated p53 and c-Jun bind to the specific sites on the
promoters of the Bcl-2 family proteins, such as Bcl-2 and Bax, to
increase the Bax/Bcl-2 ratio (26). In this study, we observed the
decreases in the phosphorylation of p53 and c-Jun that may be due
to the down-regulation of p38 and JNK activity, as a result of
inhibiting the p38 and JNK pathways with GK treatment.
In summary, GK reduced the activities of p38 and
p-JNK, decreased the phosphorylation of p53 and c-Jun, inhibited
the mitochondria-mediated apoptosis pathway and protected against
OGD-induced apoptosis in SH-SY5Y cells (Fig. 7). Taking into account the above
results, GK may be a potential compound for rescuing neurons from
ischemic stroke-induced apoptosis, however, its underlying
molecular mechanisms must be further explored.
Acknowledgements
Not applicable.
Funding
This study was supported by the grants from National
Major Scientific and Technological Special Project for ‘Significant
New Drugs Development’ during the Twelfth Five-year Plan Period
(2013ZX09402203).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QL, XL, LL, ZX, JZ and WX designed the study. QL, XL
and LL performed the experiments. QL, XL, LL, ZX and JZ analyzed
data and drafted the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shu ZM, Shu XD, Li HQ, Sun Y, Shan H, Sun
XY, Du RH, Lu M, Xiao M, Ding JH and Hu G: Ginkgolide B protects
against ischemic stroke via modulating microglia polarization in
mice. CNS Neurosci Ther. 22:729–739. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Writing Group Members, . Mozaffarian D,
Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de
Ferranti S, Després JP, et al: Heart disease and stroke
statistics-2016 update: A report from the American heart
association. Circulation. 133:e38–e360. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fann DY, Lee SY, Manzanero S, Chunduri P,
Sobey CG and Arumugam TV: Pathogenesis of acute stroke and the role
of inflammasomes. Ageing Res Rev. 12:941–966. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun J and Nan G: The mitogen-activated
protein kinase (MAPK) signaling pathway as a discovery target in
stroke. J Mol Neurosci. 59:90–98. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fann DY, Lim YA, Cheng YL, Lok KZ,
Chunduri P, Baik SH, Drummond GR, Dheen ST, Sobey CG, Jo DG, et al:
Evidence that NF-κB and MAPK signaling promotes NLRP inflammasome
activation in neurons following ischemic stroke. Mol Neurobiol.
55:1082–1096. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nakka VP, Gusain A, Mehta SL and Raghubir
R: Molecular mechanisms of apoptosis in cerebral ischemia: Multiple
neuroprotective opportunities. Mol Neurobiol. 37:7–38. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu X, Yan Y, Bao L, Chen B, Zhao Y and Qi
R: Ginkgolide B inhibits platelet release by blocking Syk and p38
MAPK phosphorylation in thrombin-stimulated platelets. Thromb Res.
134:1066–1073. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang S, Wang Z, Fan Q, Guo J, Galli G, Du
G, Wang X and Xiao W: Ginkgolide K protects the heart against
endoplasmic reticulum stress injury by activating the
inositol-requiring enzyme 1α/X box-binding protein-1 pathway. Br J
Pharmacol. 173:2402–2418. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma S, Liu X, Xun Q and Zhang X:
Neuroprotective effect of Ginkgolide K against H2O2-induced PC12
cell cytotoxicity by ameliorating mitochondrial dysfunction and
oxidative stress. Biol Pharm Bull. 37:217–225. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao Y, Signore AP, Yin W, Cao G, Yin XM,
Sun F, Luo Y, Graham SH and Chen J: Neuroprotection against focal
ischemic brain injury by inhibition of c-Jun N-terminal kinase and
attenuation of the mitochondrial apoptosis-signaling pathway. J
Cereb Blood Flow Metab. 25:694–712. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cheng A, Chan SL, Milhavet O, Wang S and
Mattson MP: p38 MAP kinase mediates nitric oxide-induced apoptosis
of neural progenitor cells. J Biol Chem. 276:43320–43327. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cao G, Minami M, Pei W, Yan C, Chen D,
O'Horo C, Graham SH and Chen J: Intracellular Bax translocation
after transient cerebral ischemia: Implications for a role of the
mitochondrial apoptotic signaling pathway in ischemic neuronal
death. J Cereb Blood Flow Metab. 21:321–333. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bourgain RH, Andries R and Braquet P:
Effect of ginkgolide PAF-acether antagonists on arterial
thrombosis. Adv Prostaglandin Thromboxane Leukot Res. 17B:815–817.
1987.PubMed/NCBI
|
|
14
|
Yang ZZ, Li J, Li SX, Feng W and Wang H:
Effect of ginkgolide B on striatal extracellular amino acids in
middle cerebral artery occluded rats. J Ethnopharmacol.
136:117–122. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cain K, Brown DG, Langlais C and Cohen GM:
Caspase activation involves the formation of the aposome, a large
(approximately 700 kDa) caspase-activating complex. J Biol Chem.
274:22686–22692. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim HE, Du F, Fang M and Wang X: Formation
of apoptosome is initiated by cytochrome c-induced dATP hydrolysis
and subsequent nucleotide exchange on Apaf-1. Proc Natl Acad Sci
USA. 102:17545–17550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mouw G, Zechel JL, Zhou Y, Lust WD, Selman
WR and Ratcheson RA: Caspase-9 inhibition after focal cerebral
ischemia improves outcome following reversible focal ischemia.
Metab Brain Dis. 17:143–151. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yuan J and Yankner BA: Apoptosis in the
nervous system. Nature. 407:802–809. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sulejczak D, Czarkowska-Bauch J, Macias M
and Skup M: Bcl-2 and Bax proteins are increased in neocortical but
not in thalamic apoptosis following devascularizing lesion of the
cerebral cortex in the rat: An immunohistochemical study. Brain
Res. 1006:133–149. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Poppe M, Reimertz C, Düssmann H, Krohn AJ,
Luetjens CM, Böckelmann D, Nieminen AL, Kögel D and Prehn JH:
Dissipation of potassium and proton gradients inhibits
mitochondrial hyperpolarization and cytochrome c release during
neural apoptosis. J Neurosci. 21:4551–4563. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hara A, Iwai T, Niwa M, Uematsu T, Yoshimi
N, Tanaka T and Mori H: Immunohistochemical detection of Bax and
Bcl-2 proteins in gerbil hippocampus following transient forebrain
ischemia. Brain Res. 711:249–253. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gong X, Liu A, Ming X, Deng P and Jiang Y:
UV-induced interaction between p38 MAPK and p53 serves as a
molecular switch in determining cell fate. FEBS Lett.
584:4711–4716. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bogoyevitch MA, Ngoei KR, Zhao TT, Yeap YY
and Ng DC: c-Jun N-terminal kinase (JNK) signaling: Recent advances
and challenges. Biochim Biophys Acta. 1804:463–475. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Smeal T, Binetruy B, Mercola D,
Grover-Bardwick A, Heidecker G, Rapp UR and Karin M:
Oncoprotein-mediated signalling cascade stimulates c-Jun activity
by phosphorylation of serines 63 and 73. Mol Cell Biol.
12:3507–3513. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McGahan L, Hakim AM and Robertson GS:
Hippocampal Myc and p53 expression following transient global
ischemia. Brain Res Mol Brain Res. 56:133–145. 1998. View Article : Google Scholar : PubMed/NCBI
|