Introduction
With the aging population, the number of patients
with abdominal aortic aneurysm (AAA) is increasing yearly. The
incidence of AAA may be 2–8.9% in the general population, and as
high as 19.8% among men over the age of 75 (1). The pathological and physiological
processes of AAA have been a continuous focus of research. The
typical and common pathological alterations include: i)
Infiltration of inflammatory cells (including macrophages,
neutrophil granulocytes, T cells, B cells, mastocytes and natural
killer cells) in the tunica intima, media and externa; ii)
angiogenesis in the tunica media and apoptosis of vascular smooth
muscle cells (VSMCs); and iii) degradation of the extracellular
matrix, breakage of elastic fibers and loss of collagen. All of
these factors indicate that inflammation may be the common
mechanism in AAA formation, although the degree and type may vary
in different types of AAA. Inflammation exists throughout the
entire AAA process (2).
In particular, multiple inflammatory cytokines have
been identified in aortic aneurysms. There is evidence that Th1-
and Th2-type cytokines are involved in aneurysm formation (2). Schönbeck et al (3) identified that Th2-characteristic
cytokines, including interleukin (IL)-4, IL-5 and IL-10, were
predominantly expressed in the AAA tissue; however, IL-2 and IL-15,
as Th1-characteristic cytokines, were expressed at low levels.
However, other studies have demonstrated that Th1-associated
cytokines are required for aneurysm formation. For example, it was
observed that tumor necrosis factor (TNF)-α, IL-1β, IL-8 and C-C
motif chemokine 2 have upregulated expression in AAA tissues
(2). In animal models,
interferon-γ (IFN-γ) is required in the process of aneurysm
development involving increased matrix metalloproteinase (MMP)
expression (4). These differences
may be attributed to differences in the source of the control
tissue, patient demographics, lesion characteristics, the stage of
aneurysm formation, and tissue preservation or treatment conditions
(5). There is a hypothesis that
persistent inflammatory conditions initiate the promotion of
Th1-type inflammation, leading to atherosclerosis and fibrotic
lesions; and the secondary trigger, including smoking, reactive
oxygen species, autoimmune factors or genetic predisposition, may
lead to conversion to a Th2-type cytokine profile, causing matrix
degradation and ultimately, aneurysm formation (6).
Peroxisome proliferator-activated receptors (PPARs)
are members of the nuclear receptor superfamily. In total, three
types of PPARs have been discovered: PPAR-α, β and γ. They serve an
important role in regulating energy metabolism and the circadian
rhythm (7). PPARγ activators,
thiazolidinediones (TZDs), are used in pharmaceutical therapy for
diabetes. However, PPARγ has additionally been observed to be
involved in the shift in cytokine production and inflammation. TZDs
may reduce TNF-α expression in sepsis, ischemia/reperfusion,
gastric injury and spinal trauma models (8). PPARγ agonists may reduce the
expression of TNF-α and MMP-9 in the aortic aneurysmal wall and
retroperitoneal periaortic fat (9). The expression of other Th1-associated
cytokines, including IL-6 and IL-1β, is additionally downregulated
by PPARγ agonists in sepsis or ischemia/reperfusion models
(8). However, PPARγ affects the
expression of Th2-type inflammatory cytokines. Rosiglitazone (RGZ),
a PPARγ agonist, induces IL-10 expression in colitis and
Parkinson's disease models (10,11).
PPARγ has been demonstrated in certain studies to be
involved in AAA formation and development. TZDs reduced the maximum
diameter and rupture of the aorta in an Ang-II-induced experimental
aneurysm model of apolipoprotein E (ApoE)−/− mice
(7,12). However, in aneurysms of other parts
of the body, including cerebral aneurysm, pioglitazone was not able
to reduce the incidence of aneurysm (13).
The distribution of Th1 and Th2-type inflammation in
the process of aortic aneurysm formation and development has not
yet been confirmed, and one of the aims of the present study was to
demonstrate the spatial and temporal distributions of different
types of inflammatory cytokines. Therefore, TNF-α and IL-10 were
selected for examination, as they are typical examples of Th1- and
Th2-type cytokines. Additionally, the present study attempted to
confirm the effect of PPARγ on the incidence of AAA formation and
on the distribution of inflammation in the disease process.
Materials and methods
Animals and treatment
Male ApoE−/− mice (8–10 weeks old; weight
17–23 g; n=80) were obtained from the Beijing Vital River
Laboratory Animal Technology Co., Ltd. (Beijing, China), housed in
clean barrier conditions (22–25°C, 1 bar pressure, 12-h light/dark
cycle, and free accessing to water and food), and were administered
a standard laboratory diet in the Peking Union Medical College
Hospital (Beijing, China) animal house. The experiments in the
present study were approved by the Ethics Committee of Peking Union
Medical College Hospital. All of the 80 mice were divided into
eight groups (n=10 mice/group): i) Ang-II 7 days group; ii) Ang-II
14 days group; iii) Ang-II 21 days group; iv) Ang-II 28 days group;
v) Ang-II 42 days group; vi) Ang-II+RGZ 28 days group; vii)
Ang-II+RGZ 42 days group; and viii) saline control 42 days group.
Osmotic pumps (ALZET Osmotic Pumps, Cupertino, CA, USA) delivering
1,000 ng/kg/min Ang-II (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) for 7, 14, 21, 28 or 42 days were implanted subcutaneously
in the seven groups of mice. Ang-II was replaced by saline in
osmotic pumps in the saline control group. In groups 6 and 7,
intragastric administration of RGZ (3 mg/kg/day) was begun 7 days
prior to osmotic pump implantation and ended when the mice were
sacrificed. When it was time for tissue harvesting, the mice were
sacrificed. The tissues of the suprarenal aortas were removed and
prepared for further analysis. According to a previous study
(7), AAA was able to develop in 28
days in this animal model. For analysis, 7–21 days was defined as
the early stage, and 28–42 days as the late stage. For AAA rupture,
a rupture that occurred in the first 7 days following Ang-II
pumping was defined as an early rupture and the remainder as late
ruptures.
Histology
The maximum diameters of the suprarenal abdominal
aortas were measured, and the widest parts of suprarenal abdominal
aortas were fixed in 10% formalin for 24 h at room temperature. If
aneurysm formation (compared with the normal regions, dilation
≥50%) was observed, the aneurysm neck (defined as 0.5 cm of the
normal aorta above the aneurysm) was additionally reserved. The
fixed samples were embedded in paraffin, sectioned into 10-µm-thick
slices and then stained with hematoxylin and eosin to confirm the
formation of AAA. The hematoxylin and eosin staining was performed
according to standard protocols (14), and the stained slides were observed
using a DMI4000 B light microscope (Leica Microsystems, Inc.,
Buffalo Grove, IL, USA). Images were captured 50 times
(magnification, ×50).
Western blot analysis
A portion of the widest part of the suprarenal
abdominal aorta of each mouse was stored at −80°C, in addition to
the aneurysm neck if there was aneurysm formation. The aortic
proteins were subsequently extracted with protein lysis buffer
(Ukzybiotech., Ltd., Beijing, China; cat. no. P0001). The protein
concentration was standardized with a Bio-Rad protein assay
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Equal amounts (15
µg) of aortic extracts from mice in the different groups were
loaded onto a 15% SDS-PAGE gel and transferred to polyvinylidene
difluoride membranes. The membranes were blocked with 5% skim milk
in Tris-buffered saline with Tween 20 (0.01 mol/l Tris-HCl, pH 7.6,
0.15 mol/l NaCl, 0.05% Tween 20) for 1 h at room temperature. The
membranes were incubated with a mouse anti-TNF-α monoclonal
antibody (Ab; ProteinTech Group, Inc., Chicago, IL, USA; cat. no.
60291; 1:500) or rabbit anti-IL-10 polyclonal Ab (Lifespan
Biosciences, Inc., Seattle, WA, USA; cat. no. LS-C331959; 1:500)
overnight at 4°C. GAPDH was selected as the internal control (Cell
Signaling Technology, Inc., Danvers, MA, USA; cat. no. 5174;
1:1,000). The bound primary Ab was detected with horseradish
peroxidase-linked goat anti-mouse immunoglobulin G (IgG; Jackson
ImmunoResearch Laboratories, Inc., West Grove, PA, USA; cat. no.
115-035-003; 1:10,000) or anti-rabbit IgG (Jackson ImmunoResearch
Laboratories, Inc.; cat. no. 111-035-003; 1:10,000), respectively.
The secondary incubation was conducted at room temperature for 40
min. The enhanced chemiluminescent method (EMD Millipore,
Billerica, MA, USA; cat. no. WBKLS0500) was subsequently used to
develop the bands. The gray value of TNF-α or IL-10 was divided by
the gray value of the internal reference to calculate the relative
gray value. The gray value was calculated using Image J 1.50i
software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All of the data were analyzed using SPSS version
19.0.0 (IBM Corp., Armonk, NY, USA) and GraphPad Prism version 6.0
(GraphPad Software, Inc., La Jolla, CA, USA). The results are
presented as the mean ± standard error. Differences were analyzed
using Student's unpaired t-test or analysis of variance followed by
the Newman-Keuls multiple comparisons test. P<0.05 was
considered to indicate a statistically significant difference. All
experiments were repeated at least three times.
Results
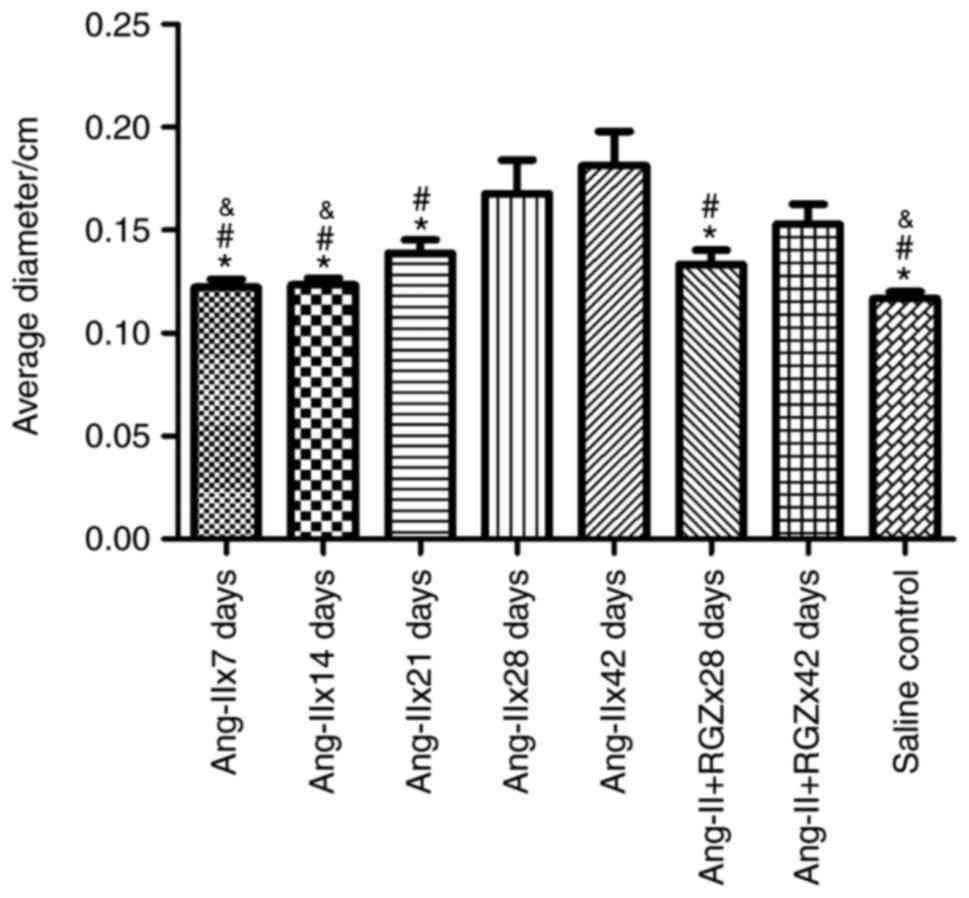
RGZ reduces the diameters of the
aortas in Ang-II-treated ApoE−/− mice
A mouse succumbed to pneumorrhagia during
intragastric gavage in the Ang-II+RGZx42 days group, and one mouse
in each of the Ang-IIx14 days, Ang-IIx42 days and saline control
groups succumbed to anesthetization. There was one case of the pump
falling out in the Ang-IIx7 days group. Compared with the saline
control group, the maximum diameters of the Ang-IIx7 days,
Ang-IIx14 days and Ang-IIx21 days groups were not significantly
different (P=0.676, 0.616 and 0.098, respectively). Treatment with
Ang-II resulted in dilation of the aortas in ApoE−/−
mice, primarily in the late-stage groups; the Ang-IIx28 days
(P<0.001) and Ang-IIx42 days groups (P<0.001). RGZ reduced
the maximum diameters of the aortas. In the 28 days group, RGZ
significantly prevented dilation of the aortas (Ang-IIx28 days,
0.17±0.05 cm vs. Ang-II+RGZx28 days, 0.13±0.02 cm; P=0.012); in the
42 days group, use of RGZ had a trend of reducing the maximum
diameters (Ang-IIx42 days, 0.18±0.05 cm vs. Ang-II+RGZx42 days,
0.15±0.03; P=0.055). When comparing the entire late stage, RGZ
significantly reduced the maximum diameters of the aortas in the
Ang-II-induced AAA model of ApoE−/− mice (without RGZ,
0.17±0.05 cm vs. with RGZ, 0.14±0.02 cm; P=0.021; Fig. 1 and Table I).
 | Table I.Aortic diameters and ruptures in
mice. |
Table I.
Aortic diameters and ruptures in
mice.
| Group | I | II | III | IV | V | VI | VII | VIII |
|---|
| Maximum diameter,
cm | 0.12±0.01 | 0.12±0.01 | 0.14±0.02 | 0.17±0.05 | 0.18±0.05 | 0.13±0.02 | 0.15±0.03 | 0.12±0.04 |
| Aneurysm formation,
no. cases | 0 | 0 | 1 | 3 | 5 | 1 | 2 | 0 |
| Early rupture, no.
cases | 0 | 0 | 1 | 1 | 1 | 1 | 2 | 0 |
| Late rupture, no.
cases | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 |
However, there was no aneurysm formation in the
control group or in the early stage, with the exception of one case
that was identified in the Ang-IIx21 days group. In the late stage,
aneurysm formation was significant compared with the control and
early stage (late stage 47.1% vs. control 0%; P=0.012; and vs.
early stage 3.7%; P=0.002; Table
I). Early rupture was not influenced by RGZ (Ang-II, 10.5% vs.
Ang-II+RGZ, 15.8%; P=0.631; Table
II). There was also no statistically significant difference for
aneurysm formation or rupture in the late stage of aneurysm
development between the groups with RGZ and without RGZ. The rates
of aneurysm formation in the late stage were 47.1 and 18.8% in
Ang-II without RGZ groups and Ang-II with RGZ groups, respectively
(P=0.141). Except for two mice that succumbed and one mouse that
had claudication in the Ang-IIx42 days group, no mice succumbed or
presented with lower extremity ischemia due to the late rupture of
aneurysms in the other groups. In the late stage, the incidences of
rupture were 17.6% and 0 in the groups without RGZ and with RGZ,
respectively (P=0.227).
| Table II.Contrast between the late stages
without or with RGZ. |
Table II.
Contrast between the late stages
without or with RGZ.
| Group | Late stage without
RGZ (%) | Late stage with RGZ
(%) | P-value |
|---|
| Aneurysm
formation | 47.1 | 18.8 | 0.141 |
| Early rupture | 10.5 | 15.8 | 0.631 |
| Late rupture | 17.6 |
0.0 | 0.227 |
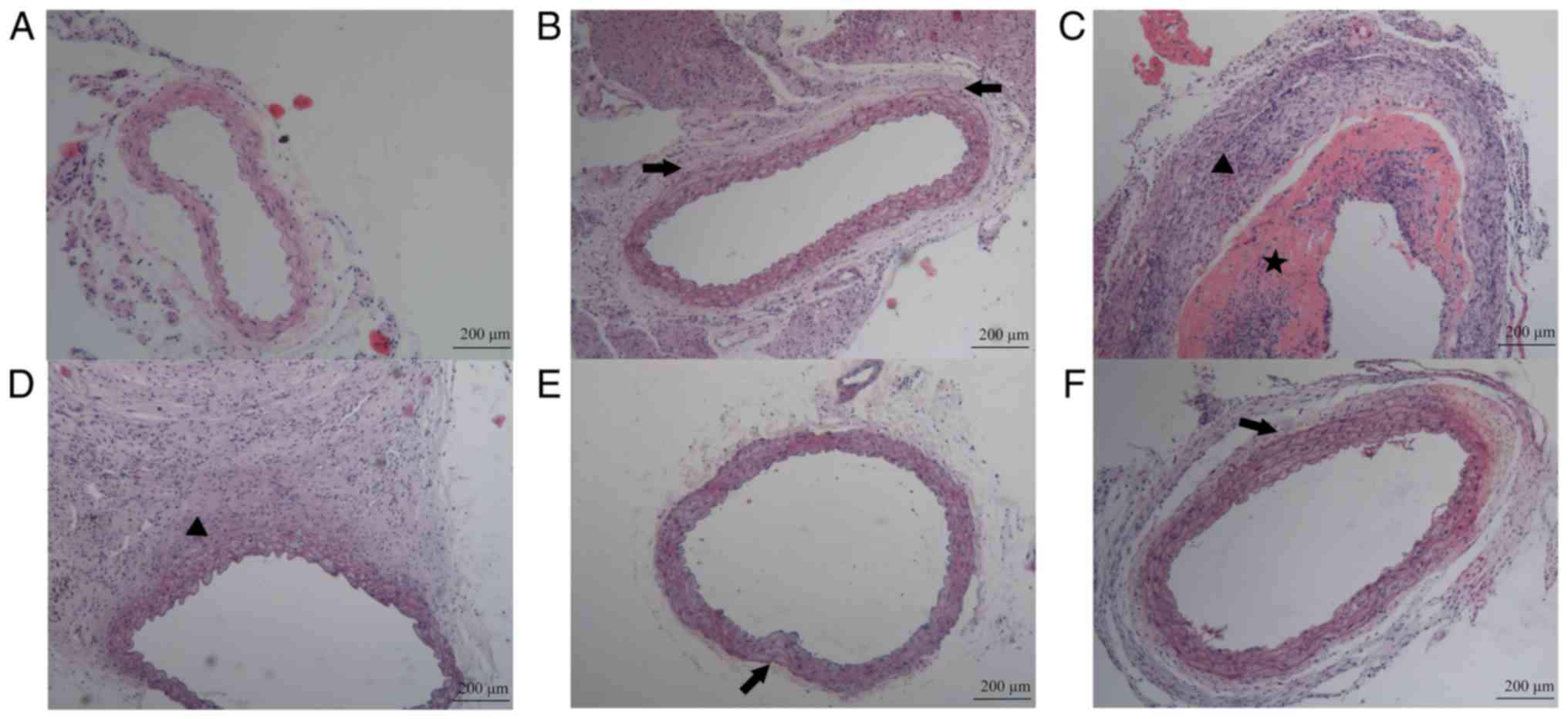
Pathomorphology of the aorta in
Ang-II-treated ApoE−/− mice
The saline-infused control mice had a normal aortic
structure without aortic wall thickening, fiber breakage or
inflammatory cell infiltration (Fig.
2A). However, early alterations in Ang-II-treated mice
primarily focused on the fiber arrangement. The elastic fibers
became partially straight and the space between the fibers
increased (Fig. 2B). By the late
stage of aneurysm formation, the aortic structure at the aneurysmal
body was notably damaged, and the boundaries of the intima, media
and adventitia became indistinct. The aortic wall was thickened;
inflammatory cells infiltrated markedly, particularly in the
adventitia; and the VSMCs became disordered or disappeared. The
fibers, or even the whole walls, were broken down. Blood clots
formed in the lumen of the aorta (Fig.
2C and D). However, the structure at the aneurysmal neck was
quite similar to that in the early stage of Ang-II infusion
(Fig. 2E). RGZ may inhibit the
occurrence of the late pathological alterations. Although the wave
shape of the elastic fibers may have altered to a linear type, the
aortic structure remained intact and the inflammatory cell
infiltration was unremarkable (Fig.
2F).
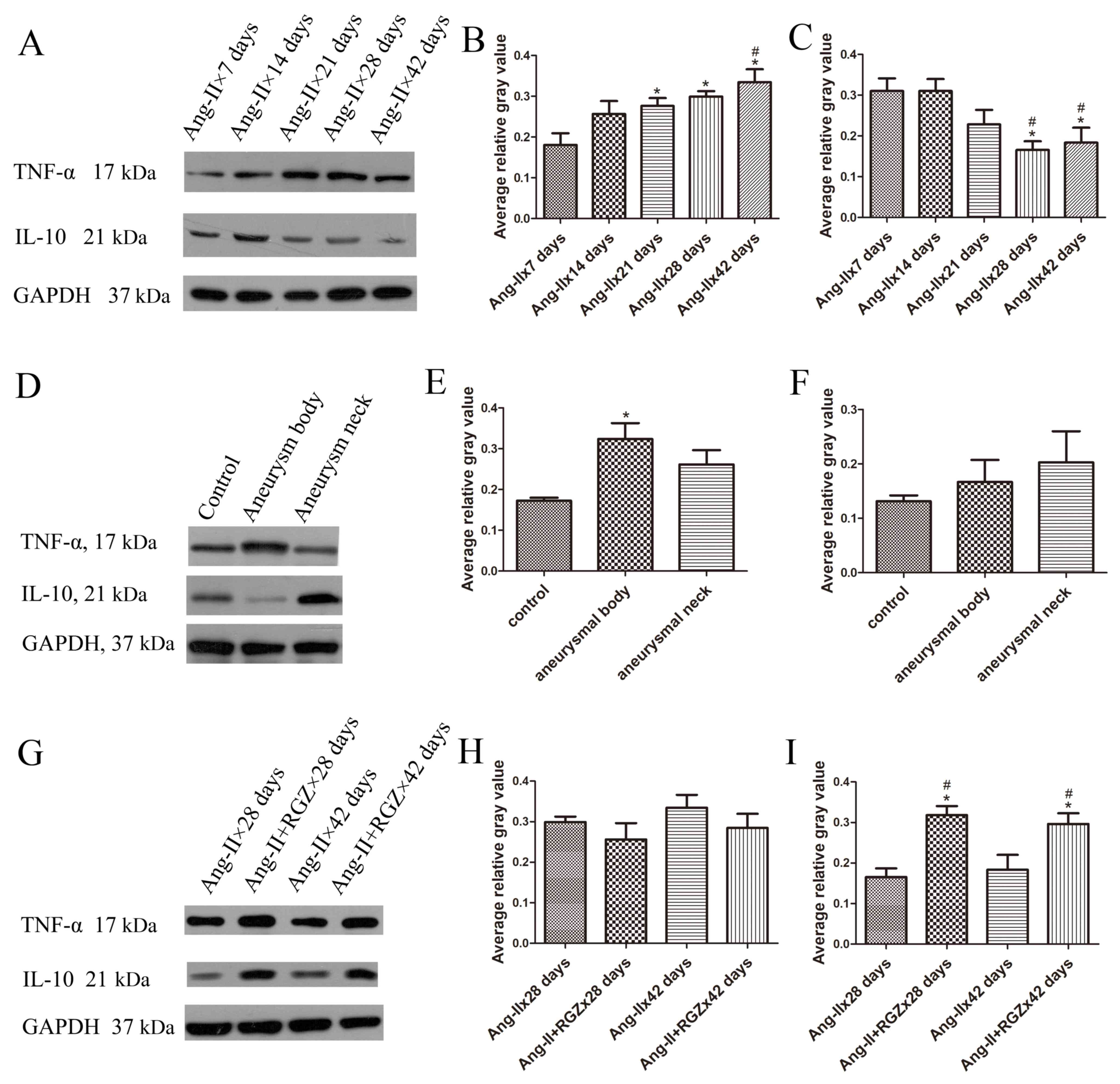
Temporal distribution of inflammatory
types during AAA development
In the present study, it was observed that the
expression of Th1- and Th2-associated inflammatory factors altered
during the development of aortic aneurysms in Ang-II-induced
ApoE−/− mice. Th1-associated cytokine TNF-α expression
increased in the aortic tissue over time; whereas, Th2-associated
cytokine IL-10 expression decreased (Fig. 3A). Therefore, Th2-type inflammation
served a dominant role in the early stage of aneurysm formation,
and Th1-type in the late stage.
The average relative gray values of TNF-α (Fig. 3B) in the Ang-IIx21 days group,
Ang-IIx28 days group and Ang-IIx42 days group were significantly
increased compared with the Ang-IIx7 days group (P=0.018, 0.004 and
<0.001, respectively); however, compared with the Ang-IIx14 days
group, the average relative gray values of TNF-α were not
significantly different, apart from in the Ang-IIx42 days group
(P=0.602, 0.260 and 0.047, respectively). There was no significant
difference between the Ang-IIx7 days group and Ang-IIx14 days group
(P=0.053), or between the Ang-IIx28 days group and Ang-IIx42 days
group (P=0.349). The average relative gray values of IL-10
(Fig. 3C) in the late-stage groups
were significantly decreased compared with the early-stage groups
(28 vs. 7 days, P=0.004; 28 vs. 14 days P=0.004; 42 vs. 7 days,
P=0.010; 42 vs. 14 days, P=0.010). However, there were no
significant differences between the average relative gray values of
IL-10 of the Ang-IIx7 days group and Ang-IIx14 days group (P=0.996)
or between the Ang-IIx28 days group and the Ang-IIx42 days group
(P=0.690). There was no significant difference between Ang-IIx21
days and other groups.
Spatial distribution of inflammatory
types
In mice with aneurysm formation in the late stage,
the differential expression of TNF-α and IL-10 in the aneurysmal
body tissue and aneurysmal neck tissue was compared. As
demonstrated in Fig. 3D, the
expression of TNF-α was increased in the aneurysmal body compared
with the saline control, and decreased in the aneurysm neck
compared with the saline control. The opposite results were
observed for the expression of IL-10.
The expression of TNF-α was increased at the site of
the aneurysmal body compared with the control (P=0.016): However,
there was no significant difference between the body and neck sites
(P=0.211), or between the neck site and the control (P=0.109;
Fig. 3E). The expression level of
IL-10 at the site of the aneurysmal neck exhibited increased
expression compared with the site of the aneurysmal body (P=0.573)
or in the control (P=0.312). There was no marked difference between
the expression level of IL-10 in the aneurysmal body and the
control (P=0.607; Fig. 3F).
PPARγ agonist RGZ promotes
Th2-associated inflammation in AAA
RGZ was able to reduce the TNF-α expression level
and increase the expression level of IL-10 in the aortic tissue
(Fig. 3G). During the development
of AAA, the expression level of TNF-α increased over time as
mentioned above; however, RGZ was able to reverse this trend of
upregulation in the late stage of AAA development. The expression
level of TNF-α in the Ang-II+RGZ groups was decreased compared with
the Ang-II groups (Ang-IIx28 days vs. Ang-II+RGZx28 days, P=0.350;
and Ang-IIx42 days vs. Ang-II+RGZx42 days, P=0.282; Fig. 3H). The effect of RGZ on the
expression of IL-10 was more marked (Fig. 3I). Compared with the Ang-II groups,
the IL-10 expression level in the Ang-II+RGZ groups was increased
(Ang-IIx28 days vs. Ang-II+RGZx28 days, P=0.001; and Ang-IIx42 days
vs. Ang-II+RGZx42 days, P=0.010).
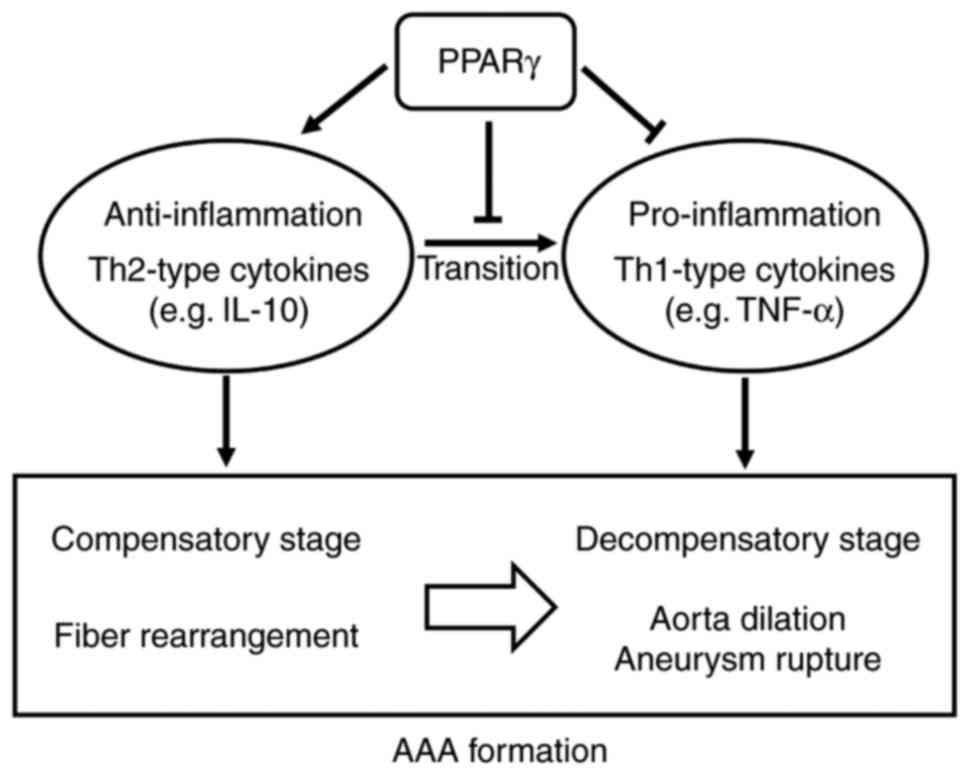
The results above demonstrated that: i) Th2-type and
Th1-type inflammatory cytokines are involved in AAA formation and
they dominate, in turn, during AAA development in Ang-II-induced
ApoE−/− mice; ii) the inflammatory types in the
aneurysmal body and neck are different; iii) the PPARγ agonist,
RGZ, may reduce the dilation of aortas induced by Ang-II in
ApoE−/− mice and may prevent AAA late rupture to a
certain degree; and iv) the shift in inflammatory types may be one
of the mechanisms that allows RGZ to prevent the formation of AAA
(Fig. 4).
Discussion
There has been evidence indicating that the renin
Ang system serves an important role in human AAA (15,16).
More than a decade ago, it was confirmed that Ang-II was able to
promote atherosclerotic lesions and abdominal aortic aneurysm
formation in ApoE−/− mice (17). The pathological alterations in
abdominal aortas in Ang-II-infused ApoE−/− mice were
consistent with a previous report (18). In the past, it was thought that
Ang-II promoted pathological processes in the aorta via two primary
mechanisms: By increasing arterial blood pressure, and by
stimulating monocyte recruitment and activating macrophages
(19–21). However, certain previous studies
reported that the effect of producing AAA by Ang-II is independent
of elevations in blood pressure, or alterations in plasma lipid and
cholesterol concentrations (17,22,23).
However, increasing evidence has demonstrated that an Ang-II
infusion may promote leucocyte infiltration, inflammatory
responses, extracellular matrix degradation and vascular oxidative
stress (24), and that these
effects of Ang-II may occur primarily through the type I Ang-II
receptor (25).
As key mediators of inflammation, cytokines serve an
important role in a number of chronic diseases; however, they have
not been sufficiently studied in the context of AAA. Traditionally,
T cell-derived cytokines are classified into two types, the Th1
type and Th2 type. Th1-type cytokines may activate macrophages and
T cells and produce strong immune responses, while Th2-type
cytokines produce relatively milder immune responses (26). IFN-γ and TNF-α, termed
pro-inflammatory factors, are typical examples of Th1-type
cytokines, and their effects on AAA are unclear. Xiong et al
(4) reported that IFN-γ was able
to promote AAA formation in mice lacking CD4+ cells;
however, King et al (27)
identified that an IFN-γ deficiency was able to protect
Ang-II-treated mice from AAA formation. It was additionally
demonstrated that in the calcium chloride model, mice with a TNF-α
deficiency were resistant to AAA formation (28); however, AAA formation was not
significantly affected in Ang-II-treated low-density lipoprotein
receptor−/− mice with a TNF-α receptor deficiency
(29). Th2-type cytokines may
additionally serve different roles in AAA. IL-4 and IL-5 were
revealed to promote AAA (30,31),
although they have the ability to prevent atherosclerosis (32,33).
IL-10 is considered to be an anti-inflammatory cytokine and has the
ability to suppress the activation of Th1 cells and macrophages
(26). In IL10−/− mice,
AAA was induced more easily by Ang-II (34). However, in an early study of human
AAAs, Th2-type cytokines, including IL-10, IL-4 and IL-5, were
expressed more predominantly in the AAA tissues compared with
Th1-type cytokines (IL-2, IL-12, IL-15 and IL-18) (3).
Based on the above, there may be some confusion
regarding these complex results. In reality, considering this
question in a static way is not appropriate. As AAA development is
a gradual, altering process, inflammation, including cells and
cytokines, may alter. The two types of vascular diseases,
atherosclerosis and aneurysm, are usually associated, and there is
certain speculation regarding the mechanisms of transition from
atherosclerosis to aneurysm, as previously mentioned (6). However, data from human or mouse
studies has demonstrated that in atherosclerosis and AAA pathology,
in the majority of cases, these conditions co-exist (26), and specific additional key factors,
including genetics or smoking, may be required to trigger the
formation of AAA (2,26). When considering AAA alone, the
temporal and spatial distributions of inflammatory cytokines remain
unclear. The present study demonstrated that in the Ang-II-induced
AAA model of ApoE−/− mice, IL-10 was expressed
predominantly in the early stage of AAA development; however, it
was attenuated in the late stage. This is consistent with the
results of a previous study (32).
Conversely, the expression of TNF-α significantly increased between
the early and late stages of AAA formation. Considering the
alteration that was observed, it was hypothesized that
anti-inflammatory Th2-type cytokines may be produced to resist the
inflammation caused by the Ang-II infusion in the early stage and
that this process may be regarded as the ‘compensatory stage’.
IL-10 may suppress TNF-α expression during this stage, and the
infiltration of inflammatory cells may not be substantial. With
inflammation-enhancing inflammatory cells, including macrophages,
lymphocytes accumulate and pro-inflammatory Th1-type cytokines are
increasingly expressed. In turn, TNF-α promotes inflammation in the
local region, the production of MMPs, (35) and the destruction of aortic walls,
leading to AAA formation. This action may be considered the
‘decompensatory stage’. As the aneurysmal neck is the marginal area
of AAA, there may be a ‘delayed effect’ in inflammation at the site
of the aneurysmal neck. When the inflammatory type shifts to the
‘decompensatory stage’ in the aneurysmal body, the inflammatory
type remains as that of the ‘compensatory stage’ in the aneurysmal
neck. This is consistent with the previous opinion that different
sampling sites may impact the inflammatory types of AAA (36).
The PPARγ agonist RGZ reduced aortic dilation and
late rupture in the present study. This result is consistent with
the results of a previous study conducted by Jones et al
(7). However, in this early study,
the administration of RGZ reduced the expression of TNF-α induced
by Ang-II; however, it exerted no marked effect on IL-10. In other
models, PPARγ may induce the production of IL-10 (8,37,38).
The difference between the present results and those of Jones et
al (7) may be due to the
experimental conditions, including the age of the mice (6–8 weeks
in the present study compared with 12 months in the study conducted
by Jones et al (7)).
Different previous studies have demonstrated that PPARγ may
influence inflammation, and the biggest focus has been the function
of the shifting of monocyte subtypes. PPARγ may promote
transformation to M2 macrophages, and TZDs are the most potent
PPARγ ligands to induce M2 polarization (8,39).
ILs, including IL-4, IL13 and IL-10, as Th2 types, may stimulate M2
macrophages, and M2 macrophages, in turn, may promote the
production of anti-inflammatory cytokines and jointly create a
microenvironment of tissue repair and wound healing (8). In the present study, the PPARγ
agonist RGZ significantly upregulated IL-10 in the late stage and
the expression level of IL-10 was comparable with the early stage.
Considering the pathological alterations together, PPARγ may
maintain the aortic inflammatory conditions in the ‘compensatory
stage’ and delay the development and rupture of AAA. These results
demonstrated that PPARγ serves a protective role in AAA formation
induced by Ang-II in ApoE−/− mice.
In human AAA studies, research is limited to the
late-stage of this disease. Although there are specific differences
between the Ang-II-infused mouse model and humans, a number of
primary pathological features of AAA in humans have been reproduced
in this model (18). The present
study preliminarily demonstrated that the overexpression of Th1- or
Th2-associated cytokines is altered during the process of AAA
formation and that they predominantly express, in turn, in the
early compensatory stage anti-inflammatory factors, including
IL-10, to suppress pro-inflammatory factors; whereas, in the
decompensatory stage, pro-inflammatory cytokines, including TNF-α,
are upregulated. PPARγ may aid the maintenance of the expression
levels of anti-inflammatory cytokines and extend the compensatory
stage (Fig. 4).
There are limitations to the present study. The
small number of samples may influence the significance of the
statistical differences, and only a few results demonstrated
variation trends. Further research is required to inform a broader
audience regarding pathology and inflammation during AAA formation,
including inflammatory cell infiltration and the downstream factors
of these cytokines. Another limitation of the present study is the
lack of a direct mechanism through which PPARγ may act on the
temporal and spatial distribution of inflammation. Additional
research is required to focus on the direct mechanism, and
PPARγ-knockout mice may be useful.
In conclusion, aortic inflammation during AAA
formation is dynamic and variable when considering the temporal and
spatial distributions. The overexpression and dominance of the
relevant cytokines require examination at different stages.
Protective anti-inflammatory cytokines are upregulated in the early
compensatory stage; however, pro-inflammatory cytokines are
dominant in the late decompensatory stage. When focusing on the
inflammatory alterations in AAA, PPARγ is likely to continue to
upregulate anti-inflammatory cytokines and decelerate the process
of AAA development and rupture.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81300235).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YC and WW conceived and designed the experiments. WW
and RS performed the animal experiments. WW produced the
manuscript. WW and RS conducted data analysis. All authors have
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The experiments in the present study were approved
by the Ethics Committee of Peking Union Medical College Hospital
(Beijing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Singh K, Bønaa KH, Jacobsen BK, Bjørk L
and Solberg S: Prevalence of and risk factors for abdominal aortic
aneurysms in a population-based study: The Tromsø study. Am J
Epidemiol. 154:236–244. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eagleton MJ: Inflammation in abdominal
aortic aneurysms: Cellular infiltrate and cytokine profiles.
Vascular. 20:278–283. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schönbeck U, Sukhova GK, Gerdes N and
Libby P: T(H)2 predominant immune responses prevail in human
abdominal aortic aneurysm. Am J Pathol. 161:499–506. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xiong W, Zhao Y, Prall A, Greiner TC and
Baxter BT: Key roles of CD4+ T cells and IFN-gamma in the
development of abdominal aortic aneurysms in a murine model. J
Immunol. 172:2607–2612. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shimizu K, Libby P and Mitchell RN: Local
cytokine environments drive aneurysm formation in allografted
aortas. Trends Cardiovasc Med. 15:142–148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shimizu K, Mitchell RN and Libby P:
Inflammation and cellular immune responses in abdominal aortic
aneurysms. Arterioscler Thromb Vasc Biol. 26:987–994. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jones A, Deb R, Torsney E, Howe F, Dunkley
M, Gnaneswaran Y, Gaze D, Nasr H, Loftus IM, Thompson MM and
Cockerill GW: Rosiglitazone reduces the development and rupture of
experimental aortic aneurysms. Circulation. 119:3125–3132. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Croasdell A, Duffney PF, Kim N, Lacy SH,
Sime PJ and Phipps RP: PPARγ and the innate immune system mediate
the resolution of inflammation. PPAR Res. 2015:5496912015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Motoki T, Kurobe H, Hirata Y, Nakayama T,
Kinoshita H, Rocco KA, Sogabe H, Hori T, Sata M and Kitagawa T:
PPAR-γ agonist attenuates inflammation in aortic aneurysm patients.
Gen Thorac Cardiovasc Surg. 63:565–571. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Celinski K, Dworzanski T, Korolczuk A,
Piasecki R, Slomka M, Madro A and Fornal R: Effects of peroxisome
proliferator-activated receptors-gamma ligands on dextran sodium
sulphate-induced colitis in rats. J Physiol Pharmacol. 62:347–356.
2011.PubMed/NCBI
|
|
11
|
Pisanu A, Lecca D, Mulas G, Wardas J,
Simbula G, Spiga S and Carta AR: Dynamic changes in pro- and
anti-inflammatory cytokines in microglia after PPAR-γ agonist
neuroprotective treatment in the MPTPp mouse model of progressive
Parkinson's disease. Neurobiol Dis. 71:280–291. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Golledge J, Cullen B, Rush C, Moran CS,
Secomb E, Wood F, Daugherty A, Campbell JH and Norman PE:
Peroxisome proliferator-activated receptor ligands reduce aortic
dilatation in a mouse model of aortic aneurysm. Atherosclerosis.
210:51–56. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hasan DM, Starke RM, Gu H, Wilson K, Chu
Y, Chalouhi N, Heistad DD, Faraci FM and Sigmund CD: Smooth muscle
peroxisome proliferator-activated receptor γ plays a critical role
in formation and rupture of cerebral aneurysms in mice in vivo.
Hypertension. 66:211–220. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Apgar JM, Juarranz A, Espada J, Villanueva
A, Cañete M and Stockert JC: Fluorescence microscopy of rat embryo
sections stained with haematoxylin-eosin and Masson's trichrome
method. J Microsc. 191:20–27. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hackam DG, Thiruchelvam D and Redelmeier
DA: Angiotensin-converting enzyme inhibitors and aortic rupture: A
population-based case-control study. Lancet. 368:659–665. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jones GT, Thompson AR, van Bockxmeer FM,
Hafez H, Cooper JA, Golledge J, Humphries SE, Norman PE and van Rij
AM: Angiotensin II type 1 receptor 1166C polymorphism is associated
with abdominal aortic aneurysm in three independent cohorts.
Arterioscler Thromb Vasc Biol. 28:764–770. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Daugherty A, Manning MW and Cassis LA:
Angiotensin II promotes atherosclerotic lesions and aneurysms in
apolipoprotein E-deficient mice. J Clin Invest. 105:1605–1612.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Daugherty A, Cassis LA and Lu H: Complex
pathologies of angiotensin II-induced abdominal aortic aneurysms. J
Zhejiang Univ Sci B. 12:624–628. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chobanian AV and Alexander RW:
Exacerbation of atherosclerosis by hypertension. Potential
mechanisms and clinical implications. Arch Intern Med.
156:1952–1956. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim JA, Berliner JA and Nadler JL:
Angiotensin II increases monocyte binding to endothelial cells.
Biochem Biophys Res Commun. 226:862–868. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yanagitani Y, Rakugi H, Okamura A,
Moriguchi K, Takiuchi S, Ohishi M, Suzuki K, Higaki J and Ogihara
T: Angiotensin II type 1 receptor-mediated peroxide production in
human macrophages. Hypertension. 33:335–339. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cassis LA, Gupte M, Thayer S, Zhang X,
Charnigo R, Howatt DA, Rateri DL and Daugherty A: ANG II infusion
promotes abdominal aortic aneurysms independent of increased blood
pressure in hypercholesterolemic mice. Am J Physiol Heart Circ
Physiol. 296:H1660–H1665. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Manning MW, Cassi LA, Huang J, Szilvassy
SJ and Daugherty A: Abdominal aortic aneurysms: Fresh insights from
a novel animal model of the disease. Vasc Med. 7:45–54. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lu H, Rateri DL, Bruemmer D, Cassis LA and
Daugherty A: Involvement of the renin-angiotensin system in
abdominal and thoracic aortic aneurysms. Clin Sci (Lond).
123:531–543. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cassis LA, Rateri DL, Lu H and Daugherty
A: Bone marrow transplantation reveals that recipient AT1a
receptors are required to initiate angiotensin II-induced
atherosclerosis and aneurysms. Arterioscler Thromb Vasc Biol.
27:380–386. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Peshkova IO, Schaefer G and Koltsova EK:
Atherosclerosis and aortic aneurysm-is inflammation a common
denominator? FEBS J. 283:1636–1652. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
King VL, Lin AY, Kristo F, Anderson TJ,
Ahluwalia N, Hardy GJ, Owens AP III, Howatt DA, Shen D, Tager AM,
et al: Interferon-gamma and the interferon-inducible chemokine
CXCL10 protect against aneurysm formation and rupture. Circulation.
119:426–435. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xiong W, MacTaggart J, Knispel R, Worth J,
Persidsky Y and Baxter BT: Blocking TNF-alpha attenuates aneurysm
formation in a murine model. J Immunol. 183:2741–2746. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xanthoulea S, Thelen M, Pöttgens C,
Gijbels MJ, Lutgens E and de Winther MP: Absence of p55 TNF
receptor reduces atherosclerosis, but has no major effect on
angiotensin II induced aneurysms in LDL receptor deficient mice.
PLoS One. 4:e61132009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Davenport P and Tipping PG: The role of
interleukin-4 and interleukin-12 in the progression of
atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol.
163:1117–1125. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Binder CJ, Hartvigsen K, Chang MK, Miller
M, Broide D, Palinski W, Curtiss LK, Corr M and Witztum JL: IL-5
links adaptive and natural immunity specific for epitopes of
oxidized LDL and protects from atherosclerosis. J Clin Invest.
114:427–437. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu J, Ehrman B, Graham LM and Eagleton MJ:
Interleukin-5 is a potential mediator of angiotensin II-induced
aneurysm formation in apolipoprotein E knockout mice. J Surg Res.
178:512–518. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shimizu K, Shichiri M, Libby P, Lee RT and
Mitchell RN: Th2-predominant inflammation and blockade of IFN-gamma
signaling induce aneurysms in allografted aortas. J Clin Invest.
114:300–308. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ait-Oufella H, Wang Y, Herbin O, Bourcier
S, Potteaux S, Joffre J, Loyer X, Ponnuswamy P, Esposito B, Dalloz
M, et al: Natural regulatory T cells limit angiotensin II-induced
aneurysm formation and rupture in mice. Arterioscler Thromb Vasc
Biol. 33:2374–2379. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
de Araújo RF Jr, Reinaldo MP, Brito GA,
Cavalcanti Pde F, Freire MA, de Medeiros CA and de Araújo AA:
Olmesartan decreased levels of IL-1β and TNF-α, down-regulated
MMP-2, MMP-9, COX-2, RANK/RANKL and up-regulated SOCs-1 in an
intestinal mucositis model. PLoS One. 9:e1149232014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shimizu K, Libby P and Mitchell RN: Local
cytokine environments drive aneurysm formation in allografted
aortas. Trends Cardiovasc Med. 15:142–148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Celinski K, Dworzanski T, Korolczuk A,
Piasecki R, Slomka M, Madro A and Fornal R: Effects of peroxisome
proliferator-activated receptors-gamma ligands on dextran sodium
sulphate-induced colitis in rats. J Physiol Pharmacol. 62:347–356.
2011.PubMed/NCBI
|
|
38
|
Pisanu A, Lecca D, Mulas G, Wardas J,
Simbula G, Spiga S and Carta AR: Dynamic changes in pro- and
anti-inflammatory cytokines in microglia after PPAR-γ agonist
neuroprotective treatment in the MPTPp mouse model of progressive
Parkinson's disease. Neurobiol Dis. 71:280–291. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chawla A: Control of macrophage activation
and function by PPARs. Circ Res. 106:1559–1569. 2010. View Article : Google Scholar : PubMed/NCBI
|