Introduction
Chronic obstructive pulmonary disease (COPD) has
become a principal international health problem with a rise in
prevalence and mortality (1), as
there was a 44.2% increase of the number of prevalent COPD cases
between 1990 and 2015 (2). The
most common symptoms of COPD include, chronic cough, expectoration,
shortness of breath and difficulty in breathing (3). COPD is generally considered to be an
abnormal inflammatory disorder leading to airflow limitation
associated with the response of lungs to toxic and noxious
particles, and gases, and is primarily caused by tobacco smoking
(4). Inflammation, oxidative
stress and imbalance between levels of proteinases and
antiproteinases within the lungs are also the predisposing factors
to COPD (5).
The pathological alterations may be identified in
the large airways, small airways, lung parenchyma and pulmonary
vascular of patients with COPD (6–8).
Multiple alterations in the airway are closely associated with
COPD, and the symptoms are similar to vascular remodeling in
patients with atherosclerosis (9).
Airway wall has been demonstrated to be thickened in patients with
COPD, primarily as a result of hyperplasia and hypertrophy of
airway smooth muscle cells (ASMCs) and accumulation of
extracellular matrix (ECM) components (10). The ECM of the lung consists of a
number of macromolecules, including collagens, fibronectin, laminin
(LN), proteoglycans, glycosaminoglycan, elastin and tenascin, which
form a complex, dynamic network (11,12).
In patients with COPD, the levels of elastin (13) and proteoglycans (14) were deceased, while LN and total
collagen content were elevated (15). Collagen I (COL-1) and collagen-III
(COL-3) are two types of lung interstitium components that serve a
role in establishing normal lung architecture (16). LN has been reported to be the
primary component of basement membranes and to regulate cell
behavior in bronchiolar epithelium of patients with COPD (17). Furthermore, ECM proteins are
associated with biological properties of human ASMCs including
growth, survival, migration and adhesion (12). To date, a limited number of studies
investigating the role of deposition of COL-1, COL-3 and LN on the
biological behavior of ASMCs in patients with COPD. Airway smooth
muscle (ASM) preserves the dynamic homeostasis of
synthesis/degradation of ECM though secretion of various ECM
proteins, including matrix metalloproteinases (MMPs) (18), tissue inhibitors of
metalloproteinases (TIMPs) (19),
cytokines (12), chemokines
(12) and growth factors (20). Evidence accumulated from previous
studies indicated that ECM proteins are implicated directly or
indirectly in multiple signaling pathways to influence various
biological processes (21–23). Khatiwala et al (24) demonstrated that ECM modulates the
mitogen-activated protein kinase signaling pathway and acts
downstream of transforming protein RhoA and Rho-associated protein
kinase 1 for the regulation of osteogenesis. In a previous study,
COL-1 was associated with morphological alterations of mouse NMuMG
cells and upregulation of cadherin-2 though the phosphoinositide
3-kinase (PI3K)-Rac1-JNK pathway (24). Integrin α6β1 and FAK-MEK/ERK
signaling promote mesenchymal stem cell growth induced by LN-1
under growth factors in the absence of serum and differentiation
factors (25). However, few
studies have evaluated the association between ECM proteins and
signaling pathways in the airways of patients with COPD.
In the present study, a rat model of COPD was
established to assess the impact of ECM components (COL-1, COL-3
and LN) on proliferation, migration and attachment of ASMCs.
Cytokines, chemokines, growth factors, MMP-9 and its inhibitor,
TIMP1, were detected. The molecular signaling that may be involved
the thickening of airway wall was further elucidated.
Materials and methods
COPD animal model construction
A total of 24 healthy male Sprague Dawley (SD; 10–12
weeks; 180–220 g; five per cage) rats were purchased from Shanghai
Laboratory Animal Research Center (Shanghai, China) and housed in
the animal room (25±3°C; 50% humidity; 12/12 h light and dark
cycle) with free access to water and food. The rat model of COPD
was established as described in previous studies (26–28).
Briefly, 12 SD rats were anesthetized with 0.45% pentobarbital
sodium (50 mg/kg) via intraperitoneal injection, followed by
injection of lipopolysaccharide (LPS; 200 mg/200 µl) through the
trachea and inhalation of fresh smoke once a day for 30 days, then
kept at −20°C for 5 min/day for the next 30 days to establish
animal models of COPD. The remaining 12 rats served as the normal
group and received no treatment. All experimental protocols were
approved by the Animal Research Committee of Hubei University of
Medicine (Suizhou, China). The care and use of animals was based on
the Guidelines by the National Institutes of Health.
Isolation of rat airway smooth muscle
cells (ASMCs)
Rat ASMCs were isolated as described in previous
studies (29–31). Briefly, entire rat tracheae were
rapidly dissected from normal and COPD rats in ice-cold PBS
solution. Subsequently, the mucosal and connective tissues were
removed and muscle was gently separated in small bundles. Separated
muscle was placed in Ham's F12 digestion solution containing 0.5%
fetal bovine serum (FBS; cat. no. 16000-044; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with collagenase
IV (2 mg/ml; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and
papain (1 mg/ml; Sigma-Aldrich; Merck KGaA). Following
centrifugation at 500 × g for 15 min at 4°C, cell suspension was
collected and isolated ASMCs were treated with 0.25% trypsin and
cells from passages 3–5 were selected for the subsequent
experiments.
Cell culture and treatment
All ASMCs were maintained in Ham's F12 medium (cat.
no. A1526_9010; AppliChem, Inc., St. Louis, MO, USA) with 10% FBS
in a humidified atmosphere containing 5% CO2 at 37°C.
For cell functional analysis, ASMCs were seeded in 24-well plates
and each well was treated with 500 µl ECM components, including
COL-1 [cat. no. CLS001654; Sangon Biotech (Shanghai) Co., Ltd.,
Shanghai, China], LN (cat. no. L4544; Sigma-Aldrich; Merck KGaA) or
COL-3 (cat. no. ab7535; Abcam, Cambridge, UK) at a concentration of
10 mg/l each, except for the blank control cells, which were
treated with 500 µl bovine serum albumin (BSA; 10 ml/l; cat. no.
A8020; Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China) in a humidified atmosphere containing 5%
CO2 at 37°C for 2 h.
Immunofluorescence (IF)
The morphology of the isolated ASMCs was assessed by
IF staining. Briefly, ASMCs at a density of 106 were
washed twice with PBS and fixed with 4% paraformaldehyde for 10 min
at room temperature. The cells were subsequently treated with
pre-cooled 100% methanol for 20 min at −20°C and subsequently
blocked in PBS containing 3% BSA for 1 h at room temperature. The
cells were then incubated with α-smooth muscle actin (α-SMA) mouse
primary antibody (1:200; Abcam; cat. no. ab8211) at 4°C overnight,
followed by incubation with Alexa Fluor 647 (red)-conjugated
secondary antibody (1:500; cat. no. Z25008; Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C for 30 min in the dark. DAPI (100
ng/ml; Invitrogen; Thermo Fisher Scientific, Inc.) was used to
stain the nuclei (10 min at room temperature). Finally, the
location of α-SMA was detected using a laser scanning confocal
microscope (LSM 710; Carl Zeiss AG, Oberkochen, Germany).
Cell Counting kit-8 (CCK-8) assay
The proliferation of ASMCs was determined by CCK-8
assay (cat. no. C0037; Beyotime Institute of Biotechnology, Haimen,
China). Briefly, cells were seeded into 96-well plates at a density
of 3×104 cells/well. Following different treatments, the
supernatant was removed and 10 µl CCK-8 solution was added to each
well at 24, 48 or 72 h, and the mixture was incubated for 1 h at
37°C. The absorbance was measured at a wavelength of 450 nm using a
microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
The experiment was repeated three times.
5-Ethynyl-2′-deoxyuridine (EdU)
proliferation analysis
Following different treatments, ASMCs
(105 cells/well) were seeded in triplicate in 96-well
plates and their proliferation was measured using the Cell-Light
EdU DNA Cell Proliferation kit (Guangzhou RiboBio Co., Ltd.,
Guangzhou, China) according to the manufacturer's protocol. In
brief, cells were treated with 50 mM EdU for 4 h at 37°C and fixed
with 4% formaldehyde for 15 min at room temperature. Subsequently
cells were incubated with 100 µl Apollo reaction cocktail for 30
min at room temperature and stained with DAPI (50 ng/ml) at room
temperature for 30 min. The images of stained cells were observed
using a fluorescent microscope (Olympus Corporation, Tokyo, Japan;
magnification, ×120). Each sample was analyzed in triplicate in
three independent experiments. The experiment was repeated three
times.
Cell cycle detection
ASMCs (105 cell/well) were treated with
0.25% trypsin and seeded into 24-well plates at a density of
5×105 cells/well. Following washing with PBS, cells were
fixed with 75% ethanol at room temperature for 24 h, washed again
with PBS and stained with 100 ng/ml propidium iodide for 30 min at
room temperature (BD Biosciences, San Jose, CA, USA). Subsequently,
cell cycle analysis was performed on FACSCalibur flow cytometer (BD
Biosciences). Cell Quest software 1.1 (BD Biosciences) was used to
calculate the percentage of cell population in each phase,
including G0/G1, S and G2/M.
Wound healing analysis
The ASMCs (105 cells/well) were incubated
overnight following treatment with different ECM components until
grown to ~90% confluence. On the following day, a standard 200 µl
plastic filter tip was drawn across the well to produce a wound to
evaluate the directional motility of cells. The wound areas were
observed and images were captured at 0 and 48 h with a fluorescent
microscope under bright field illumination (Axio Observer A1; Zeiss
AG, Oberkochen, Germany; magnification, ×4). The wound healing
effect was determined by calculating the radio of the wound area to
the total area using ImageJ bundled with Java 1.8.0_112 (National
Institutes of Health, Bethesda, MD, USA). The degree of wound
closure was analyzed in three randomly selected fields of view. The
experiment was repeated three times.
Cell adhesion assay
Following different treatments, ASMCs were
trypsinized and seeded at a density of 5×104 cells/well
on Matrigel-precoated 96-well culture plates. The cells were
incubated for 2 h at 37°C and non-adherent cells were removed by
washing with PBS. Cells were fixed with 4% paraformaldehyde at 4°C
for 24 h, followed by 0.5% toluidine blue staining for 20 min at
room temperature and 1% sodium dodecyl sulfate (2 h at room
temperature). The optical density values were determined using a
microplate reader (Bio-Rad Laboratories, Inc.) at a wavelength of
595 nm. The experiment was repeated two times.
ELISA
The supernatants were collected from ASMCs following
treatment with different ECM components. The levels of total MMP-9,
C-X-C motif chemokine ligand 1 (CXCL1), C-X-C motif chemokine
ligand 8 (CXCL8) and interleukin-6 (IL-6) in the supernatants were
measured using MMP-9 ELISA kit (cat. no. CSB-E08008r), CXCL1 ELISA
kit (cat. no. CSB-E12997r), CXCL8 ELISA kit (cat. no. ml002885) and
IL-6 ELISA kit (cat. no. CSB-E04640r), according to the
manufacturer's protocols (all Cusabio Technology, LLC, College
Park, MD, USA). The concentrations are expressed in pg/ml. The
experiment was repeated three times.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from ASMCs using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and reverse transcribed into cDNA using the PrimeScript RT
Reagent kit (Takara Bio, Inc., Otsu, Japan), according to the
manufacturer's protocol. The RT-qPCR reactions were performed using
ABI7500 real-time RCR System (Applied Biosystems; Thermo Fisher
Scientific, Inc.). SYBR-Green Master Mix kit and the following
thermocycling conditions were used for qPCR: 30 cycles of 30 sec at
94°C, 30 sec at 55°C and 30 sec at 72°C. The following primer
sequences were used for PCR: MMP-9 forward,
5′-TCATCCAGTTTGGTGTCGC-3′ and reverse, 5′-AGTGGGCATCTCCCTGAAT-3′;
TIMP1 forward, 5′-GCCTCTGGCATCCTCTTG-3′ and reverse,
5′-CTGCGGTTCTGGGACTTG-3′; transforming growth factor β-1 (TGFβ1)
forward, 5′-AGGCGGTGCTCGCTTTG-3′ and reverse,
5′-TGCGTTGTTGCGGTCCA-3′; fibroblast growth factor 1 (FGF-1)
forward, 5′-GATGGCACAGTGGATGGGAC-3′ and reverse,
5′-AAGCCCGTCGGTGTCCATGG-3′; and GAPDH forward,
5′-CCTCGTCTCATAGACAAGATGGT-3′ and reverse,
5′-GGGTAGAGTCATACTGGAACATG-3′. The fold-change in the expression of
each target mRNA relative to GAPDH was calculated using the
2−ΔΔCq method (32).
Western blot analysis
All ASMCs (106) were lysed in 120 µl
radioimmunoprecipitation assay lysis buffer (cat. no. P0013B;
Beyotime Institute of Biotechnology) with phosphatase and protease
inhibitors (BIOSS, Beijing, China) for 2 h at 4°C. Following
protein quantitation using a bicinchoninic acid protein assay kit
(Thermo Fisher Scientific, Inc.), proteins (30 µg/lane) were
separated by 10% SDS-PAGE and transferred to polyvinylidene
fluoride membranes. Following blocking with 5% non-fat milk for 1 h
at room temperature, the membranes were incubated at 4°C overnight
with primary antibodies, including anti-TGFβ1 (1:800; cat. no.
bs-0086R; BIOSS), anti-FGF-1 (1:800; cat. no. 10-P1311-1; American
Research Specialty Products; Palos Verdes Estates, CA, USA),
anti-MMP-9 (1:800; cat. no. 10-P1357-1; American Research Specialty
Products), anti-TIMP1 (1:800; cat. no. 28-60319P; American Research
Specialty Products), anti-PI3K (1:800; cat. no. 05-212-K; Merck
KGaA), anti-p (phosphorylated)-RAC-α serine/threonine-protein
kinase (AKT; 1:800; cat. no. 28101-025; AnaSpec; Eurogentec, Liège,
Belgium), anti-p-serine/threonine-protein kinase mTOR (mTOR; 1:800;
cat. no. BS4706; Bioworld Technology, Inc., St. Louis Park, MN,
USA), anti-AKT (1:800; cat. no. MA514916; Thermo Fisher Scientific,
Inc.), anti-mTOR (1:800; cat. no. 17-18594; American Research
Specialty Products) and anti-GAPDH (1:800; cat. no. sc-47724; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA), followed by incubation
with horseradish peroxidase-conjugated goat anti-mouse secondary
antibody (1:2,000; cat. no. A12000-1; Epigentek Group, Inc.,
Farmingdale, NY, USA) for 1 h at room temperature. The protein
bands were detected using a Chemiluminescence Exposure Screen kit
(cat. no. 62242; Pierce; Thermo Fisher Scientific, Inc.). The
experiment was repeated three times.
Signal transduction pathways
COL-3-mediated adhesion and migration of ASMC was
studied using specific pharmacological antagonists of PI3K. The
PI3K inhibitor LY294002 (500 nM; cat. no. S1105; Selleck Chemicals,
Houston, TX, USA) and activator insulin-like growth factor-1 (50
mM; IGF-1; Sigma-Aldrich; Merck KGaA) were added to the COPD ASMCs
30 min prior to the addition of COL-3 and remained in contact with
the cells for the duration of the present study. The experiment was
repeated three times.
Statistical analysis
All data are presented as the mean ± standard
deviation. The differences between groups were analyzed using
GraphPad Prism (version 7; GraphPad Software, Inc., La Jolla, CA,
USA) using one-way analysis of variance with Tukey's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
ECM components promote cell
proliferation and cell cycle progression of ASMCs
ASMC were successfully isolated from normal and COPD
rats. Following inoculation of ASMC into culture plates at 37°C for
3 days, the cells presented spindle shape (round centered nuclei).
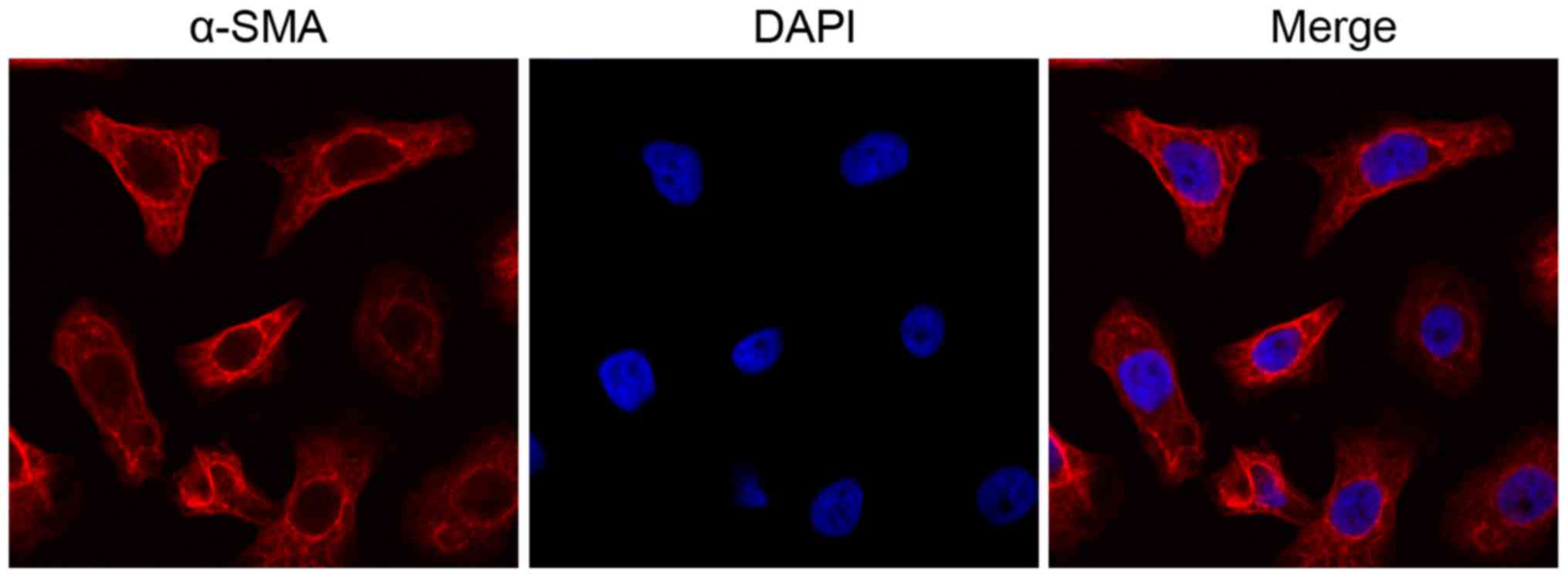
Subsequently, the expression of α-SMA in normal and COPD rat ASMCs
was determined by IF. The ASMCs exhibited abundant expression of
α-SMA (Fig. 1) indicating that
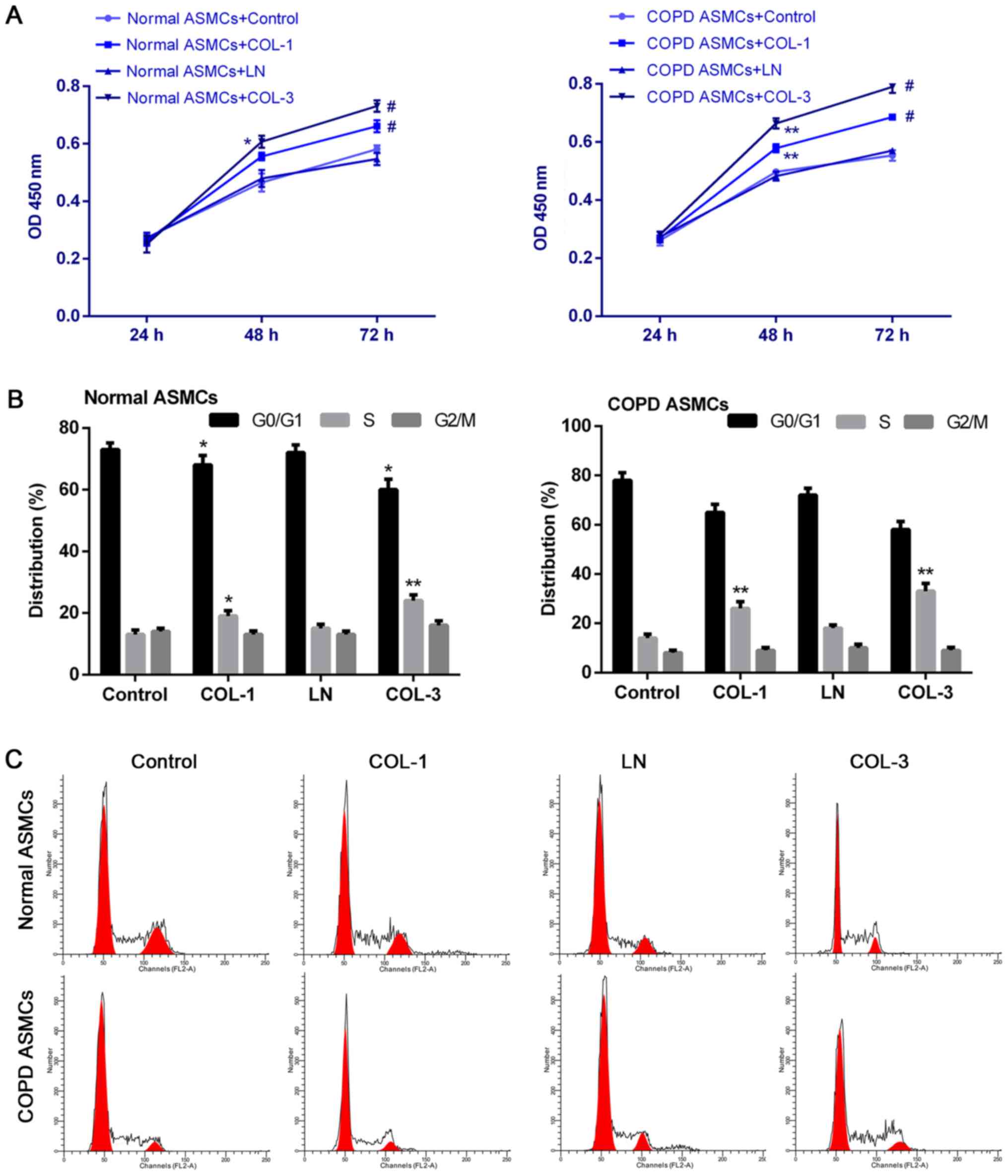
ASMCs were isolated in high purity. To determine the role of ECM
components in proliferative kinetics of ASMCs, a CCK-8 assay was
performed to determine cell proliferation ability of ASMCs. COL-1
and COL-3 significantly promoted proliferation of control and COPD
ASMCs (P<0.05; Fig. 2A), while
LN exhibited no significant effect on cell proliferation ability.
The percentage of cells in S phase was significantly increased
following treatment of normal and COPD ASMCs with COL-1 and COL-3
compared with the respective control groups (P<0.05 and
P<0.01; Fig. 2B).
Representative histograms of cell cycle distribution are presented
in Fig. 2C. Promotion of cell
cycle progression induced by ECM components was more significant in
ASMCs derived from COPD rats compared with normal ASMCs.
ECM components promote migration and
adhesion of ASMCs
Wound-healing assay was performed to determine the
effect of ECM components on cell migration ability. Deceleration in
the wound closure rate was observed in ASMCs derived from normal
rats following treatment with ECM components compared with the
control group (P<0.01, P<0.001; Fig. 3A and B). Treatment with COL-1 and
COL-3 significantly elevated cell migration ability of ASMCs
derived from rats with COPD. Furthermore, adhesion ability of ASMCs
was assessed by cell adhesion assay. The results indicated that the
adhesive ability of ASMCs was stronger in groups treated with ECM
components compared with the control cells in normal ASMCs and COPD
ASMCs (P<0.05 and P<0.01; Fig.
3C).
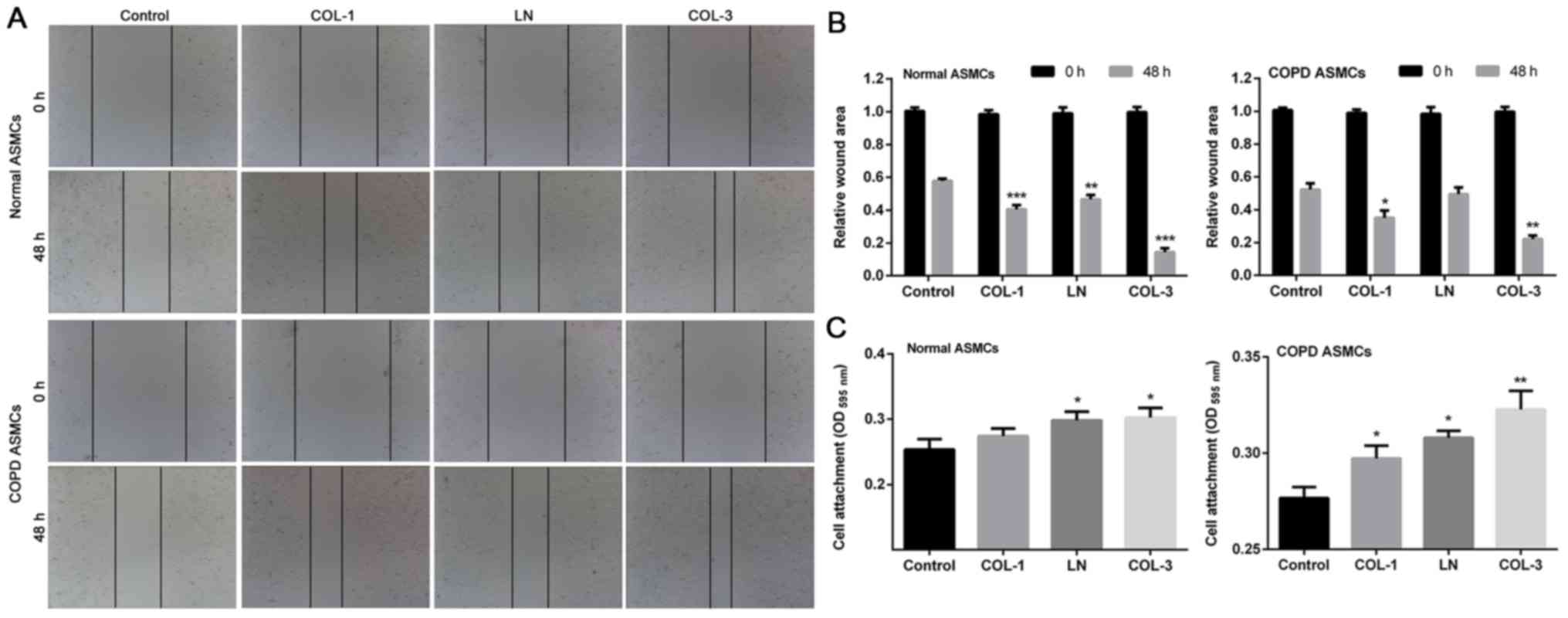 | Figure 3.Effects of ECM components on
migration and adhesion of ASMCs. Wound-healing assay was used to
evaluate motility of ASMCs following treatments ECM components,
including COL-1, LN and COL-3 in normal rat ASMCs and ASMCs form
rat models of COPD. The wound area was (A) visualized and (B)
calculated at 0 and 48 h. (C) Cell adhesion assay was performed in
ASMCs following treatment with COL-1, LN and COL-3. Data are
presented as the mean ± standard deviation in triplicate.
*P<0.05, **P<0.01 and ***P<0.001 vs. the control group.
ECM, extracellular matrix; COL, collagen; LN, laminin; ASMCs,
airway smooth muscle cells; COPD, chronic obstructive pulmonary
disease; OD, optical density. |
Effect of ECM components on factors
associated with inflammation, growth and migration of ASMCs
It is widely accepted that inflammation serves a
critical role in the pathogenesis of COPD (33,34).
Expression levels of factors associated with inflammation were
determined using ELISA. ECM components, including COL-1, LN and
COL-3, altered the levels of CXCL1, CXCL8 and IL-6 in ASMCs derived
from normal rats (Fig. 4A) and rat
models of COPD (Fig. 4B).
Expression of factors associated with growth and migration in ASMCs
was also determined by RT-qPCR and western blotting assays. The
results indicated that expression levels of TGFβ1 and FGF-1, both
associated with cell growth, were significantly elevated following
treatment with ECM components in ASMCs from normal rats and COPD
model rats (Fig. 5). Consistent
with the promoted cell migration ability, treatment with ECM
components markedly decreased expression of MMP-9 and increased
expression of TIMP1 in ASMCs derived from normal rats and rat
models of COPD. The above data suggests that ECM may promote
inflammation, proliferation and migration ability of ASMCs.
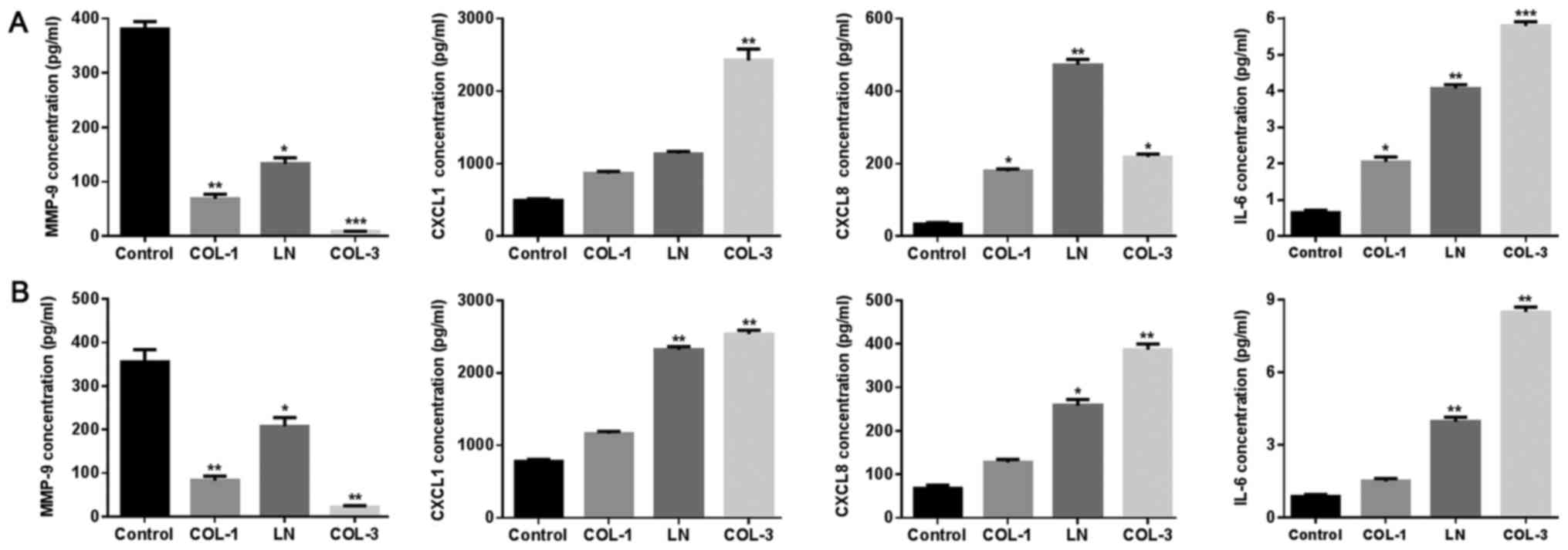 | Figure 4.Effects of ECM components on
cytokines in ASMCs. The expression of MMP-9, CXCL1, CXCL8 and IL-6
was determined following treatment with ECM components, including
COL-1, LN and COL-3 in (A) normal ASMCs and (B) ASMCs from rat
model of chronic obstructive pulmonary disease using ELISA assay.
Data are presented as the mean ± standard deviation in triplicate.
*P<0.05, **P<0.01 and ***P<0.001 vs. the control group.
ECM, extracellular matrix; ASMCs, airway smooth muscle cells;
MMP-9, matrix metalloproteinase-9; COL, collagen; LN, laminin;
CXCL, C-X-C motif chemokine ligand; IL-6, interleukin-6. |
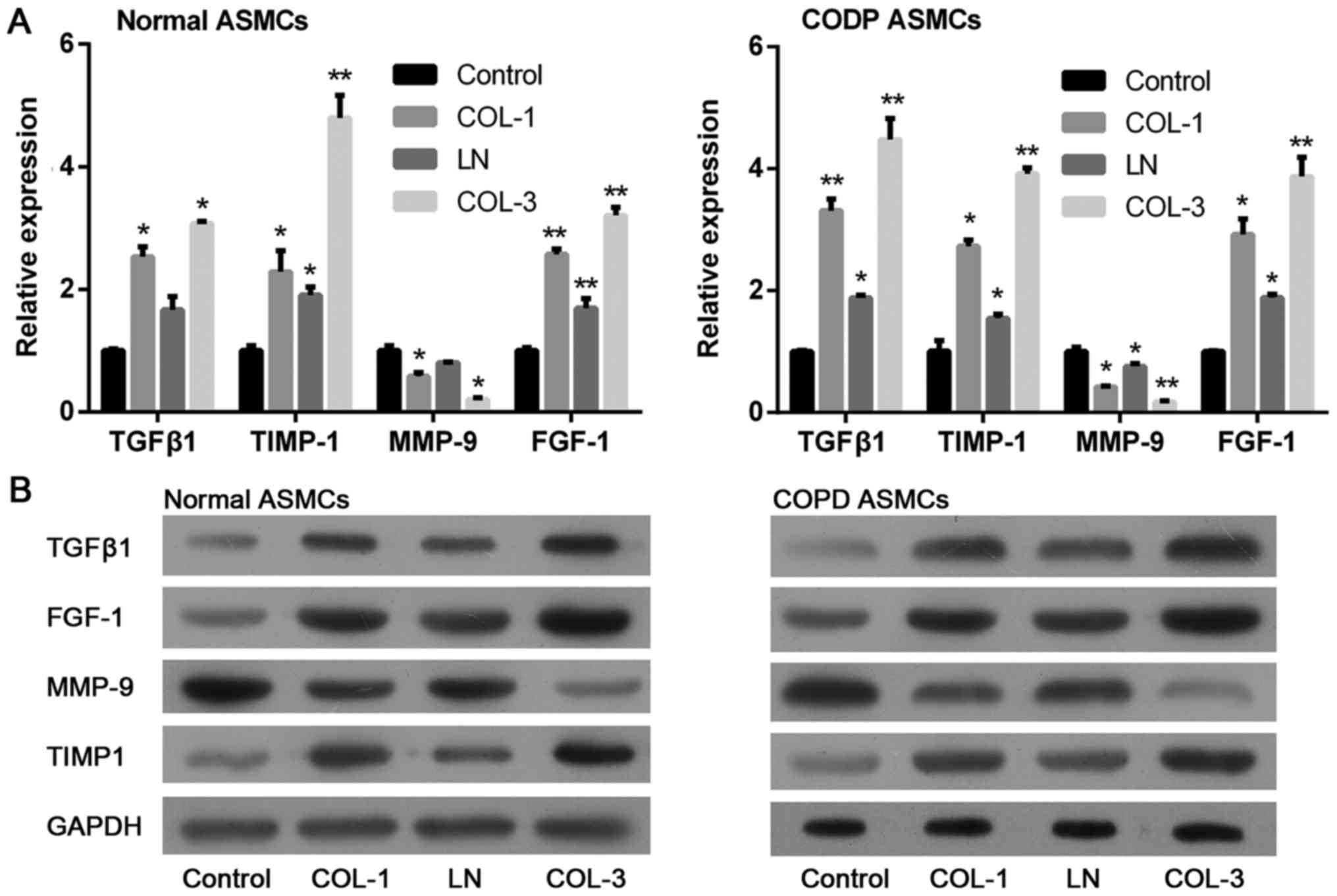 | Figure 5.Effects of ECM components on the
downstream molecules in ASMCs. Relative expression of TGFβ1, FGF-1,
MMP-9 and TIMP1 was determined in ASMCs following treatment with
ECM components, including COL-1, LN and COL-3 by (A) reverse
transcription-quantitative polymerase chain reaction and (B)
western blotting. Data are presented as the mean ± standard
deviation in triplicate. *P<0.05 and **P<0.01 vs. the control
group. ECM, extracellular matrix; ASMCs, airway smooth muscle
cells; COL, collagen; LN, laminin; TGFβ1, transforming growth
factor β-1; FGF-1, fibroblast growth factor-1; MMP-9, matrix
metalloproteinase-9; TIMP1, metalloproteinase inhibitor 1; COPD,
chronic obstructive pulmonary disease. |
Effects of ECM components of ASMCs in
rat models of COPD are mediated by the PI3K/AKT signaling
pathway
PI3K is a signaling pathway associated with cell
proliferation and migration (35,36).
Based on the above results, which indicated that ECM components
promoted migration and adhesion of ASMCs, the subsequent
experiments aimed to determine whether PI3K signaling pathway
mediated the effects of the ECM components. The above results
indicated that COL-3 was more efficient at promoting cell
proliferation and migration, compared with COL-1 and LN. Therefore,
COL-3-mediated promotion of adhesion and migration of ASMCs was
studied using specific PI3K inhibitor LY294002 or activator IGF-1.
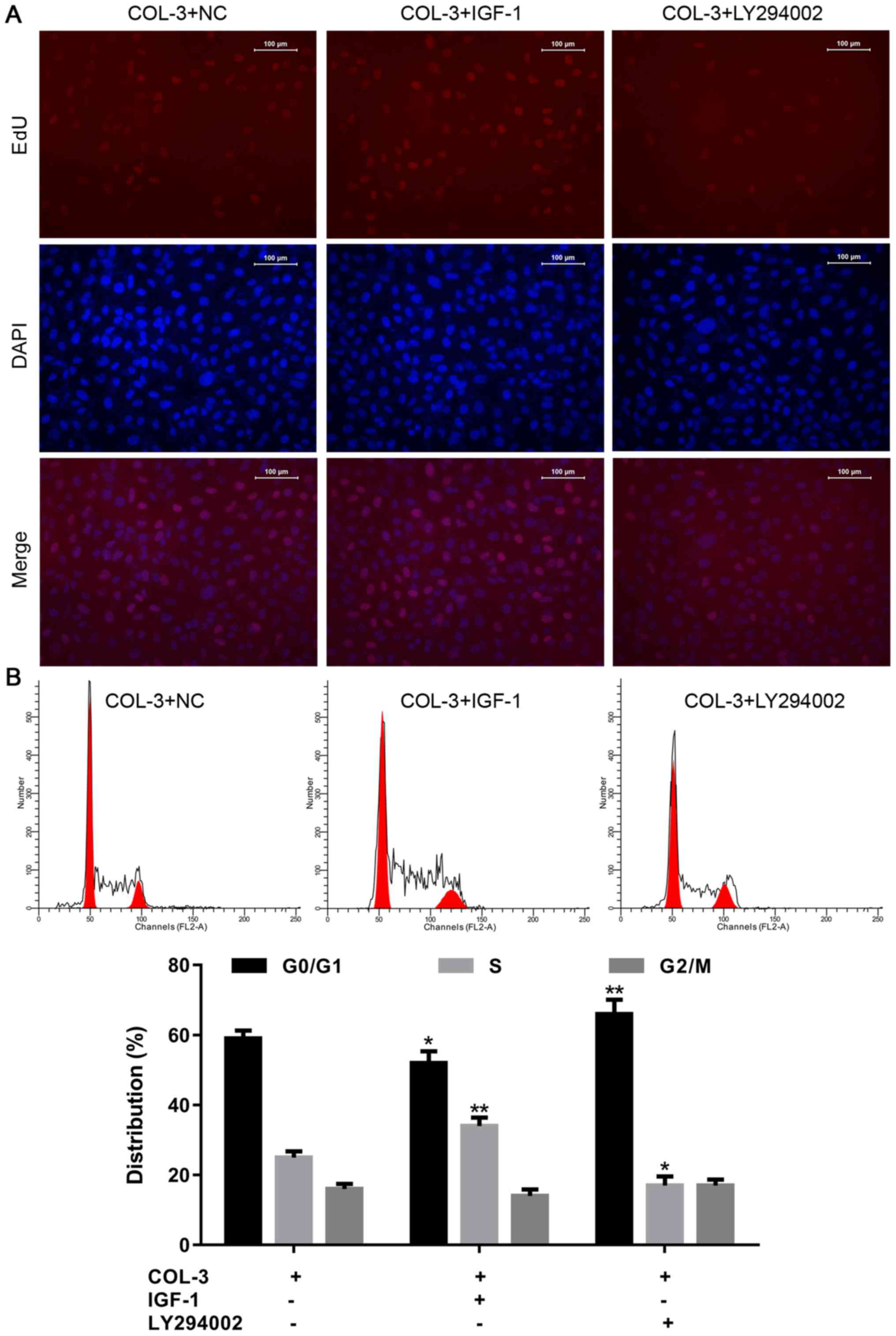
As demonstrated in Fig. 6A,
incubation of ASMCs form rat models of COPD with COL-3 and IGF-1
increased the proportion of EdU-positive cells, compared with cells
incubated with COL-3 only, indicating enhanced cell proliferation.
By contrast, incubation with COL-3 and LY294002 resulted in the
opposite effect. In the cell cycle assay, treatment with IGF1
significantly reduced the percentage of cells in
G0/G1 phase and increased the percentage of
cells in S phase. The opposite was the case following incubation
with LY294002 (Fig. 6B).
Furthermore, cell migration ability was enhanced by incubation with
IGF-1 and impaired following treatment with LY294002 in ASMCs
derived from rat models of COPD (Fig.
7A and B; P<0.01). At the molecular level, western blot
analysis confirmed that treatment with IGF1 markedly elevated the
level of TGFβ1, FGF-1, TIMP1, PI3K, p-AKT and p-mTOR, and decreased
the expression of MMP-9 (Fig. 7C).
Consistently, treatment with LY294002 induced the opposite effects
on these molecules. These results further demonstrated that the
effects of ECM components on ASMCs are associated with the
activation of the PI3K signaling pathway.
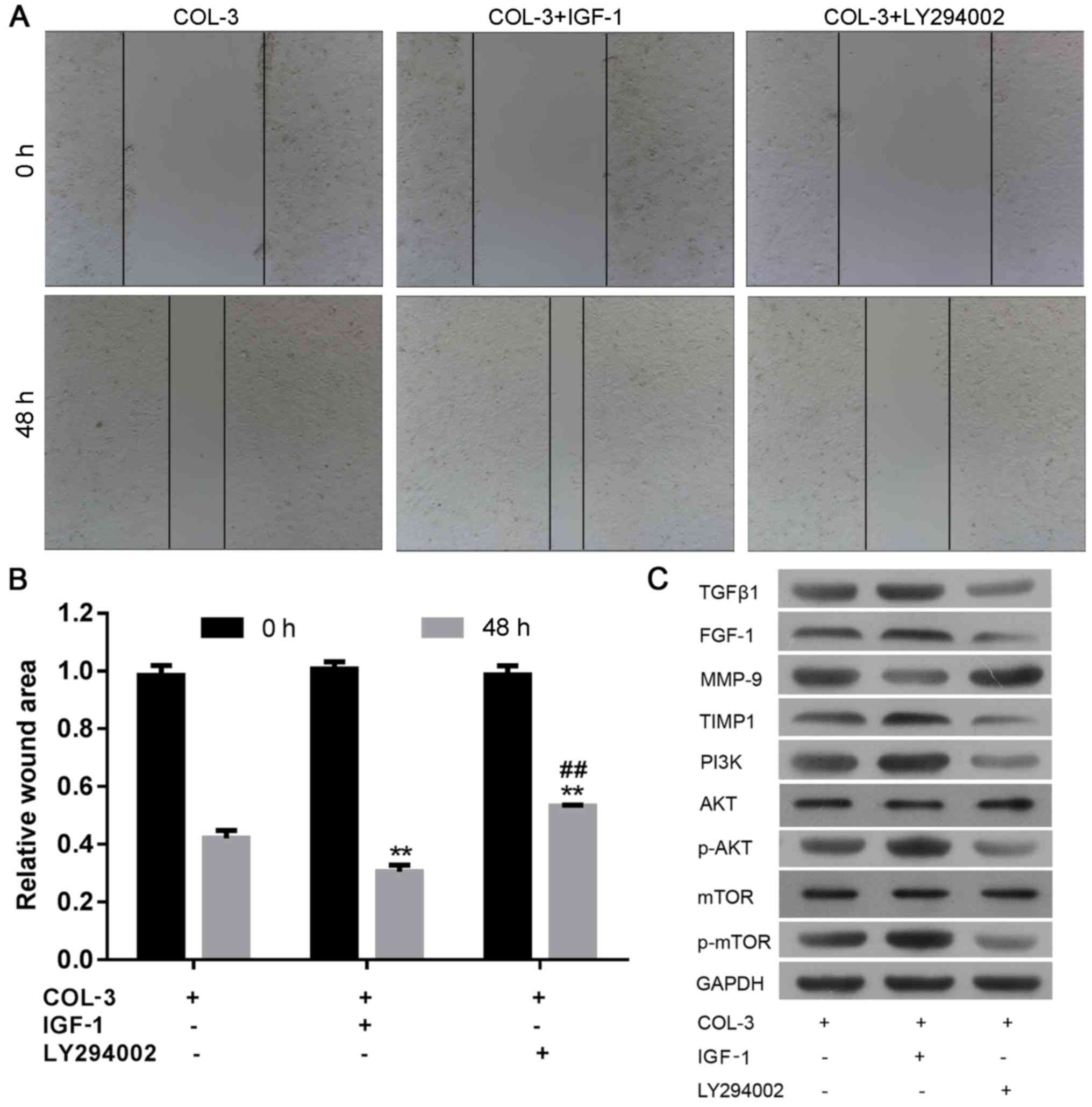 | Figure 7.Effects of extracellular matrix
components of ASMCs in rat models of chronic obstructive pulmonary
disease are mediated by the PI3K/AKT signaling pathway. The wound
area was (A) visualized and (B) calculated to evaluate motility of
ASMCs following combined treatment with COL-3 and PI3K inhibitor
LY294002 or activator IGF1. (C) Western blot analysis of components
of the PI3K/AKT signaling pathway in ASMCs following combined
treatment with COL-3 and PI3K inhibitor LY294002 or activator IGF1.
Data are presented as the mean ± standard deviation in triplicate.
**P<0.01 vs. the control group and ##P<0.01 vs.
the IGF1 treatment group. ASMCs, airway smooth muscle cells; COL,
collagen; IGF-1, insulin-like growth factor-1; TGFβ1, transforming
growth factor β-1; FGF-1, fibroblast growth factor-1; MMP-9, matrix
metalloproteinase-9; TIMP1, tissue inhibitors of
metalloproteinase-1; PI3K, phosphoinositide 3-kinase; p-, phospho-;
mTOR, serine/threonine-protein kinase mTOR. |
Discussion
COPD is a disorder typically caused by inhalation of
toxic gases and particles, which leads to impairments of lung
function, including emphysema, chronic bronchitis, mucus production
and irreversible airway obstruction (37). Alteration of ECM deposition is a
characteristic of COPD and provides a suitable environment for
airway wall remodeling, especially airway wall thickening (18). The present study aimed to determine
the response of ASMCs to the accumulation of components of ECM,
including COL-1, COL-3 or LN. COL-1 and COL-3 markedly increased
proliferation of ASMCs, whereas no alterations were detected in
cells treated with LN, indicating that the proliferative response
of ASMCs varied depending exposure to different components of ECM.
Consistently, the present study revealed similar results to
previous studies; components of ECM differentially affected
proliferation of vascular smooth muscle cells in cell culture
studies (38,39). Following treatment with COL-1 and
COL-3, there was an increased percentage of ASMCs in the S phase.
Migration of cells is controlled by multiple stages, including
extension of the lamellipodium, formation of new adhesions and
retraction of protrusions (40).
Correct modulation of adhesion and de-adhesion are necessary for
cell migration (40). In the
present study, migratory responses to treatment with ECM substrates
were estimated using a wound-healing assay. Culture dishes treated
with COL-1, COL-3 or LN exhibited relatively decreased wound area
in ASMCs from normal rats and rat models of COPD, compared with the
respective controls. The data suggests that these three ECM
components promote migration of ASMCs in culture. Compared with the
control, ASMCs from normal rats and the COPD model exhibited
enhanced attachment following stimulation with COL-1, LN or COL-3,
compared with the control. Increased cell adhesion may contribute
to migration of ASMCs induced by treatment with COL-1, COL-3 or
LN.
COPD results in aberrant thickening of generalized
airway walls, particularly in smokers, with airway walls thickened
throughout the lung (41).
Hyperplasia and hypertrophy of the smooth muscle are associated
with the pathology of COPD (42).
Induction of proliferation, migration and attachment by ECM
components in the present study lead to investigation of the
effects on cytokines, growth factors, MMPs, TIMPs and chemokines
synthesized by ASMCs. The level of MMP-9 was downregulated, while
the concentration of its inhibitor TIMP1 was upregulated in ASMCs
from normal rats and the COPD model rats following treatment with
the ECM molecules. Previous studies demonstrated that the activated
and precursor forms of MMP-9 may bind to its inhibitor TIMP-1 to
form a complex, thus the ratio of MMP-9 and TIMP-1 is closely
associated with the degradation and accumulation ECM (43). An increased TIMP-1/MMP-9 ratio
facilitates TGFβ1 induced synthesis of ECM substrates, including
COL-1, COL-2, COL-5 and fibronectin in human ASMCs. Furthermore,
TGFβ has been demonstrated to promote expression of certain other
ECM proteins, including perlecan, elastin, LN, COL-3, COL-4 and
thrombospondin (18,44). In the present study, mRNA and
protein expression levels of TGFβ1 were evaluated following
treatment with COL-1, COL-3 or LN, thus it was suggested that TGFβ1
induced over production of multiple ECM proteins in ASMCs in COPD
rats. The results of the present study suggest that there may be an
association between ECM and TGFβ1 in rats. Cell proliferation and
migration are promoted by ECM proteins (12). In the cells treated with COL-1,
COL-3 and LN, an upregulation of TGFβ1 and alteration of ECM was
identified, which may result in ASMC hyperplasia and migration.
It has been previously reported that cytokines,
chemokines and growth factors may influence the secretion of ECM
proteins by ASMCs and therefore impact biological behavior of ASMCs
(12). ECM proteins are considered
to promote production of cytokines and chemokines by ASMCs, which
are associated with the development of COPD pathophysiology
(12). In the present study, the
pro-inflammatory chemokines CXCL1 and CXCL8, cytokine IL-6, and
FGF-1 were increased in ASMCs by treatment with ECM molecules. The
imbalance in ECM homeostasis causes hyper proliferative and
migratory phenotypes in ASMCs.
PI3K is a signaling pathway associated with
proliferation and migration of ASMCs (45,46).
Incubation of rat ASMCs with PI3K agonist IGF-1 induced progression
to S-phase and enhanced migration, whereas the opposite effect was
observed following treatment with PI3K inhibitor LY294002. Protein
expression levels of PI3K, p-AKT and p-mTOR, which have roles in
PI3K signaling, were upregulated by treatment with IGF-1 and
decreased following treatment with LY294002. These results indicate
that activation of the PI3K signaling promotes proliferation and
migration of ASMCs. Additionally, protein expression levels of
TGFβ1, FGF-1 and TIMP1 were upregulated by treatment of IGF-1,
while the effect was reversed by treatment with LY294002. Stewart
et al (47) demonstrated
that FGFs can stimulate the PI3K signaling pathway to induce
proliferation of ASMCs in COPD. In addition, TGF and collagen
proteins also regulate the PI3K signaling pathway in human ASMCs
(48). Following treatment with
IGF-1, alterations in the levels of TGFβ1, FGF-1, MMP-9 and TIMP1
may result in ECM imbalance, and the PI3K signaling may be
activated by TGFβ1 and FGF-1, leading to induction of proliferation
and migration.
In conclusion, the present study demonstrated that
COL-1, COL-3 and LN are effective stimulators of proliferation,
migration and attachment of rat AMSCs. Treatment with ECM
components enhanced the expression of factors associated with
inflammation, growth and migration. COL-3 induced proliferation,
migration and progression to S-phase in ASMCs, partially though
activation of the PI3K signaling pathway. The present study may
contribute to further understanding of the molecular mechanisms
underlying the COPD airway wall thickening.
Acknowledgements
Not applicable.
Funding
The present study was supported by Hubei University
of Medical Sciences Graduate Student Project (Shiyan, China; grant
no. 2011QDZR-28), Hubei Province Students Innovation Training
Project (grant no. 201610929022) and Research Project Guidance
Project founded by Hubei Provincial Department of Education (Wuhan,
China; grant no. B2017486).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZW designed the experiment, performed the
statistical analysis and interpretation of data and wrote the
manuscript. RL and RZ conducted the experiments and made
substantial contributions to conception and design, and acquisition
of data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Animal Research Committee of Hubei University of Medicine (Suizhou,
China). The care and use of animals was based on the Guidelines of
the National Institutes of Health.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Asia Pacific CRG: Global Initiative for
Chronic Obstructive Lung Disease strategy for the diagnosis,
management and prevention of chronic obstructive pulmonary disease:
An Asia-Pacific perspective. Respirology. 10:9–17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
GBD 2015 Chronic Respiratory Disease
Collaborators: Global, regional, and national deaths, prevalence,
disability-adjusted life years, and years lived with disability for
chronic obstructive pulmonary disease and asthma, 1990–2015: A
systematic analysis for the Global Burden of Disease Study 2015.
Lancet. Respir Med. 5:691–706. 2017.
|
|
3
|
Celli BR, MacNee W and Force AET:
Standards for the diagnosis and treatment of patients with COPD: A
summary of the ATS/ERS position paper. Eur Respir J. 23:932–946.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Agusti A, MacNee W, Donaldson K and Cosio
M: Hypothesis: Does COPD have an autoimmune component? Thorax.
58:832–834. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van Eeden SF and Sin DD: Oxidative stress
in chronic obstructive pulmonary disease: A lung and systemic
process. Can Respir J. 20:27–29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saetta M, Di Stefano A, Turato G, Facchini
FM, Corbino L, Mapp CE, Maestrelli P, Ciaccia A and Fabbri LM: CD8+
T-lymphocytes in peripheral airways of smokers with chronic
obstructive pulmonary disease. Am J Respir Crit Care Med.
157:822–826. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Peinado VI, Barbera JA, Abate P, Ramírez
J, Roca J, Santos S and Rodriguez-Roisin R: Inflammatory reaction
in pulmonary muscular arteries of patients with mild chronic
obstructive pulmonary disease. Am J Respir Crit Care Med.
159:1605–1611. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Celli BR: Pathophysiology of chronic
obstructive pulmonary disease. Respir Care Clin N Am. 4:359–370.
1998.PubMed/NCBI
|
|
9
|
Aoshiba K and Nagai A: Differences in
airway remodeling between asthma and chronic obstructive pulmonary
disease. Clin Rev Allergy Immunol. 27:35–43. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chung KF: The role of airway smooth muscle
in the pathogenesis of airway wall remodeling in chronic
obstructive pulmonary disease. Proc Am Thorac Soc. 2:347–354. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ge Q, Chen L, Jaffar J, Argraves WS, Twal
WO, Hansbro P, Black JL, Burgess JK and Oliver B: Fibulin1C peptide
induces cell attachment and extracellular matrix deposition in lung
fibroblasts. Sci Rep. 5:94962015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Parameswaran K, Willems-Widyastuti A,
Alagappan VK, Radford K, Kranenburg AR and Sharma HS: Role of
extracellular matrix and its regulators in human airway smooth
muscle biology. Cell Biochem Biophys. 44:139–146. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Merrilees MJ, Ching PS, Beaumont B, Hinek
A, Wight TN and Black PN: Changes in elastin, elastin binding
protein and versican in alveoli in chronic obstructive pulmonary
disease. Respir Res. 9:412008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
van Straaten JF, Coers W, Noordhoek JA,
Huitema S, Flipsen JT, Kauffman HF, Timens W and Postma DS:
Proteoglycan changes in the extracellular matrix of lung tissue
from patients with pulmonary emphysema. Modern Pathol. 12:697–705.
1999.
|
|
15
|
Vlahovic G, Russell ML, Mercer RR and
Crapo JD: Cellular and connective tissue changes in alveolar septal
walls in emphysema. Am J Respir Crit Care Med. 160:2086–2092. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Annoni R, Lancas T, Tanigawa Yukimatsu R,
de Medeiros Matsushita M, de Morais Fernezlian S, Bruno A, da Silva
Fernando Ferraz L, Roughley PJ, Battaglia S, Dolhnikoff M, et al:
Extracellular matrix composition in COPD. Eur Respir J.
40:1362–1373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Johansson O, Erjefält J, Bjermer L,
Löfdahl C-G, Westergren-Thorsson G and Hallgren O: Aberrant
intracellular expression of laminin α-2 and −5 in bronchiolar
epithelium of COPD patients. Eur Respir J. 14:39552014.
|
|
18
|
Johnson PR, Black JL, Carlin S, Ge Q and
Underwood PA: The production of extracellular matrix proteins by
human passively sensitized airway smooth-muscle cells in culture:
The effect of beclomethasone. Am J Respir Crit Care Med.
162:2145–2151. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Elshaw SR, Henderson N, Knox AJ, Watson
SA, Buttle DJ and Johnson SR: Matrix metalloproteinase expression
and activity in human airway smooth muscle cells. Br J Pharmacol.
142:1318–1324. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
McKay S, de Jongste JC, Saxena PR and
Sharma HS: Angiotensin II induces hypertrophy of human airway
smooth muscle cells: Expression of transcription factors and
transforming growth factor-beta1. Am J Respir Cell Mol Biol.
18:823–833. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Berezin V, Walmod PS, Filippov M and
Dityatev A: Targeting of ECM molecules and their metabolizing
enzymes and receptors for the treatment of CNS diseases. Prog Brain
Res. 214:353–388. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hinderer S, Layland SL and Schenke-Layland
K: ECM and ECM-like materials-Biomaterials for applications in
regenerative medicine and cancer therapy. Adv Drug Deliv Rev.
97:260–269. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Trapani V, Bonaldo P and Corallo D: Role
of the ECM in notochord formation, function and disease. J Cell
Sci. 130:3203–3211. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Khatiwala CB, Kim PD, Peyton SR and Putnam
AJ: ECM compliance regulates osteogenesis by influencing MAPK
signaling downstream of RhoA and ROCK. J Bone Miner Res.
24:886–898. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mruthyunjaya S, Manchanda R, Godbole R,
Pujari R, Shiras A and Shastry P: Laminin-1 induces neurite
outgrowth in human mesenchymal stem cells in serum/differentiation
factors-free conditions through activation of FAK-MEK/ERK signaling
pathways. Biochem Biophys Res Commun. 391:43–48. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nie YC, Wu H, Li PB, Luo YL, Zhang CC,
Shen JG and Su WW: Characteristic comparison of three rat models
induced by cigarette smoke or combined with LPS: To establish a
suitable model for study of airway mucus hypersecretion in chronic
obstructive pulmonary disease. Pulm Pharmacol Ther. 25:349–356.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Geng L, Chen Z, Ren H, Niu X, Yu X and Yan
H: Effects of an early intervention using human amniotic epithelial
cells in a COPD rat model. Pathol Res Pract. 212:1027–1033. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang C, Feng L, Li M, Dong C and Zhang W:
Effects of Xiaoqinglong decoction on gene expression profiles in a
rat chronic obstructive pulmonary disease model. Biosci Trends.
6:262–269. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Placeres-Uray FA, Febres-Aldana CA,
Fernandez-Ruiz R, de Alfonzo Gonzalez R, de Becemberg Lippo IA and
Alfonzo MJ: M2 muscarinic acetylcholine receptor modulates rat
airway smooth muscle cell proliferation. World Allergy Organ J.
6:222013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Durand-Arczynska W, Marmy N and Durand J:
Caldesmon, calponin and alpha-smooth muscle actin expression in
subcultured smooth muscle cells from human airways. Histochemistry.
100:465–471. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
He F, Li B, Zhao Z, Zhou Y, Hu G, Zou W,
Hong W, Zou Y, Jiang C, Zhao D and Ran P: The pro-proliferative
effects of nicotine and its underlying mechanism on rat airway
smooth muscle cells. PloS One. 9:e935082014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hakansson K, Konge L, Thomsen SF, Backer V
and von Buchwald C: Sinonasal inflammation in COPD: A systematic
review. Eur Res J. 42:1402–1411. 2013. View Article : Google Scholar
|
|
34
|
Loukides S, Bartziokas K, Vestbo J and
Singh D: Novel anti-inflammatory agents in COPD: Targeting lung and
systemic inflammation. Curr Drug Targets. 14:235–245. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang Y, Wan D, Zhou R, Zhong W, Lu S and
Chai Y: Geraniin inhibits migration and invasion of human
osteosarcoma cancer cells through regulation of PI3K/Akt and ERK1/2
signaling pathways. Anticancer dsrugs. 2017. View Article : Google Scholar
|
|
36
|
Li M, Cheng W, Luo J, Hu X, Nie T, Lai H,
Zheng X, Li F and Li H: Loss of selenocysteine insertion sequence
binding protein 2 suppresses the proliferation, migration/invasion
and hormone secretion of human trophoblast cells via the PI3K/Akt
and ERK signaling pathway. Placenta. 55:81–89. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wood AM and Stockley RA: The genetics of
chronic obstructive pulmonary disease. Respir Res. 7:1302006.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yamamoto M, Yamamoto K and Noumura T: Type
I collagen promotes modulation of cultured rabbit arterial smooth
muscle cells from a contractile to a synthetic phenotype. Exp Cell
Res. 204:121–129. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Thyberg J and Hultgardh-Nilsson A:
Fibronectin and the basement membrane components laminin and
collagen type IV influence the phenotypic properties of subcultured
rat aortic smooth muscle cells differently. Cell Tissue Res.
276:263–271. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huynh-Do U, Vindis C, Liu H, Cerretti DP,
McGrew JT, Enriquez M, Chen J and Daniel TO: Ephrin-B1 transduces
signals to activate integrin-mediated migration, attachment and
angiogenesis. J Cell Sci. 115:3073–3081. 2002.PubMed/NCBI
|
|
41
|
Hashimoto M, Tanaka H and Abe S:
Quantitative analysis of bronchial wall vascularity in the medium
and small airways of patients with asthma and COPD. Chest.
127:965–972. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hirst SJ, Twort CH and Lee TH:
Differential effects of extracellular matrix proteins on human
airway smooth muscle cell proliferation and phenotype. Am J Respir
Cell Mol Biol. 23:335–344. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Roderfeld M, Graf J, Giese B,
Salguero-Palacios R, Tschuschner A, Müller-Newen G and Roeb E:
Latent MMP-9 is bound to TIMP-1 before secretion. Biol Chem.
388:1227–1234. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Black PN, Young PG and Skinner SJ:
Response of airway smooth muscle cells to TGF-beta 1: Effects on
growth and synthesis of glycosaminoglycans. Am J Physiol.
271:L910–917. 1996.PubMed/NCBI
|
|
45
|
Liu Y, Yang K, Sun X, Fang P, Shi H, Xu J,
Xie M and Li M.: MiR-138 suppresses airway smooth muscle cell
proliferation through the PI3K/AKT signaling pathway by targeting
PDK1. Exp Lung Res. 41:363–369. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Goueffic Y, Guilluy C, Guerin P, Patra P,
Pacaud P and Loirand G: Hyaluronan induces vascular smooth muscle
cell migration through RHAMM-mediated PI3K-dependent Rac
activation. Cardiovascular Res. 72:339–348. 2006. View Article : Google Scholar
|
|
47
|
Stewart DM, Tian L and Nelson DL: Linking
cellular activation to cytoskeletal reorganization: Wiskott-Aldrich
syndrome as a model. Curr Opin Allergy Clin Immunol. 1:525–533.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Stamatiou R, Paraskeva E, Gourgoulianis K,
Molyvdas PA and Hatziefthimiou A: Cytokines and growth factors
promote airway smooth muscle cell proliferation. ISRN Inflamm.
2012:7314722012. View Article : Google Scholar : PubMed/NCBI
|