Introduction
Familial dilated cardiomyopathy (FDC) has been
identified in at least two closely related patients with dilated
cardiomyopathy (DCM) meeting the criteria for idiopathic dilated
cardiomyopathy (IDC). In addition, DCM has a complex pathogenesis.
Genetic screening methods have demonstrated that common variants in
numerous genes are present in severe DCM cases, indicating that
patients with DCM have complex multi-variant or oligogenic genetic
backgrounds (1–3). It has been suggested that up to 50%
of idiopathic DCM cases may be attributed to genetic mutations
(4). It may therefore be suggested
that family heredity has a major role in the pathogenesis of DCM.
Novel diagnosis of IDC has indicated FDC in first-degree family
members at a minimum rate of 20–35% via clinical screening. Diverse
gene ontogeny, or more specifically, point mutations in 31
autosomal and 2 X-linked genes, have been reported to be associated
with the incidence of FDC; however, such factors account for 30–35%
of the genetic causes of FDC (1).
The majority of patients with FDC exhibit a dominant inheritance
pattern; however, a small minority of patients with DCM possessing
other inheritance patterns have been reported, including autosomal
recessive, X-linked and mitochondrial modes of inheritance
(1). Hence, a published clinical
guideline strongly suggests that families with a history of
cardiomyopathy should undergo genetic testing and screening in the
clinic (4). These genes and loci
have been identified in numerous families, coding for a variety of
cardiomyocyte proteins of the cytoskeleton, nuclear membrane, the
sarcomere, ion channels and the sarcolemma. The proportion of
protein-coding genes with pathogenic mutations resulting in FDC
include that of Titin (15–27%), lamins A/C (LMNA; 6%),
β-myosin heavy chain (4.20%), myopalladin (3.50%) and cardiac
troponin T (2.90%) (5). Titin is a
large cytoskeletal protein of the muscle, which regulates muscle
extensibility and fiber elasticity; lamins A/C stabilize the
nuclear membrane and provides mechanical support. Furthermore,
β-myosin heavy chain, myopalladin and cardiac troponin T are
sarcoma proteins, all of which serve roles in regulating muscle
contraction. Phospholamban, encoded by PLN, is a
sarcoplasmic reticulum protein that can inhibit the sarcoplasmic
reticulum Ca2+-ATPase pump and regulate muscle function.
As many as 6% of patients with DCM possess mutations in
LMNA, among of which almost 30–33% of these patients with
DCM suffered from conduction system disorders (6,7). In
addition, carriers of LMNA and PLN mutations were
more likely to develop malignant ventricular arrhythmia and
end-stage heart failure (8).
LMNA is located on chromosome 1q21-22 and is
an autosomal dominant gene constituting 12 exons, and encodes two
isomers, lamin A and lamin C. Lamins A/C are transcript shears of
LMNA mRNA that are selectively translated and contain 566
similar residues proximal to the N-terminus. Selective cleavage
within exon 10 results in two different mRNAs encoding prelamin A
and lamin C. Prelamin A constitutes 664 residues, the precursor
protein of lamin; its C-terminus is modified by farnesylation.
Following the loss of 3 amino acids, C-terminal methylation and
cleavage of the internal soluble protein occur, a synthetic mature
version of lamin A is produced, constituting 646 amino acids, which
is then inserted into the nuclear lamina. Lamins A/C contain the
‘rod’ functional region of an α2 helix structure comprising 360
amino acid residues, which is composed of 7 repeating hydrophobic
amino acid sequences; the components on each end are known as the
N-terminal ‘head’ and the C-terminal ‘tail’.
Exon 1 is responsible for encoding the first region
of the N-terminal head; exons 2–6 encode the remaining central
rod-shaped region, whereas exons 7–9 encode the C-terminal region,
including the nuclear localization signal region and the binding
domain for nuclear intermediate filament protein to DNA (9,10).
Cell nuclear lamins that localize to the inner nuclear membrane
consist of lamins A/C, and B, the latter is encoded by the
LMNB gene, and provides mechanical support to maintain the
integrity of the nuclear membrane (11). In addition, lamins A/C have been
reported crucial for the function of a chromosome anchor site,
periodic nuclear degradation and assembly, as well as the
regulation cellular proliferation, differentiation and apoptosis,
cellular signal transduction, chromosome segregation, gene
regulation and DNA repair (12).
Materials and methods
Clinical data of the proband and
familial members
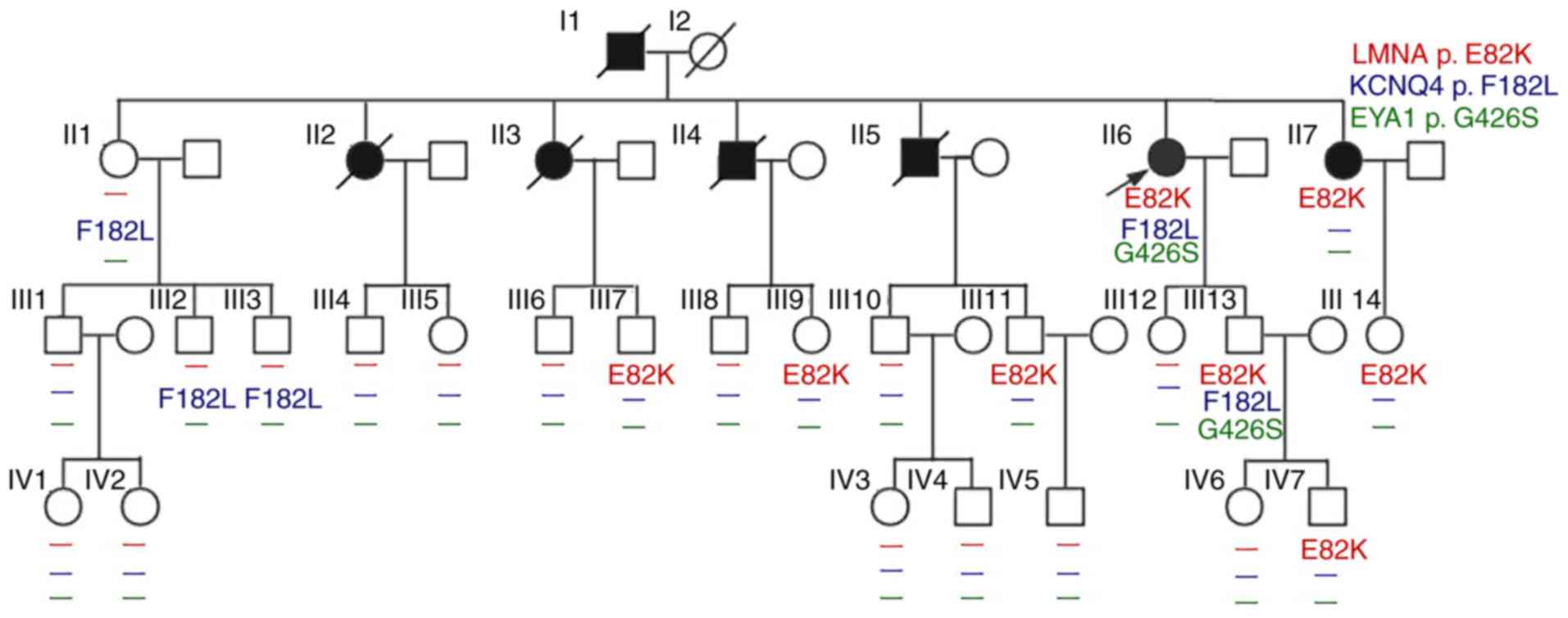
The present study investigated the clinical data of
the proband and familial members (Table I; Fig.
1). The proband (II-6) was a 54-year-old female whose main
symptoms were recurrent chest congestion and pain with shortness of
breath for >1 year. The patient was admitted to Fujian
Provincial Hospital (Fuzhou, China) in March 2008. The results of
physical examination revealed a pulse of >60 beats per minute
(BPM), blood pressure of 137/67 mmHg, a lack of jugular vein
distension and clear breathing without rales. Furthermore, the
relative cardiac dullness expanded to both sides, the heart rate
was 60 BPM and heart rhythm was uniform; a 3/6 grade systolic
murmur in accordance with the Levine classification method
(13) at the second intercostal
space of the left sternal border was reported and no lower
extremity edema was observed. Repeated electrocardiogram
examinations indicated that the patient had frequent ventricular
premature beats (bigeminy), poor R wave progression on the
V1-V4 leads, sinus bradycardia, first-degree
atrioventricular block, notable U waves, a low ST segment on part
of the leads and T-wave inversion. Echocardiographic screening
revealed that from the perspective of the parasternal long-axis
view of the left ventricle (LV), the left atrial diameter was 3.76
cm and the LV end-diastolic diameter was 5.68 cm, the right atrial
diameter (apical four-chamber view, AP4) was 4.18 cm, and the right
ventricular diameter (RVD; AP4) was 4.06 cm. Additionally, the LV
ejection fraction (LVEF) was 35% and the LV fractional shortening
was 23%, which suggested decreased overall motion of the left
ventricular wall. Furthermore, a reduced LVEF, LV enlargement with
moderate mitral valve regurgitation, and right atrium and RV
enlargement with moderate tricuspid regurgitation were observed.
There was no previous history of heart disease due to the
enlargement of heart, smoking and alcohol consumption of the
proband.
 | Table I.Clinical data of some members in a
family associated with dilated cardiomyopathy. |
Table I.
Clinical data of some members in a
family associated with dilated cardiomyopathy.
| Patient | Age (years) | Gender | Dynamic
electrocardiogram/Holter | NYHA | Cardiac symptoms at
onset (age, years) | Left ventricular
end-diastolic diameter (mm) | Left ventricular
ejection fraction (%) | LMNA p.E82K
mutation |
|---|
| II-1 | 69 | Female | N |
|
| N | N | Negative |
| II-2 | 50a | Female | N |
| 40 | N | N | – |
| II-3 | 48a | Female | N |
| 36 | N | N | – |
| II-4 | 45a | Male | N |
| 35 | N | N | – |
| II-5 | 57a | Male | Pre-pacing,
abnormal Q wave in leads V1-V4, left
ventricular high voltage, slow JR with sino-capture, CRBBB, ST-T
change, paired PVC, sinus arrest, paroxysmal SR; post-pacing,
Frequent PVC, ST-T change, AAI | III–IV | 54 | 69.6 | 45 | – |
| II-6 | 54 | Female | Pre-pacing,
frequent PVC (bigeminal rhythm), degree I AVB, slow JR, accelerated
atrioventricular rhythm, ST-T Change, poor R-wave progression in
leads V1-V4, post-pacing, paroxysmal AF,
occasional PVC, AAI | II–III | 44 | 56.8 | 35 | Heterozygous |
| II-7 | 52 | Female | Pre-pacing, slow
AF, degree III AVB, JR, ST-T change in some leads, Poor R-wave
progression in leads V1-V4, post-pacing, AF,
AAI | II–III | 47 | 50.2 | 62.3 | Heterozygous |
| III-3 | 41 | Male | N | I |
| 42.5 | 62 | Negative |
| III-7 | 35 | Male | N | II |
| 49.6 | 55 | Heterozygous |
| III-10 | 38 | Male | N | I |
| 44.3 | 72 | Negative |
| III-11 | 34 | Male | Degree I AVB | II |
| 51.5 | 56 | Heterozygous |
| III-13 | 30 | Male | SR, ST-T
change | II |
| 51.5 | 56 | Heterozygous |
| IV-6 | 8 | Female | N |
|
| 30.9 | 65 | Negative |
| IV-7 | 4 | Male | Sinus
arrhythmia |
|
| 28.6 | 73 | Heterozygous |
Myocardial perfusion imaging demonstrated that the
left ventricular cavity was slightly enlarged, the thickness of the
ventricular wall was uneven, the perfusion of blood to the LV was
decreased, and the anterior wall was close to the apex and the
posterior wall. This was consistent with the phenotypic
characteristics for DCM (14), as
determined by the New York Heart Association Standard Committee
revised cardiac function classification (15). A Holter monitor indicated a
junctional rhythm with an average heartrate of 44 BPM. The longest
R-R interval was 1.69 sec; there were 2,431 instances where the R-R
interval was longer than 1.6 sec. An occasional ventricular
premature beat (22 beats/24 h, one paired beat/24 h, dual-source),
paroxysmal ventricular tachycardia (2 times/24 h) and a ST-T change
were also observed. Subsequently, the patient underwent permanent
pacemaker implantation. After 6.5 years following hospital
admission, the patient had permanent resynchronization pacemaker
replacement surgery again as the battery of the pacemaker had worn.
After 7 months following the operation, the Holter monitor revealed
paroxysmal atrial fibrillation with the pacemaker where the fastest
ventricular rate was as high as 86 BPM (atrial fibrillation) and
the lowest ventricular rate was as low as 60 BPM (pacemaker heart
rate). The pacemaker functioned as a dual-chamber model with a
frequency of 60 BPM and a ventricular premature beat frequency of
96 beats per 24 h.
In the second generation, a total of 3 patients with
DCM (II-5, −6 and −7) underwent permanent pacemaker implantation
due to sick sinus syndrome and unstable cardiac electrophysiology.
Additionally, two sisters (II-2 and II-3) and an elder brother
(II-4) of the proband (II-6) in this family succumbed to mortality
due to heart disease or heart failure at 40–50-years-old. An
additional family member (III-3) revealed a slight decline in
hearing ability of the left ear via an audiometry examination;
however, the hearing of II-6, II-1, III-2 and III-13, and the right
ear hearing of III-3 was normal. Pedigree analysis revealed no
family members with auricle or external auditory canal deformities,
preauricular fistula, branchial cleft anomalies, renal dysplasia,
or any other symptoms.
DNA extraction, capture of target gene
and library preparation
Peripheral blood samples (2 ml) obtained from the
forearm were used to extract genomic DNA using the QIAamp DNA Blood
Mini kit (Qiagen GmbH, Hilden, Germany). The concentration and
purity of DNA were measured with a Nanodrop 1000 spectrophotometer
(NanoDrop Technologies; Thermo Fisher Scientific, Inc., Pittsburgh,
PA, USA) according to the manufacturer's protocol.
Amplification of target genes was performed using
the Ion Ampliseq™ Inherited Disease Panel (Thermo Fisher
Scientific, Inc.) as mentioned below. In order to arrange the genes
according to their sequences, a next-generation sequencing (NGS)
library was prepared. A total of 20 ng gDNA obtained from each
sample was quantified using a Qubit 2.0 fluorometer (Invitrogen;
Thermo Fisher Scientific, Inc.), and multiplex PCR amplification
using each of the two primer-pools was subsequently performed in
accordance with the manufacturer's instructions. Following the
mixing of the resultant amplicons from the two primer-pools, the
amplicons were ligated to barcodes and Ion Torrent adapters (Thermo
Fisher Scientific, Inc.). Agencourt AMPure XP beads (Beckman
Coulter, Inc., Brea, CA, USA) were then utilized for purification
of the amplified libraries using an Ion AmpliSeq Library kit 2.0,
which was performed in accordance with the manufacturer's
instructions (Thermo Fisher Scientific, Inc.) (16).
Personal Genome Machine (PGM)
sequencing and variant calling
Ion 318 chips were employed with the Ion PGM system
(Thermo Fisher Scientific, Inc.). Ion Torrent Suite Software v3.6.2
(Thermo Fisher Scientific, Inc.) was used to i) collect data of Ion
Torrent reads, ii) determine trim adapter sequences (clean data),
iii) align clean data to the hg19 human reference genome, iv)
analyze coverage of target sequence and v) variant calling
(16). Furthermore, in accordance
with the quality score system of Ion Torrent, the software scores
for the quality of the reads were computed via assignment of the
Q17 and Q20 scores. Following this, primers of overlapping
amplicons covering the coding sequence region and flanking
sequences of each target gene were designed by Ion AmpliSeq™
Ready-to-Use custom designer platform (v7.0.2; http://www.ampliseq.com/protected/dashboard.action).
The reaction system and conditions of polymerase chain reaction
(PCR) were performed using the AmpliSeq™ Inherited Disease Panel
(Thermo Fisher Scientific, Inc.) in accordance with the
manufacturer's instructions. Multiple PCR amplifications were
performed for 325 genes associated with genetic disease (61 heart
disease-associated genes, such as LMNA) and the region near
the splice sites. Mutated polymorphic sites that had a global minor
allele frequency (MAF) of ≤1% were screened, which was calculated
from the population frequency information regarding dbSNP obtained
from the 1000 genomes project (17). The identified mutation points were
compared via Ion reporter 5.0 (https://ionreporter.thermofisher.com/ir/) against the
following databases: NCBI dbSNP database (https://www.ncbi.nlm.nih.gov/snp/), the 1000-Genome
project database (http://www.internationalgenome.org/category/dbsnp/),
UCSC common SNP database (https://genome.ucsc.edu/goldenpath/gbdDescriptionsOld.html)
and the 5000 Exmoes database in Exome Sequencing Project
(https://ionreporter.thermofisher.com/ir/). The SIFT
(SIFT v4.0.3; http://provean.jcvi.org/index.php) and PolyPhen-2
(Polymorphism Phenotyping v2; http://genetics.bwh.harvard.edu/pph2/) were used to
predict missense mutation protein function, and Phenotyping v2 was
also used to determine species conserved sequences and to perform
protein homology analysis. LMNA GeneView (https://www.uniprot.org/uniprot/P02545#structure)
was used to determine LMNA gene structure.
Sanger DNA sequencing
Variants in this family were determined via Sanger
DNA sequencing. Primer Premier 5.0 software (Premier Biosoft India
Pvt., Ltd., Indore, India) was applied to design primers for the
targeted sequence. The gene sequence for LMNA (250 bp) was
obtained from GenBank (https://www.ncbi.nlm.nih.gov/genbank/) (NM_170707.3)
and was amplified using the following primers: Forward,
5′-ATGATCGCTTGGCGGTCTAC-3′ and reverse, 5′-CGAACTCACCGCGCTTTC-3′.
The gene sequence for KCNQ4 (NM_004700.3) with a target
sequence of 226 bp according to the following primers: Forward,
5′-GGTAGGCTGGCTGTGATCTC-3′ and reverse, 5′-GCAGGCCTCACCTTGCTAT-3′.
The gene sequence for EYA transcriptional coactivator and
phosphatase 1 (EYA1) was obtained from GenBank (NM_000503.5)
and its target sequence was 250 bp with the following primers:
Forward, 5′-TCACAGCAGAATAATGGCCAGT-3′ and reverse,
5′-GTTTGGCAACTGGTGTACGG-3′. The synthesized primers were obtained
from Thermo Fisher Scientific. Inc. The PCR products were purified
via an E.Z.N.A.™ Gel Extraction kit (Omega Bio-Tek, Inc., Norcross,
GA, USA) according to the manufacturer's protocols. PCR products
were sequenced by Thermo Fisher Scientific, Inc.
Results
NGS analysis
A total of 522,349,503 bases from a typical run were
obtained and 490,137,344 bases were >Q20. There were 3,533,834
reads and 3,523,608 were mapped. The mean read length was 135 bp on
average; 98% of the reads were on target and the mean depth was
324.4X. Furthermore, for 503 homozygous single nucleotide variants
(SNVs), 0 homozygous multiple nucleotide variations (MNPs), 31
homozygous insertions/deletions (INDELs), 642 heterozygous SNVs, 1
heterozygous MNP and 39 heterozygous INDELs were determined; the
present study reported SNVs/Total=0.948, INDELs/Total=0.058, Ti/Tv
ratio (SNVs)=2.562, NCBI dbSNP database concordance=0.952 and
Heterozygotes/Homozygotes=1.275.
Determination of suspected pathogenic
mutations in the proband
Following the removal of mutations that had a MAF
>1%, 6 out of 157 mutations were identified in the ClinVar
database (https://www.ncbi.nlm.nih.gov/clinvar/) as presented in
Table II.
| Table II.Mutations reported in the ClinVar
database for a proband in a family with dilated cardiomyopathy
(MAF≤0.01). |
Table II.
Mutations reported in the ClinVar
database for a proband in a family with dilated cardiomyopathy
(MAF≤0.01).
| Gene | Transcript
accession no. | Location | Function | Exon | Protein | Coding | SIFT | PolyPhen-2 | ClinVar | dbSNP | MAFa |
|---|
| KCNQ4 | NM_004700.3 | Exonic | Missense | 4 | p.Phe182Leu | c.546C>G | 1 | 0.046 | Pathogenic | rs80358273 | 0.00060 (G) |
| LMNA | NM_170707.3 | Exonic | Missense | 1 | p.Glu82Lys | c.244G>A | 0.0 | 1.0 | Pathogenic | rs59270054 |
|
| EYA1 | NM_172058.3 | Exonic | Missense | 13 | p.Gly426Ser | c.1276G>A | 0.0 | 0.891 | Pathogenic | rs121909199 |
|
| HBA2 | NM_000517.4 | utr_3 |
| 3 |
|
|
|
| Untested | rs2541640 | 0.006 |
| RYR1 | NM_000540.2 | Intronic |
|
|
|
|
|
| Untested | rs4476278 | 0.01 |
| ADA | NM_000022.2 | Exonic | Synonymous | 2 | p.(=) | c.36G>A |
|
| Untested | rs394105 | 0.01 |
The present study examined all exonic and intronic
borders of a targeted NGS panel comprising all genes known to be
associated with cardiac diseases. Common variants identified were:
c.*107A>G at untranslated regions-3 of exon 3 in hemoglobin A2
(HBA2), c.1122+52T>C in ryanodine receptor 1
(RYR1) and synonymous mutation (p.Val12=) in adenosine
deaminase (ADA). No rare nonsynonymous variants were
detected except for heterozygous c.244G>A (p.E82K) in
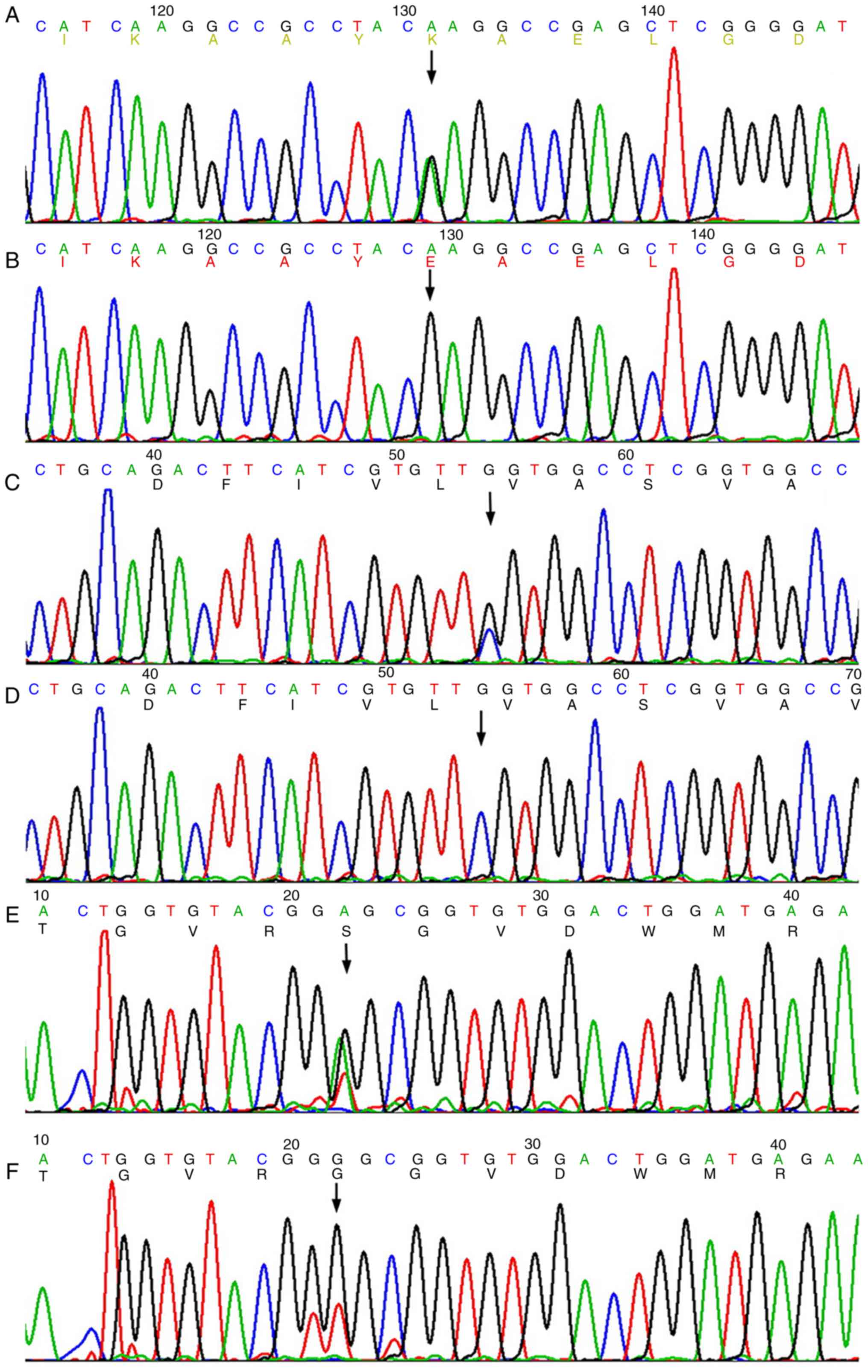
LMNA (NM_170707.3) (Fig. 2A and
B), c.546C>G (p.F182L) in KCNQ4 (NM_004700.3)
(Fig. 2C and D) and c.1276G>A
(p.G426S) in EYA1 (NM_172058.3) (Fig. 2E and F). Additionally, c.*107A>G
(NM_000517.4) was identified in the 3′untranslated region of exon 3
in HBA2. c.1122+52T>C (NM_000540.2) was located in the
intronic region of RYR1 and a synonymous (p.Val12=;
NM_000022.2) mutation was identified in ADA. These mutations
were all identified in non-coding regions and may exhibit no
effects on the onset of DCM.
These variants (p.E82K in LMNA, F182L in
KCNQ4 and G426S in EYA1) were absent in the database
of 1000 genomes project (17). The
NCBI ClinVar database revealed that the F182L mutation in
KCNQ4 has been reported to cause deafness (18,19),
and G426S in EYA1 was reported as the pathogenic gene
responsible for branchiootorenal syndrome 1 or Branchiootic
syndrome 1 (20); similar results
have been reported in the ExAC database (http://exac.broadinstitute.org/). The SIFT and
PolyPhen-2 scores for p.F182L (c.546C>G, rs80358273) of
KCNQ4 were 1.0 and 0.046, respectively; the SIFT and
PolyPhen-2 scores for p.G426S (c.1276G>A, rs121909199) of
EYA1 were 0 and 0.891, respectively. Finally, the SIFT and
PolyPhen-2 scores for p.E82K (c.244G>A, rs59270054) of
LMNA were 0 and 1, respectively.
Features viewer and functional
consequence of LMNA p.E82K
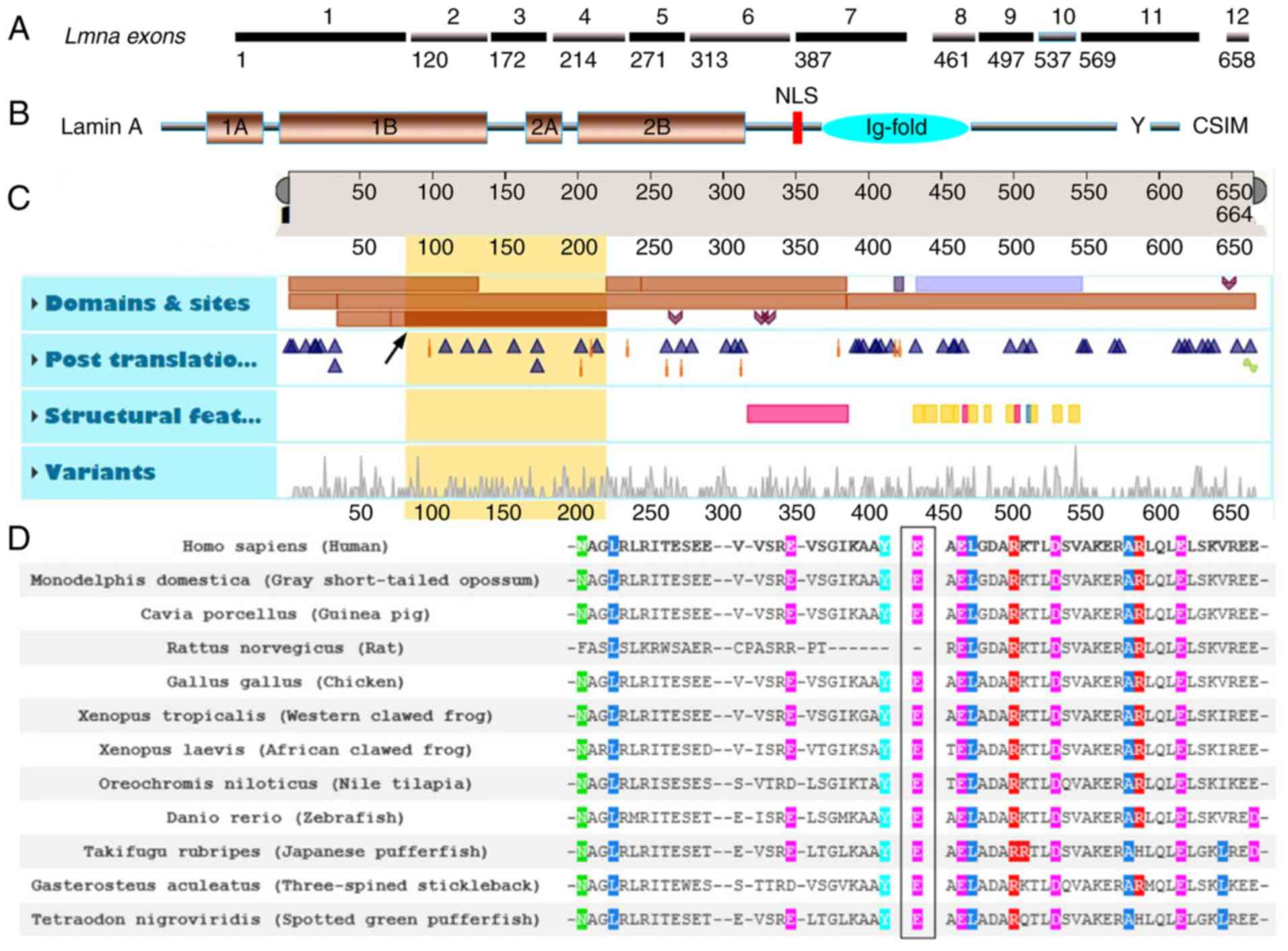
LMNA p.E82K was located in the first exon of
LMNA and close to the 1B structural domain margin in the rod
area of the Lamin A/C N-terminus (Fig.
3A-C, yellow region of interest 81–218 and description of Coil
1B in LMNA; Table II). The
analysis of LMNA p.E82K across numerous species revealed
that LMNA Glu82 was conserved among all species examined
with the exception of Rattus norvegicus (Rat) as presented
in Fig. 3D.
DNA samples could not be obtained from the patients
who had succumbed to heart disease-associated mortality; however,
the proband (II-6) was identifiable in the present study. II-7
exhibited clinical features of DCM; the son (III-11) of II-5
(succumbed to mortality due to DCM), the son (III-7) of II-3
(succumbed to mortality due to heart disease), and the daughter
(III-9) of II-4 (succumbed to mortality due to heart disease),
along with III-13, −14, −11 and IV-7 were heterozygous for p.E82K
in LMNA. However, no clinical data for DCM were obtained
until the end of the present study. Furthermore, II-1 and their
daughters (III-2 and III-3), II-6 and their son (III-13) were
heterozygous for p.F182L in KCNQ4, while II-6 and III-13
were heterozygous for p.G426S in EYA1. Excluding a family
member (III-3) who possessed the F182L mutation in KCNQ4 and
had slight hearing decline in the left ear as demonstrated via an
audiometry examination, the hearing of II-6, II-1, III-2, and
III-13, as well as that for the right ear of III-3 were all normal.
The pedigree analysis demonstrated no family members with symptoms
of branchiootorenal syndrome 1 or branchiootic syndrome 1.
Discussion
LMNA is one of the most common pathogenic
genes responsible FDC (3). At
birth, no significant differences in the growth status between
LMNA knockout mice (LMNA−/−) and wild type
mice were reported (16). After
few weeks post-partum, knockout mice gradually exhibited severe
growth delays, as well as muscle and heart atrophy; the
LMNA−/− mice were generated by Sullivan et
al (21). Additionally, it has
been demonstrated that these mice expressed a truncated lamin A
product at the transcriptional and translational levels (22); however, the novel LMNA null
mouse (LMNAGT−/−) created via gene trapping
exhibited severe growth retardation and developmental defects of
the heart, which resulted in mortality at 2–3 weeks post-partum
without the occurrence of DCM or any notable progeroid phenotypes
(23). At present, >400
mutations of LMNA have been identified, including deletions,
missense mutations, insertions and nonsense mutations, which have
been associated with a wide spectrum of diseases, including
Emery-Dreifuss muscular dystrophy, accelerated aging disorders,
such as the premature aging syndrome, Hutchinson-Gilford progeria,
DCM, lipodystrophy and neurological disorders. These disorders have
been classified as the ‘laminopathies’ (24,25).
Such conditions are defined as serious diseases resulting from
mutations in genes encoding lamins, particularly LMNA, which
encodes lamin A (12), or by
mutations in genes that directly or indirectly encode
lamin-associated proteins (26).
Mutations in LMNA mainly result in diseases,
which feature nerve and muscle-associated symptoms with cardiac
involvement; the first manifestations may include delayed
electrical activities of the atrium and ventricle, followed by
conduction system diseases, involving the sinoatrial node and
atrioventricular node conduction disorders. Ventricular
enlargement, heart failure, or sudden mortality may arise (24). Therefore, LMNA has been
reported as the most common pathogenic gene for inhibiting FDC
(6,27). This may be associated with
mutations in LMNA that cause abnormalities in expression and
the localization of gap junction proteins (28). The rate of sudden mortality of
patients with DCM resulting from mutations in LMNA was
reported to be higher compared with other causes of DCM9 (29). A 2005 meta-analysis of the clinical
characteristics of 299 carriers of LMNA mutations revealed a
sudden mortality rate of 46% (29). Furthermore, >40 mutations in
LMNA may lead to FDC with an abnormal conduction system as
suggested by the ClinVar database. The majority of mutation sites
were located in the ‘rod’ functional area of the lamin A/C
proteins; via the ‘rod’-like functional area and chromatin, the
inner nuclear protein is connected as a whole, in which the two
nucleolar proteins exhibit chain interactions, forming a
coiled-coil dimer structure in the inner layer of the nuclear
membrane, reported as the isα2 helix structure (30). Therefore, it may be speculated that
a mutation in the ‘rod’-like functional area may likely affect the
α2 helix structure. Thus, the formation of pre-lamin A may occur
and be unable to fulfil the functions of lamin A, which may lead to
structural disorder of nuclear fibers.
The pathophysiological basis of cardiomyopathies
caused by mutations in LMNA may involve abnormal signal
transduction. Abnormal activation of extracellular signal-regulated
kinase (ERK) and c-Jun N-terminal kinase, which belong to the
mitogen-activated protein kinases (MAPK) family, can affect
intracellular signaling. ERK and MAPK were pharmacologically
inhibited in LMNA mutant mice to prevent left ventricular
dilatation and myocardial contractility (31,32).
In addition, it may be that lamins A/C interact with transcription
factors associated with transcriptional regulation (28). Lamins aid the sequestration of
heterochromatin at the nuclear envelope; mutations in LMNA
can also cause an increase in heterochromatin, thereby affecting
gene expression, DNA self-repair and increased sensitivity to DNA
damage (33).
At present, only three families with DCM resulting
from similar mutations have been identified; the detection of the
responsible mutation sites in the investigated family of the
present study supports the hypothesis that p.E82K in LMNA
may exhibit strong pathogenic effects that results in DCM (34–36).
Additionally, studies with larger populations have not been
conducted; however, functional studies in vitro have
indicated a significant effect on protein function. For example,
mice exhibiting LMNA p.E82K demonstrated significantly
suppressed Cx43 expression levels as well as cell cycle arrest at
the G0/G1 phase, and presented with clinical features of DCM
(28,37,38).
It was also reported that the p.E82K mutation in LMNA may
result in Charcot-Marie-Tooth hereditary neuropathy type 2, an
axonal (non-demyelinating) peripheral neuropathy characterized by
heart weakness and atrophy, mild sensory loss, and normal or
almost-normal nerve conduction velocities (https://www.ncbi.nlm.nih.gov/clinvar/variation/66882/#clinical-assertions).
This variant also segregated with the disease in the family with
DCM in the present study. The other two pathogenic variants
(p.F182L in KCNQ4 and p.G426S in EYA1) appeared to
have no effects on this family with DCM; branchiootorenal syndrome
1 or branchiootic syndrome 1 was not observed in the present study.
The phenotype and these two mutations did not segregate, suggesting
that variation-associated information may not be effective to
investigate pathological and quantitative risk assessments; these
problems present a great challenge for accurate diagnosis (39). In summary, the p.E82K variant of
LMNA was deemed to be pathogenic based upon segregation
studies, its absence in controls and functional evidence, as
demonstrated by a systematic approach in assessing the clinical
significance of genetic variants (40).
The underlying mechanism by which the LMNA
p.E82K variant results in DCM remains unknown. In 2010, Sun et
al (28) demonstrated that
connexin 43 (Cx43) protein expression was reduced by 40% in cells
transfected with LMNA p.E82K compared with in cells
transfected with wild type (WT) LMNA cDNA. Confocal imaging
demonstrated that Cx43 localized to the cell interior via LMNA
p.E82K, whereas WT LMNA localized to gap junctions, which mediates
electrochemical connections between cells and determines the
velocity of cardiac conduction (28). Conduction abnormalities caused by
abnormal distributions and defects in lamins A/C may lead to the
occurrence of reentry arrhythmias, such as ventricular fibrillation
(41). C-Jun N-terminal kinase and
ERK1/2 associated with MAPK signaling were reported to regulate
Cx43 protein expression (42,43).
Suppressing the activity of ERKs can prevent damage
to the heart due to mutations in LMNA (44). LMNA p.E82K can result in
errors in the topology of lamins A/C and its interacting protein
emerin, which can damage cell membranes (45). The fragility of cell membranes can
lead to abnormal chromatin condensation, which may be another
mechanism underlying the onset of DCM induced by LMNA p.E82K
(45). Cells transfected with
LMNA p.E82K were arrested at the G0/G1
phase, whereas cell groups without the mutation arrested at the
G2/M phase in the presence of H2O2
(37). In addition, LMNA
p.E82K may have a tendency to promote cell immature transformation
(46). These experiments indicated
that LMNA p.E82K may promote cell apoptosis, increase DNA
synthesis and facilitate robust cell division. Such characteristics
may lead to an abnormal number of cardiac cells, decreased muscle
contractile function and weakened cardiac ejection function, thus
resulting in DCM (45,46). In addition, FDC-associated
mutations of LMNA resulting in FDC have distinct clinical
features, and ~25% of LMNA carriers do not experience any
notable symptoms during childhood (47); however, the penetrance of mutant
LMNA within carriers increases with age, attaining a rate of
almost 100% at 60 years of age (48). This was supported by the results of
the present study, which revealed that patients exhibiting clinical
symptoms of DCM belonged to the I and II generations, while the
younger III and IV generation carriers of p.E82K did not yet
exhibit typical clinical manifestations of DCM. Mutations in lamins
A/C can lead to poor prognoses of DCM, as well as a high incidence
of sudden cardiac mortality and severe heart failure (49).
The traditional definition of cardiomyopathy, which
was defined by the American Heart Association and the European
Society of Cardiology, did not account for the genetic basis of
cardiomyopathy (14). It was not
until 2013 that a descriptive genotype-phenotype nosology system,
MOGE (S), was proposed by the World Heart Federation, increasing
awareness for the genetic etiology of DCM. MOGE(S) may serve as a
comprehensive diagnostic, management and treatment tool for
myocardial diseases, similar to the tumor node metastasis
classification system for malignancy (50).
Collectively, the p.E82K mutation in LMNA may
contribute to the pathogenesis of FDC with concomitant
atrioventricular block as reported in the family investigated in
the present study. The detection of the responsible mutation sites
in the investigated family supports the hypothesis that LMNA
p.E82K may exhibit a strong pathogenic effect on DCM. Considering
that the p.E82K mutation can cause a severe DCM clinical phenotype,
this mutation may represent a therapeutic target or biomarker for
the future diagnosis and or prognosis of DCM.
Acknowledgements
The authors would like to thank all participants in
the present study for their cooperation.
Funding
The present study was supported by the Science and
Technology Program of Fujian (grant nos. 2018Y0012, 2016J01501 and
2016Y0012) and the Financial Scheme for Young Talents Training
Programme of Fujian Health Industry (grant no. 2015-ZQN-ZD-7).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JWL conceived and designed the experiments. XFL,
JWL, GL, YBZ and ZJ wrote the manuscript and performed the
experiments. JWL, GL, XL and YBZ analyzed and interpreted the data.
JWL and GL were responsible for critical revision of the content.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was performed in accordance with
the Declaration of Helsinki and was approved by the Ethics
Committee of Fujian Provincial Hospital, (Fuzhou, China). All
participants and legal guardians of minors included in the study
provided written informed consent.
Patient consent for publication
Consent for publication was obtained from all
subjects involved in the present study.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DCM
|
dilated cardiomyopathy
|
|
FDC
|
familial dilated cardiomyopathy
|
|
LMNA
|
lamins A/C
|
|
MAF
|
global minor allele frequency
|
|
EYA1
|
EYA transcriptional coactivator and
phosphatase 1
|
|
LV
|
left ventricular
|
|
AP4
|
apical four chamber view
|
|
RVD
|
right ventricular diameter
|
|
LVEF
|
left ventricular ejection fraction
|
References
|
1
|
Hershberger RE and Siegfried JD: Update
2011: Clinical and genetic issues in familial dilated
cardiomyopathy. J Am Coll Cardiol. 57:1641–1649. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hershberger RE, Hedges DJ and Morales A:
Dilated cardiomyopathy: The complexity of a diverse genetic
architecture. Nat Rev Cardiol. 10:531–547. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zahr HC and Jaalouk DE: Exploring the
Crosstalk Between LMNA and Splicing Machinery Gene Mutations in
Dilated Cardiomyopathy. Front Genet. 9:2312018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Skrzynia C, Berg JS, Willis MS and Jensen
BC: Genetics and heart failure: A concise guide for the clinician.
Curr Cardiol Rev. 11:10–17. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang J, Xu WW and Hu SJ: Heart failure:
advanced development in genetics and epigenetics. Biomed Res Int.
2015:3527342015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arbustini E, Pilotto A, Repetto A, Grasso
M, Negri A, Diegoli M, Campana C, Scelsi L, Baldini E, Gavazzi A
and Tavazzi L: Autosomal dominant dilated cardiomyopathy with
atrioventricular block: A lamin A/C defect-related disease. J Am
Coll Cardiol. 39:1–990. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
van Tintelen JP, Hofstra RM, Katerberg H,
Rossenbacker T, Wiesfeld AC, du Marchie Sarvaas GJ, Wilde AA, van
Langen IM, Nannenberg EA, van der Kooi AJ, et al: High yield of
LMNA mutations in patients with dilated cardiomyopathy and/or
conduction disease referred to cardiogenetics outpatient clinics.
Am Heart J. 154:1130–1139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van Spaendonck-Zwarts KY, van Rijsingen
IA, van den Berg MP, Lekanne Deprez RH, Post JG, van Mil AM,
Asselbergs FW, Christiaans I, van Langen IM, Wilde AA, et al:
Genetic analysis in 418 index patients with idiopathic dilated
cardiomyopathy: Overview of 10 years' experience. Eur J Heart Fail.
15:628–636. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hutchison CJ and Worman HJ: A-type lamins:
guardians of the soma? Nat Cell Biol. 6:1062–1067. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Scaffidi P and Misteli T: Lamin
A-dependent nuclear defects in human aging. Science. 312:1059–1063.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Al-Haboubi T, Shumaker DK, Köser J,
Wehnert M and Fahrenkrog B: Distinct association of the nuclear
pore protein Nup153 with A- and B-type lamins. Nucleus. 2:500–509.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Worman HJ and Bonne G: ‘Laminopathies’: A
wide spectrum of human diseases. Exp Cell. 313:2121–2133. 2007.
View Article : Google Scholar
|
|
13
|
Freeman AR and Levine SA: Clinical
significance of systolic murmurs: Study of 1000 consecutive
‘noncardiac’ cases. Ann Intern Med. 6:1371–1379. 1933. View Article : Google Scholar
|
|
14
|
Mestroni L, Maisch B, McKenna WJ, Schwartz
K, Charron P, Rocco C, Tesson F, Richter A, Wilke A and Komajda M:
Guidelines for the study of familial dilated cardiomyopathies.
Collaborative research group of the European human and capital.
mobility project on familial dilated cardiomyopathy. Eur Heart J.
20:93–102. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
AHA Medical/Scientific Statement, . 1994
revisions to classification of functional capacity and objective
assessment of patients with diseases of the heart. Circulation.
90:644–645. 1994.PubMed/NCBI
|
|
16
|
Li Z, Huang J, Zhao J, Chen C, Wang H,
Ding H and Wang DW and Wang DW: Rapid molecular genetic diagnosis
of hypertrophic cardiomyopathy by semiconductor sequencing. J
Transl Med. 12:1732014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
1000 GenomesProject Consortium, ; Abecasis
GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang
HM, Marth GT and McVean GA: An integrated map of genetic variation
from 1,092 human genomes. Nature. 491:56–65. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Smith RJH and Hildebrand M: DFNA2
Nonsyndromic Hearing LossGeneReviews® [Internet]. Pagon
RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD,
Fong CT, Mefford HC, Smith RJH and Stephens K: University of
Washington; Seattle, WA: pp. 1993–2016. 2008
|
|
19
|
Su CC, Yang JJ, Shieh JC, Su MC and Li SY:
Identification of novel mutations in the KCNQ4 gene of patients
with nonsyndromic deafness from Taiwan. Audiol Neurootol. 12:20–26.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Azuma N, Hirakiyama A, Inoue T, Asaka A
and Yamada M: Mutations of a human homologue of the Drosophila eyes
absent gene (EYA1) detected in patients with congenital cataracts
and ocular anterior segment anomalies. Hum Mol Genet. 9:363–366.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sullivan T, Escalante-Alcalde D, Bhatt H,
Anver M, Bhat N, Nagashima K, Stewart CL and Burke B: Loss of
A-type lamin expression compromises nuclear envelope integrity
leading to muscular dystrophy. J Cell Biol. 147:913–920. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jahn D, Schramm S, Schnölzer M, Heilmann
CJ, de Koster CG, Schütz W, Benavente R and Alsheimer M: A
truncated lamin A in the Lmna −/− mouse line: Implications for the
understanding of laminopathies. Nucleus. 3:463–474. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kubben N, Voncken JW, Konings G, van
Weeghel M, van den Hoogenhof MM, Gijbels M, van Erk A,
Schoonderwoerd K, van den Bosch B, Dahlmans V, et al: Post-natal
myogenic and adipogenic developmental: Defects and metabolic
impairment upon loss of A-type lamins. Nucleus. 2:195–207. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kang SM, Yoon MH and Park BJ:
Laminopathies; Mutations on single gene and various human genetic
diseases. BMB Rep. 51:327–337. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McNally EM, Golbus JR and Puckelwartz MJ:
Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin
Invest. 123:19–26. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Worman HJ, Ostlund C and Wang Y: Diseases
of the nuclear envelope. Cold Spring Harb Perspect Biol.
2:a0007602010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bilińska ZT, Sylvius N, Grzybowski J,
Fidziańska A, Michalak E, Walczak E, Walski M, Bieganowska K,
Szymaniak E, Kuśmierczyk-Droszcz B, et al: Dilated cardiomyopathy
caused by LMNA mutations. Clinical and morphological studies.
Kardiol Pol. 64:812–821. 2006.PubMed/NCBI
|
|
28
|
Sun LP, Wang L, Wang H, Zhang YH and Pu
JL: Connexin 43 remodeling induced by LMNA gene mutation Glu82Lys
in familial dilated cardiomyopathy with atrial ventricular block.
Chin Med J (Engl). 123:1058–1062. 2010.PubMed/NCBI
|
|
29
|
van Berlo JH, de Voogt WG, van der Kooi
AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T,
Heidbüchel H, de Visser M, et al: Meta-analysis of clinical
characteristics of 299 carriers of LMNA gene mutations: do lamin
A/C mutations portend a high risk of sudden death? J Mol Med
(Berl). 83:79–83. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Meune C, Van Berlo JH, Anselme F, Bonne G,
Pinto YM and Duboc D: Primary prevention of sudden death in
patients with lamin A/C gene mutations. N Engl J Med. 354:209–210.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu W, Shan J, Bonne G, Worman HJ and
Muchir A: Pharmacological inhibition of c-Jun N-terminal kinase
signaling prevents cardiomyopathy caused by mutation in LMNA gene.
Biochim Biophys Acta. 1802:632–638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu W, Muchir A, Shan J, Bonne G and Worman
HJ: Mitogen-activated protein kinase inhibitors improve heart
function and prevent fibrosis in cardiomyopathy caused by mutation
in lamin A/C gene. Circulation. 123:53–61. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mattout A, Pike BL, Towbin BD, Bank EM,
Gonzalez-Sandoval A, Stadler MB, Meister P, Gruenbaum Y and Gasser
SM: An EDMD mutation in C. elegans lamin blocks muscle-specific
gene relocation and compromises muscle integrity. Curr Biol.
21:1603–1614. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Novelli G and D'Apice MR: The strange case
of the ‘lumper’ lamin A/C gene and human premature ageing. Trends
Mol Med. 9:370–375. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang H, Zheng WY, Wang JZ, Wang XJ, Zhen
YS, Song L, Zou YB and Hui RT: A novel LMNA gene mutation E82K
associated with familial dilated cardiomyopathy. Zhonghua Xin Xue
Guan Bing Za Zhi. 33:875–879. 2005.(In Chinese). PubMed/NCBI
|
|
36
|
Wu X, Wang QK, Gui L, Liu M, Zhang X, Jin
R, Li W, Yan L, Du R, Wang Q, et al: Identification of a new lamin
A/C mutation in a Chinese family affected with atrioventricular
block as the prominent phenotype. J Huazhong Univ Sci Technolog Med
Sci. 30:103–107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang H, Song XD, Wang SX, Fu CY, Sun K and
Hui RT: Effects of a novel familial dilated cardiomyopathy
associated LMNA gene mutation E82K on cell cycle of HEK293 cells.
Zhonghua Xin Xue Guan Bing Za Zhi. 35:21–23. 2007.(In Chinese).
PubMed/NCBI
|
|
38
|
Lu D, Lian H, Zhang X, Shao H, Huang L,
Qin C and Zhang L: LMNA E82K mutation activates FAS and
mitochondrial pathways of apoptosis in heart tissue specific
transgenic mice. PLoS One. 5:e151672010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Manrai AK, Ioannidis JP and Kohane IS:
Clinical genomics: from pathogenicity claims to quantitative risk
estimates. JAMA. 315:1233–1234. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Duzkale H, Shen J, McLaughlin H, Alfares
A, Kelly MA, Pugh TJ, Funke BH, Rehm HL and Lebo MS: A systematic
approach to assessing the clinical significance of genetic
variants. Clin Genet. 84:453–463. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Severs NJ, Bruce AF, Dupont E and Rothery
S: Remodelling of gap junctions and connexin expression in diseased
myocardium. Cardiovasc Res. 80:9–19. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Petrich BG, Eloff BC, Lerner DL, Kovacs A,
Saffitz JE, Rosenbaum DS and Wang Y: Targeted activation of c-Jun
N-terminal kinase in vivo induces restrictive cardiomyopathy and
conduction defects. J Biol Chem. 279:15330–15338. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cho JH, Cho SD, Hu H, Kim SH, Lee SK, Lee
YS and Kang KS: The roles of ERK1/2 and p38 MAP kinases in the
preventive mechanisms of mushroom Phellinus linteus against the
inhibition of gap junctional intercellular communication by
hydrogen peroxide. Carcinogenesis. 23:1163–1169. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Muchir A, Shan J, Bonne G, Lehnart SE and
Worman HJ: Inhibition of extracellular signal-regulated kinase
signaling to prevent cardiomyopathy caused by mutation in the gene
encoding A-type lamins. Hum Mol Genet. 18:241–247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang H, Wang J, Zheng W, Wang X, Wang S,
Song L, Zou Y, Yao Y and Hui R: Mutation Glu82Lys in lamin A/C gene
is associated with cardiomyopathy and conduction defect. Biochem
Biophys Res Commun. 344:17–24. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang H, Zheng WY, Wang JZ, Song L, Zou YB,
Zhen YS, Wang XJ and Hui RT: The effect of cell mitosis of LMNA
gene mutation E82K associated with familial dilated cardiomyopathy.
Mol Cardiol China. 5:777–779. 2005.
|
|
47
|
Sylvius N, Bilinska ZT, Veinot JP,
Fidzianska A, Bolongo PM, Poon S, McKeown P, Davies RA, Chan KL,
Tang AS, et al: In vivo and in vitro examination of the functional
significances of novel lamin gene mutations in heart failure
patients. J Med Genet. 42:639–647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Malhotra R and Mason PK: Lamin A/C
deficiency as a cause of familial dilated cardiomyopathy. Curr Opin
Cardiol. 24:203–208. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Małek LA, Labib S, Mazurkiewicz L, Saj M,
Płoski R, Tesson F and Bilińska ZT: A new c C > G, p.R541G lamin
A/C mutation in a family with DCM and regional wall motion
abnormalities (akinesis/dyskinesis): Genotype-phenotype
correlation. J Hum Genet. 56:83–86. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Şahan E, Şahan S, Karamanlıoğlu M, Gul M
and Tufekcioğlu O: The MOGE(S) classification: A TNM-like
classification for cardiomyopathies. Herz. 41:503–506. 2016.
View Article : Google Scholar : PubMed/NCBI
|