Introduction
As estimated by the World Health Organization, 280
million individuals over 15 years of age (4.1% of the population)
are engaged in repeated excessive episodic alcohol consumption and
are affected by chronic alcoholism (1). Patients with chronic alcoholism
(i.e., Wernicke's encephalopathy) frequently exhibit motor
dysfunction, memory and cognitive impairments and mental disorders
(2,3). Of note, some disorders, such as
memory deficits in chronic alcoholism, are generally irreversible
and inclined to progress into alcoholic dementia (4). The neurological disorders of chronic
alcoholism are closely associated with neurodegeneration, synapse
loss and myelin damage probably resulting from sustained alcohol
exposure-induced neurotrophin exhaustion, oxidative stress and
neuroinflammation, in the prefrontal cortex and limbic system
structures (hippocampus and amygdala) that are particularly
vulnerable to the neurotoxic effects of alcohol (1,4–6). To
date, there is no effective therapy to reverse the permanent
deficits of chronic alcoholism.
Erythropoietin (EPO) is a 30-kDa, hypoxia-sensitive
glycoprotein with erythropoiesis-promoting properties. The EPO
receptor (EPOR) via which EPO functions is found to be expressed
ubiquitously in the mammalian central nervous system (CNS). Recent
evidence highlights the protective roles of EPO in
neurodegenerative diseases (7–9). EPO
treatment was found to reduce motor neuron degeneration and delay
the motor deterioration onset in amyotrophic lateral sclerosis
mouse models (7,8), and to alleviate learning and memory
deficits and attenuate Aβ-induced neurotoxicity in an Alzheimer's
disease mouse model (9). In
addition, data from preclinical studies have demonstrated that EPO
also contributes to the histological and functional recoveries from
various traumatic and ischemic injuries of CNS (10–12).
Mechanistically, diverse signaling pathways downstream of EPO/EPOR
(i.e., ERKs and PI3K/AKT signaling) have been implicated in the
neuroprotection of EPO (12,13).
However, the exact impacts of EPO on chronic alcoholism-related
neuropathology and neurological dysfunctions remain to be
elucidated.
Nuclear factor, erythroid 2-like 2 (Nrf2) is the
master transcription factor orchestrating antioxidant and
detoxification responses (14–16).
Under homeostatic conditions, Nrf2 is sequestered by Kelch-like
ECH-associated protein 1 (KEAP1) in the cytoplasm and is
consequently degraded by the proteasome. When cells undergo stress,
Nrf2 dissociates from KEAP1 and translocates into the nucleus to
activate the expression of its downstream target genes such as
NAD(P)H quinone oxidoreductase (NQO1), heme oxygenase 1 (HO1) and
glutamate-cysteine ligases. Intriguingly, autophagy-degraded p62
can regulate Nrf2 activity positively by inactivating KEAP1, which
has been observed during the neuroectoderm commitment of embryonic
stem cell and induced inflammatory tolerance of macrophages
(15,17). As reported recently, the elevated
Nrf2 activity can also be evoked by EPO-stimulated ERKs and
PI3K/AKT pathways, and Nrf2 can mediate the neuroprotective effects
of EPO (18–20). Nevertheless, although oxidative
stress plays a critical role in chronic alcoholism-related injuries
(21–23), whether Nrf2 activity is involved in
the autonomous and non-autonomous repair of alcoholic brain damage
remains elusive.
In this study, the therapeutic effects of EPO on
chronic alcoholism-related neural pathology and brain disorders and
as well as the involvement of Nrf2 activity were investigated on
mice subjected to chronic alcohol exposure.
Materials and methods
Animals and experimental designs
Male C57BL/6J mice at 12 weeks of age weighing 22–25
g were purchased from the Model Animal Research Center of Nanjing
University and maintained on a 12-h light/dark cycle with
controlled temperature (22–23°C) and humidity (45–55%). For all
repertoires of experiments, C57BL/6J mice were randomly assigned to
the Control, Chronic Alcohol Exposure (CAE), CAE + recombinant
human erythropoietin (rhEPO) and
CAE+rhEPO+all-trans-retinoic acid (ATRA) groups on average
(8 mice/group). The CAE, CAE+rhEPO and CAE+rhEPO+ATRA groups were
subjected to alcohol exposure for 4 weeks. Successively, the
CAE+rhEPO and CAE+rhEPO+ATRA groups were treated with rhEPO in
saline [100 IU/ml, 500 IU/kg body weight; Roche Diagnostics
(Shanghai) Ltd., China] intranasally every 12 h for 4 weeks, while
the CAE group was treated only with an equal amount of vehicle.
Simultaneously, the CAE+rhEPO+ATRA group received intraperitoneal
administration of ATRA in saline (2 mg/ml, 20 mg/kg; Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) 30 min before intranasal rhEPO
treatment for 4 weeks, while other groups received merely an equal
amount of vehicle. Animals in the Control group underwent all
procedures except chronic alcohol exposure and medication. For
neural function analyses, neurological deficits were assessed by
motor cooperation, cognition and nerve conduction tests 12 h after
rhEPO administration for the last time. For the following
histopathological analyses, animals were decapitated and the brain
specimens were dissected and subjected to immunostaining and
electron microscopy. For the independent mechanism study, the
homogenate of brain tissue of animals was subjected to western
blotting analyses for neural damage repair-related key molecules 30
min immediately after the drug treatment for the last time. All
experimental procedures on animals were approved by the Ethics
Committee of Harbin Medical University and the Ethics Committee of
Qiqihar Medical University, and followed the principles of medical
ethics.
Alcohol exposure
Chronic alcohol exposure was achieved via a chronic
intermittent ethanol (CIE) vapor inhalation procedure previously
described for C57BL/6J mice (24).
Briefly, mice were placed in standard mouse cages in Plexiglas
vapor chambers (60×36×60 cm, PlasLabs, up to six cages per chamber)
and exposed to continuous fresh air-mixed vaporized which delivered
19–22 mg of alcohol per liter of air at a rate of ~10 l/min and
produced blood alcohol levels of 175±25 mg/dl as designed. The
blood alcohol level was verified weekly via blood samples taken
from dedicated ‘sentinel’ mice exposed to alcohol simultaneously
with the test mice. To initiate intoxication and stabilize blood
alcohol levels, the alcohol groups received intraperitoneal
injections of 1 mmol/kg body weight of the alcohol dehydrogenase
inhibitor pyrazole (Sigma-Aldrich; Merck KGaA) combined with 1.5
g/kg body weight of 20% alcohol (v/v), in a volume of 10 ml/kg body
weight, before placement in the chambers. Exposure lasted for 16 h
each day (in at 17:00 h, 2 h before the start of the 12-h circadian
dark phase, out at 09:00 h), followed by an 8-h withdrawal. There
were 4 consecutive days of intermittent exposure (Monday through
Thursday) and then an 80-h withdrawal (Friday through Monday). This
complete weekly cycle was repeated four times. Controls received an
injection of 1 mmol per kg of body weight pyrazole in saline to
control for this treatment, before placement in dedicated air
chambers (located adjacent to the alcohol chambers), which received
vaporized air at the same exchange rate as the alcohol
chambers.
Intranasal administration of
rhEPO
The intranasal administration was performed as
reported previously (25). In
brief, recombinant human (rh) EPO [Roche Diagnostics (Shanghai)
Ltd.] was prepared using sterile 0.45% normal saline solution at a
concentration of 100 IU/ml and stored at −20°C. Each mouse was held
ventral side up, a small-modified 27-French catheter was inserted
into either nares, and saline or rhEPO was slowly administered
intranasally at no more than 5 µl increments 5–10 min apart for a
total of 500 IU/kg body weight. The mouse was held for 1–2 min to
ensure absorption. Drug or saline was administered every 12 h
beginning immediately after the 4-week alcohol exposure.
Motor cooperation tests
Motor cooperation of mice was assessed by rotarod
and beam walking assays. For the motor tests, the training began 3
days before alcohol exposure and the tests were conducted 1 week
after treatment. The rotarod assay was conducted using a ROTO-ROD
series 8 (IITC Inc./Life Science, Inc., Woodland Hills, CA, USA)
which could accelerate rotational speed from 4 to 40 rpm over 120
sec. The mean latency of mice to fall off the rotarod in the three
trials was recorded as measurement. For the beam walking assay, the
time mice spent in crossing a 1-m long and 3-cm thick beam was
calculated for analysis.
Cognition and activity tests
Novel location recognition and open-field tests were
used to evaluate the spatial hippocampal-dependent memory, activity
and anxiety. For the novel location recognition test, each mouse
was allowed to explore the empty arena (50-cm diameter) for 10 min
on the 1st day for familiarization. In this arena, two identical
objects were placed in the middle on the 2nd day for acquisition,
and in the same arena, one of the two objects was moved to a novel
location on the 3rd day for the test. The exploration time for the
two objects in the acquisition and test phase was recorded. The
percentage of the time mice spent exploring the displaced object in
the total exploration time for both objects was defined as the
preference index. For the open-field test, mice were placed into a
chamber measuring 50×50×38 cm for 60 min. The total number of
forelimb rears, and the ratio of time spent in the center to the
time spent in the corners were recorded as measurements.
Nerve conduction test
Somatosensory-evoked potential (SSEP) was monitored
to assess cortical synaptic transmission and nerve conduction. To
generate forepaw stimulation, 2 metal pins were inserted into the
left forepaw and a 1–5 mA 1 or 2 msec electrical pulse was
delivered by constant current stimulus output using the RM6240C
Multi-channel Bioelectric Signal Acquisition System (Chengdu
Instrument Factory, Sichuan, China). For electroencephalography
(EEG) recording, a Teflon-coated silver chloride wire (125 µm) with
an exposed tip (1 mm) as the EEG electrode was placed on the
surface of the primary somatosensory cortex corresponding to the
left forelimb within the agarose, and the reference electrode was
placed on the nasal bone under the skin. The cortical signal was
amplified, filtered and recorded using the RM6240C Multi-channel
Bioelectric Signal Acquisition System. The onset latency and the
amplitude of the large primary cortical wave were determined.
Immunofluorescence staining
Mice were anesthetized with isoflurane and carbon
dioxide as dictated by ethical guidelines and decapitated. The
brains were dissected rapidly, fixed overnight in 4%
paraformaldehyde and processed for paraffin embedding. For
immunofluorescence staining, 4-µm paraffin sections were prepared.
After deparaffinization and rehydration according to standard
procedures, sections underwent antigen retrieval in citrate buffer
(pH 6.0) using a microwave oven (750 W). Non-specific antibody
binding was blocked with 5% bovine serum albumin (BSA), 0.2% Triton
X-100 in PBS for 1 h at room temperature. Subsequently, sections
underwent incubation with primary antibodies [rabbit polyclonal
anti-human/mouse/rat synaptophysin (IgG; 1:100; catalog no.
SAB4502906; Sigma-Aldrich; Merck KGaA) and rabbit polyclonal
anti-human/mouse/rat myelin basic protein (MBP) (IgG; 1:100;
catalog no. 78896; Cell Signaling Technology, Inc., Danvers, MA,
USA)] diluted in 2% BSA, 0.1% Triton in PBS overnight at 4°C.
Following the washing out of the unbound antibodies by PBS, the
sections underwent incubation with the secondary antibody [goat
polyclonal anti-rabbit IgGs conjugated to Alexa Fluor®
488 (1:1,000; catalog no. A32723; Invitrogen, Thermo Fisher
Scientific, Inc., Waltham, MA, USA)] diluted in 2% BSA, 0.1% Triton
in PBS in dark for 1 h at room temperature, and counterstained with
DAPI (1 µg/ml; Sigma-Aldrich; Merck KGaA) in methanol for nuclear
staining. The stained sections were mounted with anti-fade reagent
(Invitrogen, Thermo Fisher Scientific) and photographed under a
BX51 fluorescence microscope (Olympus, Tokyo, Japan). The
fluorescence intensity was measured using Image-Pro Plus 6.0 (Media
Cybernetics, Inc., Rockville, MD, USA).
Transmission electron microscopy
Samples of dissected brain were fixed in 0.1 M
sodium-cacodylate-buffered mixture (5% glutaraldehyde and 4%
paraformaldehyde), post-fixed in 1% osmium tetroxide and 1.5%
potassium ferrocyanide, dehydrated and embedded in epoxy resin.
Ultrathin sections (80 nm) were then cut, collected on copper
grids, stained with lead citrate and uranyl acetate for contrast
and examined under H-7650 electron microscope (Hitachi, Osaka,
Japan) equipped with a MegaView III CCD camera (Soft Imaging
System, Lakewood, CO, USA). The synapse densities in the captured
images were quantified using Adobe Photoshop Creative Cloud.
Western blotting
The dissected brain tissue was homogenized and lysed
using RIPA lysis buffer (Beyotime, Zhejiang, China) supplemented
with protease and phosphatase inhibitors (Complete and Phosphostop,
Roche, Upper Bavaria, Germany) for 30 min on mice, and
centrifugation at 12,000 × g for 20 min at 4°C. The supernatant was
harvested, and the protein concentration was determined using the
BCA kit (Beyotime). SDS sample buffer containing β-mercaptoethanol
was added to the protein lyses. Totally 30 µg protein was loaded
per lane, resolved by 8–12% polyacrylamide gels in a
Mini-PROTEAN® Tetra Vertical Electrophoresis Cell
(Bio-Rad Laboratories, Inc., Hercules, CA, USA), electroblotted
onto polyvinylidene fluoride membranes (Merck Millipore, Darmstadt,
Germany) in a Mini Trans-Blot® Module (Bio-Rad
Laboratories), blocked with 5% skimmed milk/TBST, and incubated
with the primary antibodies diluted in the blocking buffer
overnight at 4°C. The following primary antibodies were used: mouse
monoclonal anti-human hEPO (catalog no. SAB5300475; Sigma-Aldrich;
Merck KGaA), rabbit polyclonal anti-human/mouse/rat p-EPOR (catalog
no. SAB4301492; Sigma-Aldrich; Merck KGaA), rabbit polyclonal
anti-human/mouse/rat p-ERK1/2 (catalog no. E7028; Sigma-Aldrich;
Merck KGaA), rabbit polyclonal anti-human/mouse/rat p-AKT (catalog
no. SAB4301497; Sigma-Aldrich; Merck KGaA), rabbit polyclonal
anti-human/mouse/rat Nrf2 (catalog no. SAB4501984; Sigma-Aldrich;
Merck KGaA), rabbit polyclonal anti-human/mouse/rat KEAP1 (catalog
no. AV34727; Sigma-Aldrich; Merck KGaA), rabbit polyclonal
anti-human/mouse/rat NQO1 (catalog no. N5288; Sigma-Aldrich; Merck
KGaA), rabbit polyclonal anti-human/mouse/rat p62 (catalog no.
P0067; Sigma-Aldrich; Merck KGaA), rabbit polyclonal
anti-human/mouse/rat LC3B (catalog no. 3868; Cell Signaling
Technology, Inc.) and rabbit polyclonal anti-human/mouse/rat
β-actin (catalog no. SAB2100037; Sigma-Aldrich; Merck KGaA) (IgGs;
1:1,000). β-actin expression was used as a loading control. After
the washout of the unbound primary antibodies, the membranes
underwent incubation with the secondary antibodies [horseradish
peroxidase (HRP)-conjugated goat polyclonal anti-mouse IgG
(1:5,000; catalog no. 31430; Invitrogen; Thermo Fisher Scientific,
Inc.) or HRP-conjugated goat polyclonal anti-rabbit IgG (1:5,000;
catalog no. 31460; Invitrogen; Thermo Fisher Scientific, Inc.)] for
1 h at room temperature. Protein blots were visualized using a
ChemiDoc™ Imaging System (Bio-Rad), and quantified using Image-Pro
Plus 6.0.
Data processing and statistics
Data are expressed as the mean ± standard error of
the mean (SEM). Statistical analyses were performed using GraphPad
Prism 7.0 software (GraphPad Software, Inc., La Jolla, CA, USA).
Statistical differences were determined by one-way analysis of
variance (ANOVA) followed by Bonferroni's multiple comparisons
tests (more than two experimental conditions) and Wilcoxon
signed-rank test (when compared with 50% chance level). P<0.05
was considered statistically significant.
Results
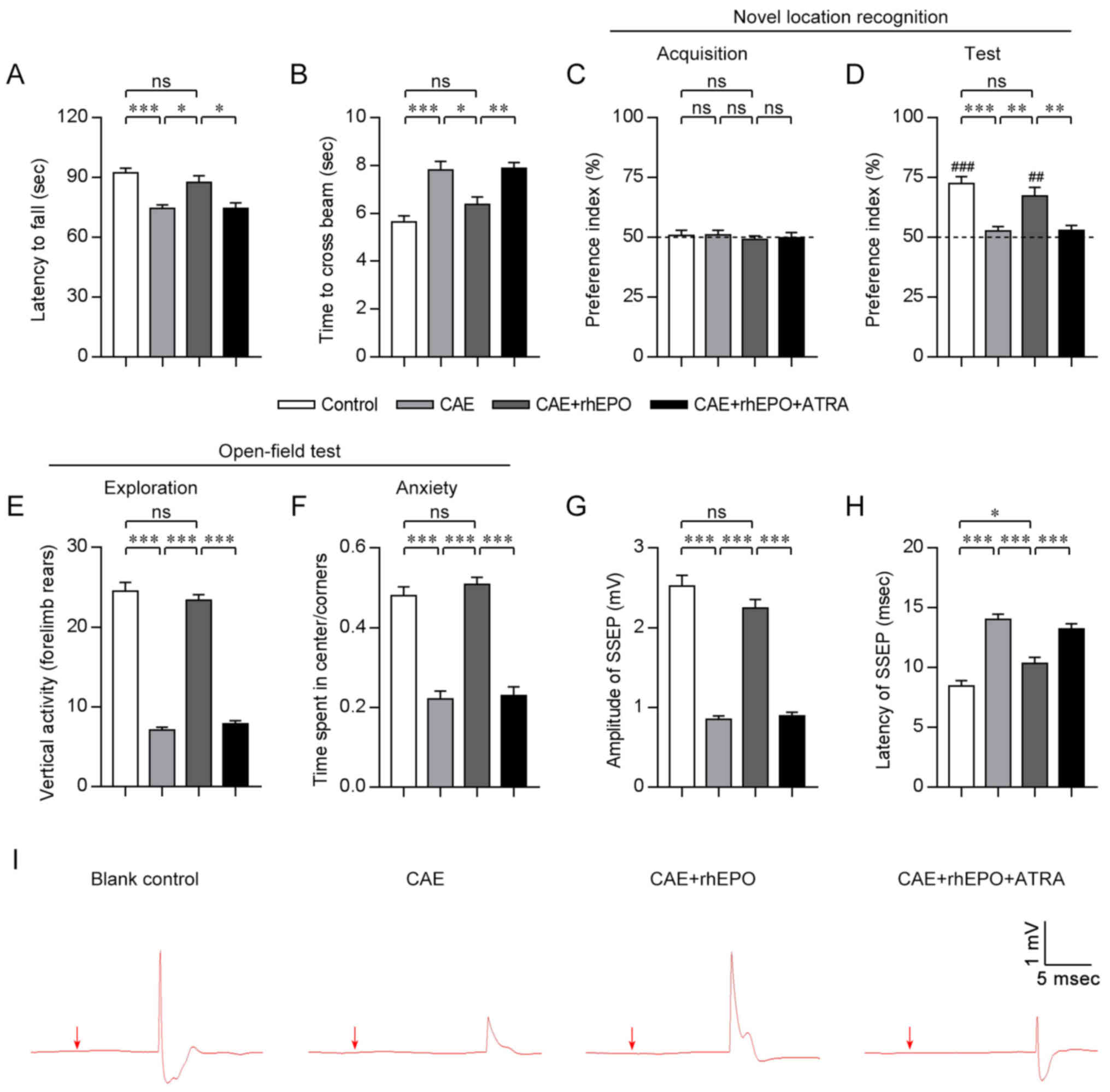
Intranasal rhEPO reverses the motor
function deficits in mice with chronic alcoholism
To evaluate the impacts of rhEPO administered via
intranasal route on the motor function of mice exposed to alcohol
for 4 weeks, rotarod and beam walk tests were conducted after a
4-week rhEPO treatment. In the rotarod test (Fig. 1A), mice exposed to sustained
alcohol in the CAE group showed dramatically shortened time
remaining on the rotarod when compared with mice in the Control
group (CAE vs. Control, P<0.001), whereas alcohol-exposed mice
receiving intranasal rhEPO administration in the CAE+rhEPO group
presented with significantly longer time maintaining on the rotarod
(CAE+rhEPO vs. CAE, P<0.05), up to the level of the Control
group (CAE+rhEPO vs. Control, P>0.05). When challenged on a 1-cm
beam, mice in the CAE group needed more time to cross the beam than
those in the Control group (CAE vs. Control, P<0.001; Fig. 1B). In contrast, mice in the
CAE+rhEPO group spent less time crossing the beam (CAE+rhEPO vs.
CAE, P<0.05) and regained similar beam-crossing capacity to that
of mice in the Control group (CAE+rhEPO vs. Control, P>0.05).
Data for the motor tests indicated that intranasal rhEPO treatment
could promote recovery from chronic alcoholism-related motor
deficits.
Intranasal rhEPO alleviates the
cognitive disorders in mice with chronic alcoholism
To examine the effects of intranasal rhEPO on
cognition and anxiety following chronic alcoholism, mice in each
group were tested in object location and open-field tasks post
rhEPO treatment (4 weeks post alcohol exposure). During the
acquisition phase in the novel location recognition task (Fig. 1C), mice in all groups spent the
same amount of time exploring the objects. During memory testing 24
h later (Fig. 1D), mice in the
Control and CAE+rhEPO groups similarly explored preferentially the
object that had been moved to a novel location (CAE+rhEPO vs.
Control, P>0.05; Control and CAE+rhEPO vs. chance level 50%,
P<0.01), but mice in the CAE group showed no exploratory
preference for the displaced object (CAE vs. Control, P<0.001;
CAE vs. CAE+rhEPO, P<0.01; CAE vs. chance level 50%, P>0.05),
indicating that the impaired discriminative memory of mice with
chronic alcoholism for the novel and familiar location could be
rescued by intranasal rhEPO. During the open-field test (Fig. 1E and F), alcohol-exposed mice
without drug treatment had sharply diminished forelimb rears (CAE
vs. Control, P<0.001), decreased time spent in the center and
consequentially increased time spent in the corners of the open
field (CAE vs. Control, P<0.001) compared to the healthy control
mice, indicative of reduced exploration and aggravated anxiety.
However, intranasal rhEPO administration remarkably restored
vertical exploration activity (CAE+rhEPO vs. CAE, P<0.001;
CAE+rhEPO vs. Control, P>0.05), increased center exploration and
relieved anxiety (CAE+rhEPO vs. CAE, P<0.001; CAE+rhEPO vs.
Control, P>0.05) in mice exposed to alcohol for 4 weeks. These
findings in cognitive and behavioral tests suggested that
intranasal rhEPO treatment could ameliorate the memory, learning
and anxiety disorders in mice with chronic alcoholism.
Intranasal rhEPO rescues the nervous
conduction impairments in mice with chronic alcoholism
To further determine the effects of intranasal rhEPO
on nervous conduction after chronic alcohol exposure,
somatosensory-evoked potential (SSEP) analysis was performed. In
this study, the amplitude and latency of SSEP were selected as the
objective measurements of nervous conduction. At week 4 after
alcohol exposure (immediately after rhEPO treatment), mice in all
groups exhibited evident SSEP waves in response to forepaw
stimulation (Fig. 1I).
Furthermore, alcohol-exposed mice with no drug treatment in the CAE
group showed an obvious decrease in amplitude of forepaw-evoked
potential (0.85±0.04 mV) when compared with those in the Control
group (Control: 2.52±0.14 mV; CAE vs. Control, P<0.001; Fig. 1G and I), indicative of impaired
ascending conduction caused by chronic alcoholism. Whereas
intranasal EPO-treated mice presented with notably recovered SSEP
amplitude (CAE+rhEPO: 2.24±0.11 mV; CAE+rhEPO vs. CAE, P<0.001)
which became comparable to that of control mice (CAE+rhEPO vs.
Control, P>0.05). Coinciding with the change in amplitude, the
SSEP latency of alcohol-exposed non-treated mice (14.00±0.45 msec)
was significantly longer than that of non-alcohol-exposed mice
(Control: 8.46±0.44 msec; CAE vs. Control, P<0.001; Fig. 1H and I), indicating the delayed
conduction of nervous impulse. Inconsistent with the results of
amplitude analysis, although the conduction velocity of
forepaw-evoked action potential was restored following intranasal
EPO treatment (CAE+rhEPO: 10.36±0.50 msec; CAE+rhEPO vs. CAE,
P<0.001), the SSEP conduction in EPO-treated mice was still
slower than that of control mice (CAE+rhEPO vs. Control,
P<0.05). The results of electrophysiological examination
verified the therapeutic effects of intranasally administered EPO
on the nervous conduction after chronic alcoholism.
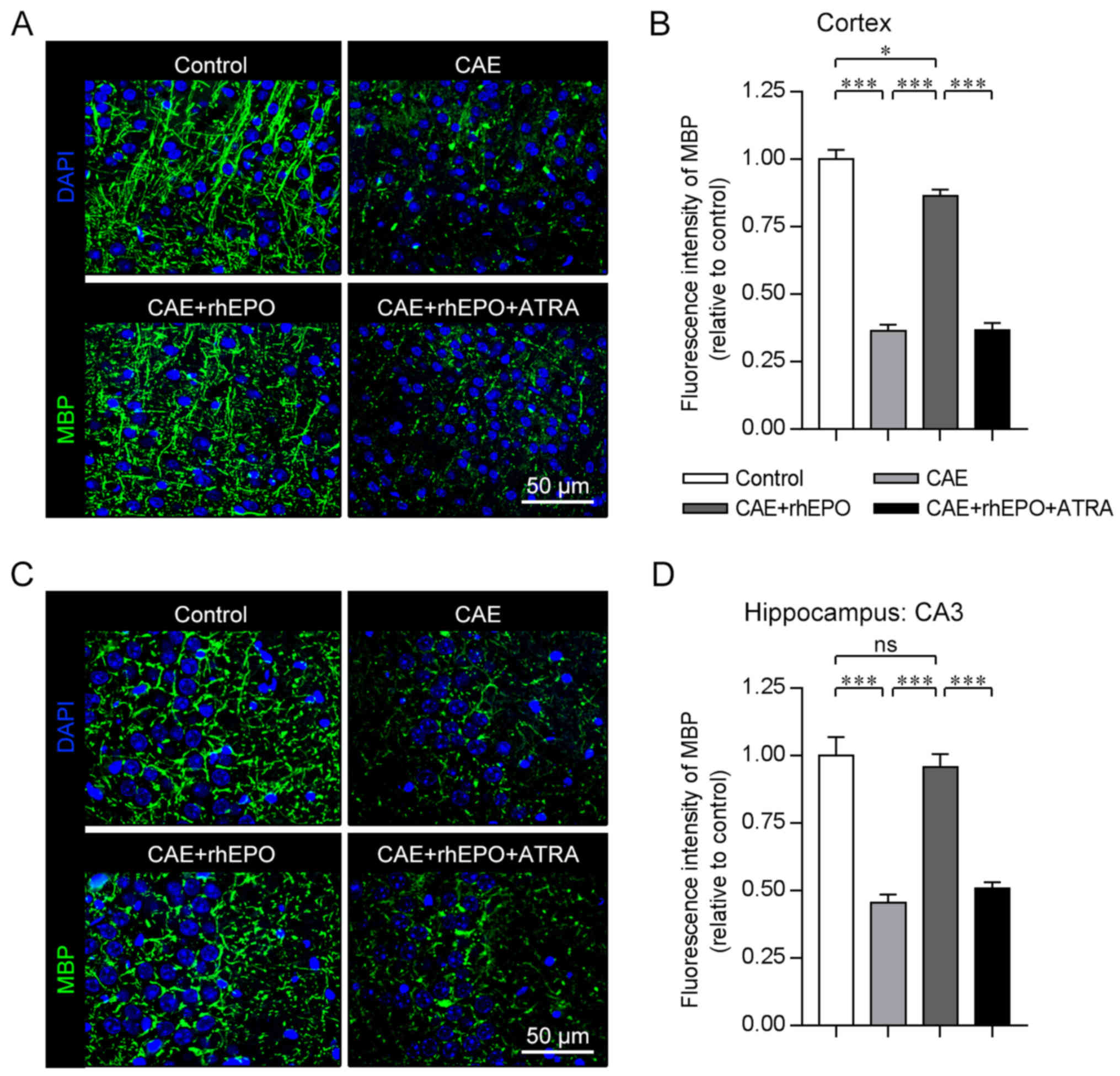
Intranasal rhEPO promotes the remyelination in mice
with chronic alcoholism. Continuous demyelination and failed
remyelination are involved in the prolonged motor coordination
impairments and progressive cognitive decline after brain injury
(26). As revealed by the
immunostaining for MBP (a major constituent of the myelin sheath)
performed 4 weeks after alcohol exposure (just after the 4-week
rhEPO treatment), the deep layers (V/VI) of the cerebral cortex of
mice in the Control group were highly myelinated (Fig. 2A), consistent with a previous
description (27). Alcohol-exposed
mice in the CAE group showed comparatively impaired myelination in
the same cortical layers, which was demonstrated by the decrease in
the MBP+ myelin sheath (CAE vs. Control, P<0.001;
Fig. 2A and B). When the
alcohol-exposed mice were subjected to intranasal EPO treatment,
the cortical MBP+ myelin level was significantly
recovered but did not attain the normal level (CAE+rhEPO vs. CAE,
P<0.001; CAE+rhEPO vs. Control, P<0.05). In the CA3 area of
the hippocampus rich in myelinated nervous fibers (28), the deteriorated myelination
occurred in mice that had been exposed to sustained alcohol (CAE
vs. Control, P<0.001), which was approximately completely
reversed in the intranasal EPO-treated mice as determined by
stereological volume analysis of MBP fluorescence intensity
(CAE+rhEPO vs. CAE, P<0.001; CAE+rhEPO vs. Control, P>0.05;
Fig. 2C and D). The data for the
immunofluorescence staining for the structural protein of myelin
sheath demonstrated the positive effects of intranasal EPO on
remyelination in chronic alcoholism-damaged brain.
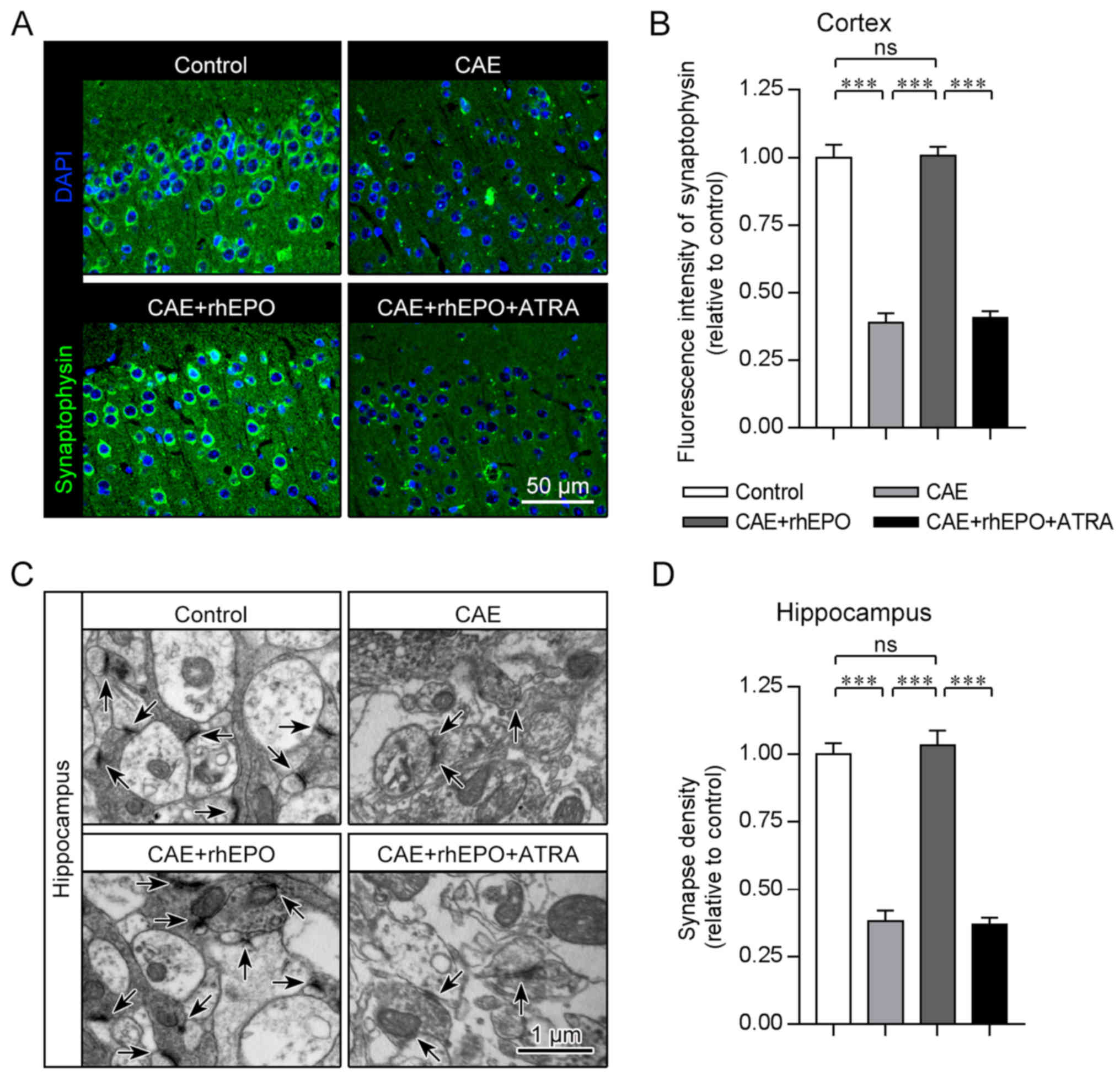
Intranasal rhEPO accelerates the
synapse regeneration in mice with chronic alcoholism
Region-specific synapse loss is closely correlated
to interrupted transmission of nervous pulse and impaired cognition
in brain injury and neurodegeneration (29–31).
To examine the cortical synapse formation which determines cortical
synaptic transmission and consequently motor and cognitive
function, the immunostaining for synaptophysin (a component of
synaptic vesicle) was performed 4 weeks post alcohol exposure (12 h
post the end of rhEPO treatment). Quantification of synaptophysin
puncta revealed substantial synapse loss in the cortex of the
chronic alcohol-exposed mice as compared to their healthy
counterpart controls (CAE vs. Control, P<0.001; Fig. 3A and B). After intranasal
administration of EPO, the chronic alcohol exposure-affected mice
showed significantly elevated fluorescence intensity of
synaptophysin in the cortex which was almost equivalent to that of
the control mice (CAE+rhEPO vs. CAE, P<0.001; CAE+rhEPO vs.
Control, P>0.05), suggestive of reconstructed cortical synapse
formation supporting the exact nervous conduction and sophisticated
neural functions. To further evaluate the cognition-related
hippocampal synaptogenesis after alcohol exposure and EPO
treatment, TEM analysis was conducted to recognize the
ultramicroscopic structures of synapse. As determined by
presynaptic (synaptic vesicle) and postsynaptic compartments
(postsynaptic density) in electron micrographs, hippocampal synapse
density obviously declined on prolonged alcohol exposure in mice of
the CAE group in contrast to that in controls (CAE vs. Control,
P<0.001; Fig. 3C and D), but
synapse amount recovered on intranasal EPO treatment and reached
the non-alcohol-affected level in the CAE+rhEPO group (CAE+rhEPO
vs. CAE, P<0.001; CAE+rhEPO vs. Control, P>0.05), which was
consistent with the tendency obverse in cortex. These data
clarified the repairing potential of intranasal EPO for brain
synaptogenesis disrupted in chronic alcoholism.
Intranasal rhEPO exerts neuroprotective effects on
mice with chronic alcoholism by regulating the autophagy-related
degradation of Nrf2. To establish the mechanisms underlying the
neuroprotective effects of intranasal rhEPO, western blot analyses
were performed on the microdissected cortex and hippocampus 30 min
after EPO administration for the last time. As expected, hEPO was
detected in the brain tissue of intranasal EPO-treated mice rather
than non-treated mice (Fig. 4A).
Correspondingly, the exogenous EPO caused a considerable increase
in phosphorylated (p)-EPOR, p-ERK1/2 and p-AKT levels in the
cerebrum of the treated mice (CAE+rhEPO vs. CAE, all P-values
<0.001; CAE+rhEPO vs. Control, P-values <0.001; Fig. 4A-D), reminiscent of the augmented
activation of EPOR, ERKs and PI3K/AKT signaling. Furthermore,
intranasally delivered EPO led to the upregulated level of cerebral
antioxidant and neuroprotective Nrf2, in parallel with the reduced
level of Nrf2 inhibitor protein KEAP1 and the elevated protein
expression of Nrf2 target gene NQO1 (CAE+rhEPO vs. CAE, all
P-values <0.001; Fig. 4A and
E-G), which suggested the increased Nrf2 activity by EPO.
However, mere chronic alcohol exposure resulted in a detectable
reduction in basal EPOR, ERK1/2, AKT and Nrf2 activity (CAE vs.
Control, all P-values <0.05). As autophagy-degraded p62 is
reported to enhance Nrf2 signaling by inactivating KEAP1 (15,17),
the relationship between Nrf2 activity and autophagy progress was
monitored. The accelerated degradation of p62 by autophagy and the
autophagy-indicative accumulation of LC3-II in alcohol-affected
brain tissue (CAE vs. Control, both P-values <0.05; Fig. 4A, H and I) was reversed by
intranasal rhEPO (CAE+rhEPO vs. CAE, all P-values <0.001),
coinciding with the EPO-activated autophagy-inhibitory ERKs and
PI3K/AKT signaling, the reduced level of Nrf2-inhibiting KEAP1, and
the rescued expression of Nrf2 and Nrf2-targeted NQO1. These
observations indicated the involvement of antioxidant Nrf2
negatively regulated by the alcohol-induced autophagic response in
the protective impacts of EPO.
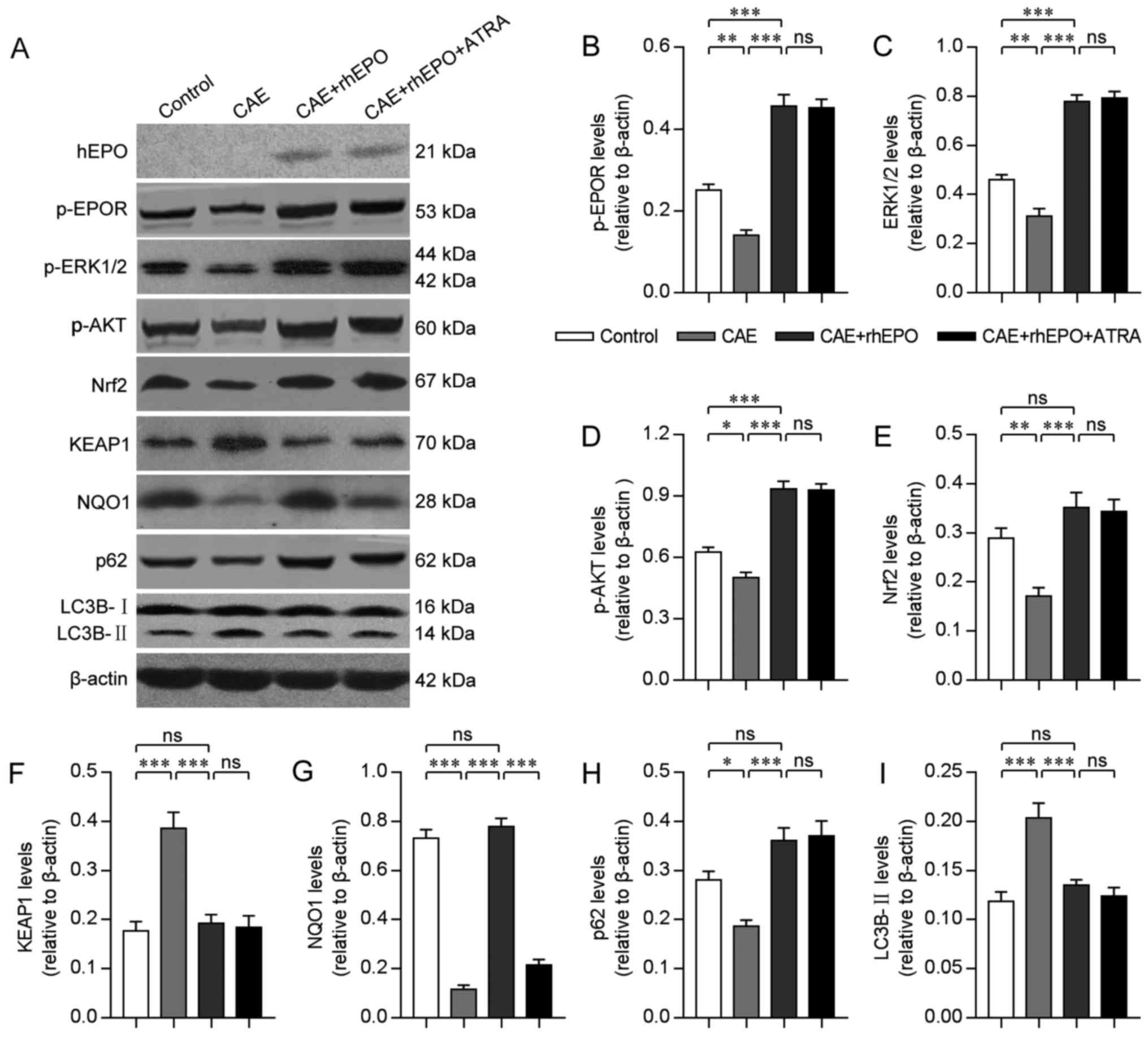 | Figure 4.Intranasal rhEPO exerts
neuroprotective effects by regulating autophagy-related Nrf2
degradation in mice with chronic alcoholism. (A) Images of western
blot analyses for hEPO, phosphorylated (p)-EPOR, p-ERK1/2, p-AKT,
Nrf2, KEAP1, NQO1, p62 and LC3-II in brain tissue of mice in each
group. (B-I) The quantitative analyses of the relative p-EPOR (B),
p-ERK1/2 (C), p-AKT (D), Nrf2 (E), KEAP1 (F), NQO1 (G), p62 (H) and
LC3-II (I) levels to β-actin in brain tissue of mice in each group
(n=8). Error bars indicate SEM. ns, non-significant; *P<0.05;
**P<0.01; ***P<0.001. Groups: Control, Chronic Alcohol
Exposure (CAE), CAE + recombinant human erythropoietin (rhEPO) and
CAE+rhEPO+all-trans-retinoic acid (ATRA). |
To further determine whether EPO functions as
neuroprotector by modulating Nrf2 activity, ATRA, the specific
antagonist of Nrf2 (16,32), was administered intraperitoneally
30 min before intranasal EPO treatment for each time. The effective
suppression of Nrf2 activity by ATRA was demonstrated by the
downregulated protein level of NQO1 (CAE+rhEPO+ATRA vs. CAE+rhEPO,
both P-values <0.001; Fig. 4A and
G). Expectedly, the levels of intracerebral hEPO, activated
EPOR and regulatory molecules (ERKs and AKT), autophagy-related
proteins (p62 and LC3-II), KEAP1 and also Nrf2 were not influenced
by ATRA-induced Nrf2 inhibition (CAE+rhEPO+ATRA vs. CAE+rhEPO, all
P-values >0.05; Fig. 4A-C and
F-H). However, if pre-treated by ATRA, the intranasal
EPO-administered mice with chronic alcohol exposure showed
significantly curtailed duration maintaining on the rotarod
(Fig. 1A), delayed beam-crossing
(Fig. 1B), abolished preference
for displaced object in novel location recognition task (Fig. 1D), impaired exploration-learning
activity and increased anxiety in open-field test (Fig. 1E and F), retarded conduction of
SSEP with amplitude loss (Fig.
1G-I), decreased MBP+ myelin sheath in cerebral
cortex (Fig. 2A and B) and
hippocampus (Fig. 2C and D),
diminished cortical synaptophysin level (Fig. 3A and B) and reduced hippocampal
synapse density (Fig. 3C and D)
(CAE+rhEPO+ATRA vs. CAE+rhEPO, all P-values <0.05), which
suggested that the EPO-induced recovery in motor cooperation,
cognition, nervous conduction and cerebral myelination and
synaptogenesis were almost abrogated by the inhibition of Nrf2
activity. These data confirmed that the therapeutic effects of
intranasal EPO on chronic alcoholism-related brain disorders were
mediated, at least partially, by impeding the autophagy-related
degradation of Nrf2.
Discussion
In the present study, we discovered the therapeutic
effects of intranasally administered erythropoietin (EPO) on the
neurological function deficits and neural pathology in chronic
alcoholism, and identified Nrf2 activity as the central mechanism
underlying the neuroprotection. EPO, a well-established
hematopoietic factor, was recently detected in the central nervous
system (CNS) where it acts as the trophic and protective factor for
neurons as well as glia (7,33).
Dysfunction of EPO-EPOR signaling is implicated in the pathogenesis
of neurogenerative diseases (such as amyotrophic lateral
sclerosis), and treatment with exogenous EPO has been reported to
rescue the functional defects and pathological alterations in the
neurodegeneration and traumatic and ischemic injuries of CNS
(7–12). However, the effects of EPO on
chronic alcoholism-related brain damages were poorly understood
previously.
To evaluate the therapeutic potential of EPO for the
neurological disorders and pathology, mice were subjected to CIE
vapor inhalation in order to recapitulate the neurological function
defects in human chronic alcoholism and the correlated
neuropathology and pathogenesis. This vapor inhalation procedure is
a canonical mouse model of chronic alcoholism and widely used in
the researches on the neural mechanisms underlying the chronic
alcohol exposure-induced behavioral and cognitive alterations
(24). In the present study,
chronic alcohol-exposed mice displayed obviously impaired motor
cooperation and velocity in rotarod and beam walk tests, reduced
discriminative memory for the displayed object in novel location
recognition task, and decreased exploration activity and
exacerbated anxiety in open field (Fig. 1A-F), reminiscent of the clinical
manifestations of motor and cognitive dysfunction in patients with
chronic alcoholism (3). In the
present study, rhEPO was administered via noninvasive intranasal
route, which allows rapid delivery of therapeutic extrinsic
cytokines and extracellular vesicles directly to the brain from the
nasal mucosa, bypasses the blood-brain barrier that limits the
distribution of systemically administered therapeutics to the CNS,
and minimizes systemic exposure (25,34).
Our findings further supported the feasibility of intranasal
administration of EPO, as evidenced by the entry of exogenous rhEPO
into mouse brain tissue and the consequent activation of EPOR and
the downstream ERKs and PI3K/AKT signaling (Fig. 4A-C). After treatment with EPO
delivered via intranasal route, alcohol-exposed animals gained the
approximately normal complex body movement and coordination,
significantly reversed spatial reference and work memory, and
notably restored exploration and learning drive and alleviated
anxiety status, which verified the therapeutic efficacy of EPO for
the sustained alcohol exposure-caused locomotor, cognitive and
emotional disorders in mice. Furthermore, somatosensory-evoked
potential (SSEP) was exploited to objectively measure the nervous
conduction of CNS as reported previously (29). Similar to the observations in the
behavioral tests, intranasal EPO led to notable recovery from the
prolonged alcohol exposure-induced amplitude loss and conduction
delay of SSEP, albeit not full restoration in the onset latency
(Fig. 1G-I).
Normal myelination is known to be essential for the
normal function of the CNS. Abnormal myelination in
neurodevelopmental defects, and also demyelination and failed
remyelination in brain injury are correlated to impaired
transmission of nervous pulse as well as the metabolic disturbance
and dysfunction of neural axon, which consequentially affects motor
and cognitive functions (26). Our
findings indicated that chronic alcohol exposure brought about poor
myelination in the deep layers (V/VI) of the cerebral cortex and
the CA3 area of hippocampus in mice, where nervous fibers are
highly myelinated as observed previously (27,28).
The repair potential of intranasal EPO for the alcohol-induced
myelin damage was demonstrated by the reversion of the MBP-positive
myelin sheath in the cortex and hippocampus to the uninjured levels
(Fig. 2), associated with the
recovery of affected nervous conduction, locomotor and cognition.
Synapse formation is another major determinant for transmission of
nervous signala, and complicatedly regulated motor and cognitive
activity. Disrupted synaptogenesis is established to be the common
mechanism mediating the neurological disorders of neurodegeneration
and damages in CNS (30,31). In the present study, sustained
alcohol generated marked synaptic toxicity in the affected cortex
and hippocampus, as revealed by the decreased level of cortical
synaptophysin (a canonical marker of synapse) and the reduced
hippocampal synapse density (Fig.
3). Following intranasal treatment with rhEPO, the EPO-provoked
regeneration of synapse was observed in the cortex, accounting for
the restored cortical synaptic transmission reflected by
forepaw-evoked potential (29).
The synaptogenesis-promoting capacity of EPO was also detected in
the hippocampus, which could be related to the improved behaviors
of treated mice in hippocampus-dependent cognitive tasks.
Therefore, our data indicated that intranasally delivered EPO
conferred profound neuroprotection in the brain against sustained
alcohol exposure. Of note, the incompletely restored SSEP
conduction suggested that the newly regenerated myelin and synapse
immediately after EPO treatment might be immature in structure and
function, which should be further studied in the future.
Nrf2 acts as the major regulator of cellular
resistance to oxidant stress and also chemical toxicity associated
with oxidative pathology by controlling the basal and induced
expression of an array of antioxidant response element-dependent
genes (i.e., NQO1 and HO-1) (14,15).
Recently, pharmacological boosting of Nrf2 activity with EPO is
reported to induce protection against neurotoxin- and aging-related
neural damages (18,20). In this study, we observed that the
level of Nrf2 and the expression of Nrf2 target gene NQO1 were
elevated by extrinsic EPO in the brain tissue of alcohol-exposed
mice (Fig. 4). Consistently, the
preservation of motor cooperation, cognition, nervous conduction,
myelination and synaptogenesis by EPO was almost abolished upon
pre-treatment with ATRA (Figs.
1–3), the well-established
blood-brain barrier-permeable potent antagonist of Nrf2 (16,32),
further demonstrating that Nrf2 signaling mediated the
neuroprotective effects of EPO in chronic alcoholism. As Nrf2 mRNA
is expressed constitutively and independently of inducers, the
protein turnover via KEAP1-ubiquitin-proteasome pathway is
suggested as the major mechanism regulating Nrf2 activation
(14). Interestingly, p62, a
substrate for autophagic degradation, can augment Nrf2 activity by
recruiting and inactivating KEAP1 in embryonic neurodevelopment and
the pharmacologically induced macrophagic tolerance to inflammatory
stimuli, indicative of the negative correlation between Nrf2
activity and autophagic reaction (15,17).
In mice with chronic alcoholism-like pathology, we detected that
intranasal rhEPO suppressed alcohol-provoked cerebral autophagic
response by stimulating ERKs and PI3K/AKT signaling, as do other
growth and neurotrophic factors (35,36).
Simultaneously, the rescued expression of p62 was closely related
to the degradation of Nrf2-sequestering KEAP1 as well as the
restored levels of Nrf2 and Nrf2-targeted NQO1. Upon the inhibition
of Nrf2 activity by ATRA, the NQO1 expression was significantly
reduced, whereas the level of intracerebral hEPO, the expression of
signaling molecules upstream of Nrf2 (the EPOR signaling as well as
autophagic progression-related proteins), the level of KEAP1
regulating the accumulation of Nrf2, and even the expression of
Nrf2, were expectedly unaffected (Fig.
4), which was consistent with the highly specific antagonism of
ATRA against Nrf2 (32). Our data
suggested that EPO could activate the neuroprotective Nrf2 activity
in chronic alcohol-affected brain tissue by hindering the
autophagy-related degradation of Nrf2 via ERKs and PI3K/AKT
signaling. This previously unanticipated mechanism may also
underlie the EPO-conferred neuroprotection against other
neurotoxins and aging.
In summary, we found that intranasal EPO effectively
rescued motor dysfunction, cognitive and emotional disorders, and
nervous conduction impairment caused by sustained alcohol exposure
in this study. Consistently, the intranasally delivered EPO
promoted the remyelination and synapse formation in chronic
alcoholism-affected neocortex and hippocampus. Moreover, we
discovered that the exogenous rhEPO, which entered the cerebrum
through the intranasal route, activated EPOR and downstream ERKs
and PI3K/AKT signaling, and suppressed autophagy-related
degradation of Nrf2. We further presented evidence to show that
Nrf2 activity mediated the intranasal EPO-exerted
neuroprotection.
Acknowledgements
The authors would like to thank Dr Caichuan Yan from
Heilongjiang Institute of Cancer Prevention and Treatment for
providing technical support.
Funding
The present study was supported by a research award
from the Health and Family Planning Commission of Heilongjiang
Province (2016–041).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HS designed the study, performed the experiments and
modified the manuscript. XN performed the CIE vapor inhalation
procedure and the related treatments, conducted the
immunofluorescence staining, analyzed the data, and was a major
contributor in writing the manuscript. WW performed the TEM and
western blotting analyses and analyzed the data. QW conducted the
motor and cognitive behavioral tests and analyzed the data. DZ
performed the nerve conduction tests and analyzed the data. All
authors helped to draft, read and approved the final
manuscript.
Ethics approval and consent to
participate
The manuscript involves no patients and human
samples. All experimental procedures on animals were approved by
the Ethics Committee of Harbin Medical University and the Ethics
Committee of Qiqihar Medical University, and followed the
principles of medical ethics.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ATRA
|
all-trans-retinoic acid
|
|
CNS
|
central nervous system
|
|
CAE
|
chronic alcohol exposure
|
|
CIE
|
chronic intermittent ethanol
|
|
EEG
|
electroencephalography
|
|
EPOR
|
erythropoietin receptor
|
|
EPO
|
erythropoietin
|
|
HO1
|
heme oxygenase 1
|
|
HRP
|
horseradish peroxidase
|
|
KEAP1
|
Kelch-like ECH-associated protein
1
|
|
MBP
|
myelin basic protein
|
|
NQO1
|
NADP, H quinone oxidoreductase
|
|
Nrf2
|
nuclear factor, erythroid 2-like 2
|
|
rhEPO
|
recombinant human erythropoietin
|
|
SSEP
|
somatosensory-evoked potential
|
References
|
1
|
Ron D and Barak S: Molecular mechanisms
underlying alcohol-drinking behaviours. Nat Rev Neurosci.
17:576–591. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fama R and Sullivan EV: Methods of
association and dissociation for establishing selective
brain-behavior relations. Handb Clin Neurol. 125:175–181. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lembke A and Stanford M: Clinical
management of alcohol use disorders in the neurology clinic. Handb
Clin Neurol. 125:659–670. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gupta S and Warner J: Alcohol-related
dementia: A 21st-century silent epidemic? Br J Psychiatry.
193:351–353. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vetreno RP and Crews FT: Current
hypotheses on the mechanisms of alcoholism. Handb Clin Neurol.
125:477–497. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Connor JP, Haber PS and Hall WD: Alcohol
use disorders. Lancet. 387:988–998. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sato K, Morimoto N, Kurata T, Mimoto T,
Miyazaki K, Ikeda Y and Abe K: Impaired response of hypoxic sensor
protein HIF-1α and its downstream proteins in the spinal motor
neurons of ALS model mice. Brain Res. 1473:55–62. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koh S, Kim Y, Kim HY, Cho GW, Kim KS and
Kim SH: Recombinant human erythropoietin suppresses symptom onset
and progression of G93A-SOD1 mouse model of ALS by preventing motor
neuron death and inflammation. Eur J Neurosci. 25:1923–1930. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rodriguez Cruz Y, Strehaiano M, Rodriguez
Obaya T, Garcia Rodriguez JC and Maurice T: An intranasal
formulation of erythropoietin (Neuro-EPO) prevents memory deficits
and amyloid toxicity in the APPSwe transgenic mouse model of
Alzheimer's disease. J Alzheimers Dis. 55:231–248. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cerri G, Montagna M, Madaschi L, Merli D,
Borroni P, Baldissera F and Gorio A: Erythropoietin effect on
sensorimotor recovery after contusive spinal cord injury: An
electrophysiological study in rats. Neuroscience. 219:290–301.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hellewell SC, Yan EB, Alwis DS, Bye N and
Morganti-Kossmann MC: Erythropoietin improves motor and cognitive
deficit, axonal pathology, and neuroinflammation in a combined
model of diffuse traumatic brain injury and hypoxia, in association
with upregulation of the erythropoietin receptor. J
Neuroinflammation. 10:1562013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma S, Chen J, Chen C, Wei N, Xu J, Yang G,
Wang N, Meng Y, Ren J and Xu Z: Erythropoietin rescues memory
impairment in a rat model of chronic cerebral hypoperfusion via the
EPO-R/JAK2/STAT5/PI3K/Akt/GSK-3β pathway. Mol Neurobiol.
55:3290–3299. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chong ZZ, Shang YC, Mu Y, Cui S, Yao Q and
Maiese K: Targeting erythropoietin for chronic neurodegenerative
diseases. Expert Opin Ther Targets. 17:707–720. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma Q: Role of nrf2 in oxidative stress and
toxicity. Annu Rev Pharmacol Toxicol. 53:401–426. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jang J, Wang Y, Lalli MA, Guzman E,
Godshalk SE, Zhou H and Kosik KS: Primary cilium-autophagy-Nrf2
(PAN) axis activation commits human embryonic stem cells to a
neuroectoderm fate. Cell. 165:410–420. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yin XP, Zhou J, Wu D, Chen ZY and Bao B:
Effects of that ATRA inhibits Nrf2-ARE pathway on glial cells
activation after intracerebral hemorrhage. Int J Clin Exp Pathol.
8:10436–10443. 2015.PubMed/NCBI
|
|
17
|
Mildenberger J, Johansson I, Sergin I,
Kjobli E, Damas JK, Razani B, Flo TH and Bjorkoy G: N-3 PUFAs
induce inflammatory tolerance by formation of KEAP1-containing
SQSTM1/p62-bodies and activation of NFE2L2. Autophagy.
13:1664–1678. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shen J, Wu Y, Xu J, Zhang J, Sinclair SH,
Yanoff M and Xu G, Li W and Xu G: ERK- and Akt-dependent
neuroprotection by erythropoietin (EPO) against glyoxal-AGEs via
modulation of Bcl-xL, Bax, and BAD. Invest Ophthalmol Vis Sci.
51:35–46. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Genc K, Egrilmez MY and Genc S:
Erythropoietin induces nuclear translocation of Nrf2 and heme
oxygenase-1 expression in SH-SY5Y cells. Cell Biochem Funct.
28:197–201. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu H, Zhao J, Chen M, Wang H, Yao Q, Fan J
and Zhang M: The anti-aging effect of erythropoietin via the
ERK/Nrf2-ARE pathway in aging rats. J Mol Neurosci. 61:449–458.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schattenberg JM and Czaja MJ: Regulation
of the effects of CYP2E1-induced oxidative stress by JNK signaling.
Redox Biol. 3:7–15. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dekeyser GJ, Clary CR and Otis JS: Chronic
alcohol ingestion delays skeletal muscle regeneration following
injury. Regen Med Res. 1:22013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Roman J: Chronic alcohol ingestion and
predisposition to lung ‘cirrhosis’. Alcohol Clin Exp Res.
38:312–315. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Holmes A, Fitzgerald PJ, Macpherson KP,
Debrouse L, Colacicco G, Flynn SM, Masneuf S, Pleil KE, Li C,
Marcinkiewcz CA, et al: Chronic alcohol remodels prefrontal neurons
and disrupts NMDAR-mediated fear extinction encoding. Nat Neurosci.
15:1359–1361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Scafidi J, Hammond TR, Scafidi S, Ritter
J, Jablonska B, Roncal M, Szigeti-Buck K, Coman D, Huang Y,
McCarter RJ Jr, et al: Intranasal epidermal growth factor treatment
rescues neonatal brain injury. Nature. 506:230–234. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Piao J, Major T, Auyeung G, Policarpio E,
Menon J, Droms L, Gutin P, Uryu K, Tchieu J, Soulet D and Tabar V:
Human embryonic stem cell-derived oligodendrocyte progenitors
remyelinate the brain and rescue behavioral deficits following
radiation. Cell Stem Cell. 16:198–210. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tomassy GS, Berger DR, Chen H, Kasthuri N,
Hayworth KJ, Vercelli A, Seung HS, Lichtman JW and Arlotta P:
Distinct profiles of myelin distribution along single axons of
pyramidal neurons in the neocortex. Science. 344:319–324. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ahn JH, Lee TK, Park JH, Cho JH, Kim IH,
Lee JC, Hong S, Jeon YH, Kang IJ, Lee YJ, et al: Age-dependent
differences in myelin basic protein expression in the hippocampus
of young, adult and aged gerbils. Lab Anim Res. 33:237–243. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen S, Mohajerani MH, Xie Y and Murphy
TH: Optogenetic analysis of neuronal excitability during global
ischemia reveals selective deficits in sensory processing following
reperfusion in mouse cortex. J Neurosci. 32:13510–13519. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Perez EJ, Tapanes SA, Loris ZB, Balu DT,
Sick TJ, Coyle JT and Liebl DJ: Enhanced astrocytic d-serine
underlies synaptic damage after traumatic brain injury. J Clin
Invest. 127:3114–3125. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hong S, Bejaglasser VF, Nfonoyim BM,
Frouin A, Li S, Ramakrishnan S, Merry K, Shi Q, Rosenthal A, Barres
BA, et al: Complement and microglia mediate early synapse loss in
Alzheimer mouse models. Science. 352:712–716. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Meng QT, Cao C, Wu Y, Liu HM, Li W, Sun Q,
Chen R, Xiao YG, Tang LH, Jiang Y, et al: Ischemic
post-conditioning attenuates acute lung injury induced by
intestinal ischemia-reperfusion in mice: Role of Nrf2. Lab Invest.
96:1087–1104. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dale EA, Satriotomo I and Mitchell GS:
Cervical spinal erythropoietin induces phrenic motor facilitation
via extracellular signal-regulated protein kinase and Akt
signaling. J Neurosci. 32:5973–5983. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dhuria SV, Hanson LR and Frey WH II:
Intranasal delivery to the central nervous system: Mechanisms and
experimental considerations. J Pharm Sci. 99:1654–1673. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Noda NN and Inagaki F: Mechanisms of
autophagy. Annu Rev Biophys. 44:101–122. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Galluzzi L, Bravo-San Pedro JM, Blomgren K
and Kroemer G: Autophagy in acute brain injury. Nat Rev Neurosci.
17:467–484. 2016. View Article : Google Scholar : PubMed/NCBI
|