Introduction
Wolf-Hirschhorn syndrome (WHS) patients display
pleiotropic phenotypes including growth delay, intellectual
disability, congenital hypotonia, distinct facial appearance,
congenital heart defects, midline defects and seizures (1,2). The
deletion of either of two critical regions (WHSCR and WHSCR-2)
within chromosome band 4p16.3 has been proposed to be necessary for
minimal clinical manifestation of WHS (3,4). A
gene within WHSCR-2, leucine zipper/EF-hand-containing
transmembrane protein 1 (LETM1), which is deleted in almost all
patients with the full phenotype, and was therefore suggested as an
excellent candidate gene for the seizure development (2,3,5).
LETM1 was proposed to contribute to the neuromuscular features of
WHS patients (6). Multiple genes
are responsible for the characteristics of WHS disorders; thus,
mouse models have been developed to complement ongoing
patient-based studies (7).
Phenotypes of a genetically modified mouse model for FGFR33, MAEA,
Sax2/Nkx1-1 and CTBP1 showed skeletal malformations, hematopoietic
dysgenesis, post-natal growth defects, and later growth defects,
respectively (7). The most severe
pathogenic phenotype of WHS is epilepsy, which has been shown in
mouse lines carrying TACC3 and FAM53A mutantions (7,8).
LETM1 is also accepted to be tightly linked to the epilepsy
pathogenesis in WHS. In Drosophila melanogaster model,
neuronal-specific knockdown of LETM1 resulted in the impairment of
locomotor behavior in the fly and reduced synaptic neurotransmitter
release. These results revealed the function of LETM1 in epilepsy:
One of the severe pathogenic phenotypes of WHS (2,9).
Decreased LETM1 levels in the neo-cortices of temporal lobe
epilepsy patients and in the hippocampi and adjacent cortices of
rats following the onset of seizures has suggested that reduction
of LETM1 contributes to the seizure phenotype. Knockdown of LETM1
leads to reduced MT-CYB expression and induction of susceptibility
to seizures, which suggested that dysfunctional LETM1 together with
mitochondrial swelling and disturbed MT-CYB expression is important
for the deteriorated behavioral phenotype of epilepsy (10). However, Nigericin, a
K+/H+ ionophore, fails to prevent epileptic
seizures, and does not reverse lentivirus-shLETM1-mediated
facilitation of epilepsy, indicating that further studies are
required to determine the detailed mechanism of LETM1 function and
delineate the involvement of LETM1 in seizure pathogenesis
(10). Therefore a detailed
understanding of LETM1 function will provide great advantages for
WHS patients.
LETM1 functions in mitochondria
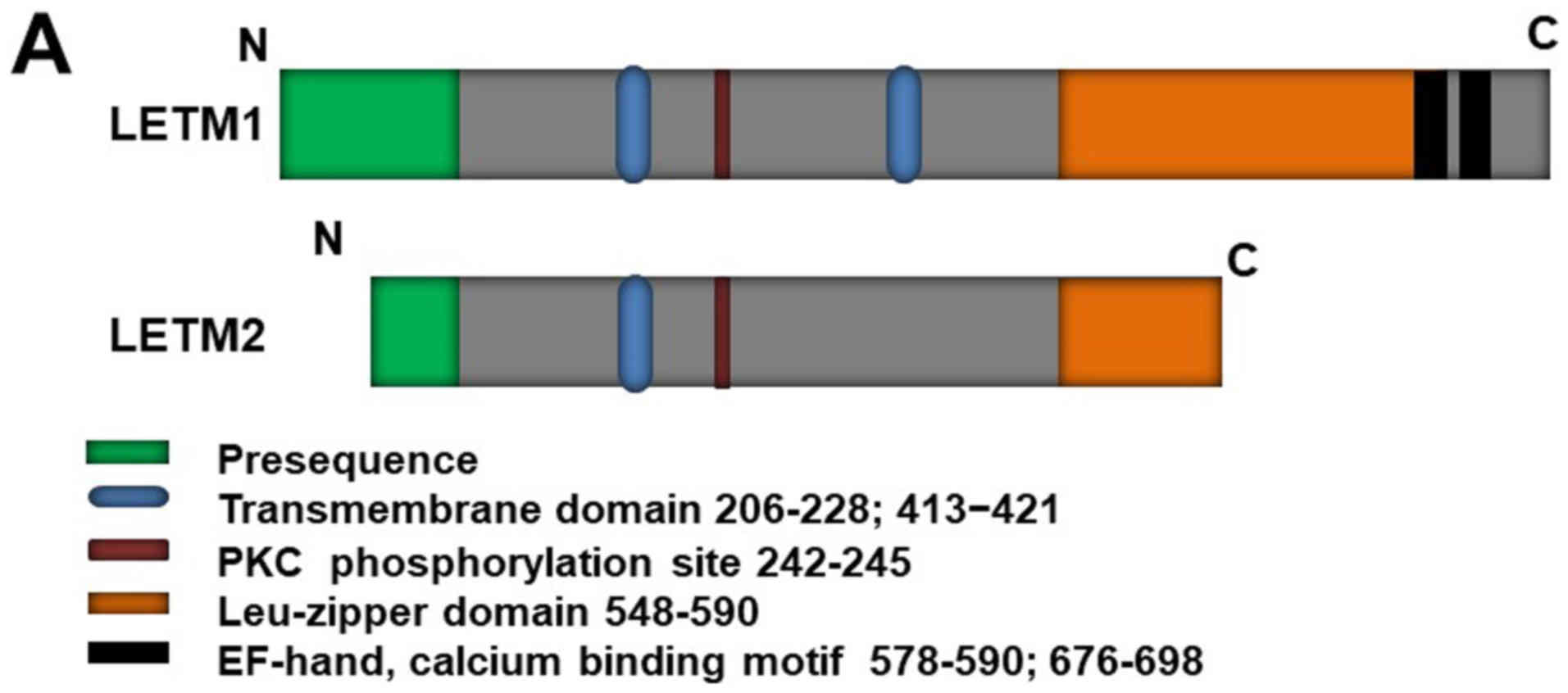
LETM1 structure and localization
LETM1 is a protein with a molecular mass of 70 kDa;
it has also been identified as a 83-kDa endogenous protein in HeLa
cells, suggesting that it is synthesized as a cytosolic precursor
with a presequence (11). LETM1,
like its yeast counterpart Mdm38 (12), is a mitochondrial inner membrane
protein (1,9,13);
it comprises a C-terminal domain bearing two EF-hand domains
(578–590 and 676–698) and an N-terminal domain bearing a protein
kinase C phosphorylation site (Fig.
1A) (6,14). LETM1 is an integral mitochondrial
inner membrane protein facing toward the matrix (Fig. 1B, right). LETM1 is co-localized
with HSP60, a mitochondria matrix protein (11). Partial digestion of LETM1 under
hypotonic conditions has suggested that LETM1 contains a small
and/or protease-resistant region exposed to the inner membrane
space (11). The N-terminus of
LETM1 is located in the inter-membrane space, connected to the
inner membrane by a transmembrane domain containing three conserved
proline residues (206–228), and the C-terminus extends to the
mitochondrial matrix (6,15) [Fig.
1B, left (1)]. However, a
recent study (16) revealed that
LETM1 contains another transmembrane domain, from residues 413 to
421. This discovery has led to controversy regarding LETM1
N-terminal localization, which was strongly suggested to face the
matrix [Fig. 1B, left (2)] (15).
LETM2, a homolog of LETM1, has already been
identified (11,17). The LETM2 gene is located at human
chromosome 8 and was also found in rat through a homolog search
using human LETM1 cDNA (17).
LETM2 is a mitochondrial inner membrane protein that shares some
features with LETM1 (e.g., they are both mitochondrial inner
membrane proteins with a leucine-zipper motif); however, it has a
function distinct from that of LETM1, as demonstrated by its
failure to suppress mitochondrial swelling caused by LETM1
knockdown (11). LETM1 has been
detected in nearly all rat tissues, whereas LETM2 is expressed
specifically in the testis, with a molecular size of 45 kDa, during
the developmental stages from spermatocyte to spermatozoon. These
expression environments indicate its probable function in inner and
crista membrane reorganization in the mitochondria during
spermatogenesis (11).
LETM1 regulates mitochondrial
ion-channels
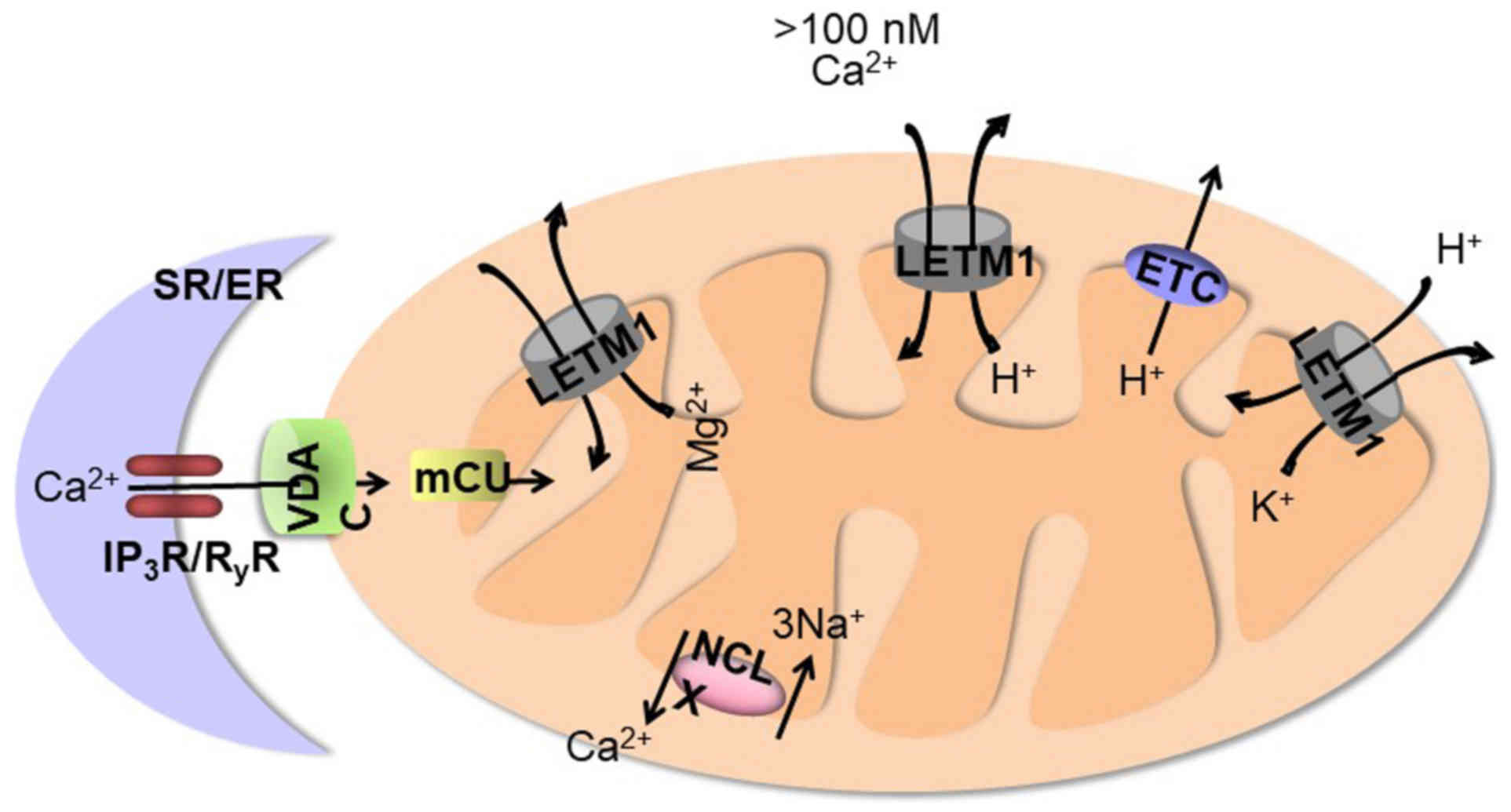
As indicated in Fig.
2, LETM1 was recently reported to function as a mitochondrial
ion channel regulator. The presence of two putative Ca2+
binding sites on LETM1 suggestes that impaired mitochondrial
Ca2+ homeostasis due to lack of LETM1 could explain the
seizures observed in some WHS patients (1). An RNAi screen was conducted to
identify the genes that control mitochondrial Ca2+
transport in Drosophila S2 cells which stably express
mitochondria-targeted ratiometric pericam. CG4589, the
Drosophila homolog of the human gene LETM1, was identified
as a gene strongly affecting (Ca2+)mito and
(H+)mito responses (18). LETM1 catalyzes the 1:1 electrogenic
exchange of Ca2+ for H+ which is driven by
the negative potential of mitochondria and by protons leaving the
mitochondrial matrix (18,19). The mitochondrial calcium unipoter
(mCU) drives rapid and massive Ca2+entry, but only at
the high cytosolic Ca2+ concentrations (>10 µM) that
are reached in microdomains near Ca2+ release channels
on the endoplasmic reticulum (ER) (20). The new
Ca2+/H+ antiporter operates at low cytosolic
Ca2+ concentrations (<100 nM) and is limited by the
pH gradient generated by the mitochondrial electron transport chain
(21). LETM1 shares a role with
mCU in catalyzing Ca2+ uptake into mitochondria; this
role can be inhibited by ruthenium red. Thus, unlike mCU, which is
critical only for Ca2+ uptake, LETM1 catalyzes
Ca2+ uptake into mitochondria in exchange for
H+, implying that proton efflux from mitochondria can
drive LETM1-dependent Ca2+ entry into mitochondria. mCU
conveys rapid Ca2+ transient from the cytosol to the
matrix but exposes mitochondria to Ca2+ overload and
alterations in ER Ca2+ handling. LETM1 is bidirectional
and can extrude Ca2+ along with the
Na+/Ca2+ exchanger during large
Ca2+ loads, allowing mitochondria to bear small
cytosolic Ca2+ elevations without risking
Ca2+ overload (21).
Knockdown and overexpression of LETM1 in cells has been shown to
alter [Ca2+]mito and [pH]mito
responses in a pattern consistent with
Ca2+/H+ exchange (18). Reconstitution of the purified
protein in liposomes confirmed that LETM1 mediates
Ca2+/H+ exchange. This transport is
electrogenic and therefore blocked by ruthenium red. Functional
data in LETM1-depleted HeLa cells have indicated that the new
antiporter can mediate both Ca2+ uptake and
Ca2+ extrusion from mitochondria; however, these
observations remain to be confirmed by simultaneous Ca2+
and pH measurements during physiological stimulation (18). LETM1 and UCP2/3 independently
contribute two molecularly distinct pathways for mitochondrial
Ca2+ uptake in endothelial cells. Knockdown of LETM1
resulted in highly depleted sequestration of entering
Ca2+, but had no effect on Ca2+ uptake at
ER-mitochondrial junctions, suggesting that LETM1 and UCP2/3
contribute Ca2+ uptake from different sources and at
different Ca2+ concentrations (22). However, it has been proposed that
LETM1 works as a Ca2+/H+ antiporter in mitochondria, but also that
LETM1 functions as a mitochondrial K+/H+
exchanger (12). Measurements in
isolated mitochondria labeled with K+ and H+
fluorescent dyes confirmed the presence of a
K+/H+ exchanger in the inner mitochondrial
membrane of yeast (12). Mutants
in the yeast LETM1 homolog Mdm38, which lacks the two Ca2+ binding
sites present in LETM1, cause loss of mitochondrial
K+/H+ exchange activity, resulting in highly
abundant K+ within the mitochondrial matrix and low
membrane potential (ΔΨm), followed by water influx and organelle
swelling (23,24). LETM1 expression restores Mdm38 and
potassium acetate indicating a defect in
K+/H+ exchange activity-induced swelling
(23). Furthermore, Mdm38
depletion resulted in an early loss of K+/H+
exchange-mediated swelling capacity of mitochondria, loss of ΔΨm
and mitophagy during the extensive interaction of mitochondria with
vacuoles (24,25). These effects strongly support the
notion that K+/H+ exchange activity is the
primary cause of morphological changes in mitochondria, which in
turn trigger the process of mitophagy (24). This evidence of mitophagy induction
via Mdm38 depletion feasibly suggests that LETM1 regulates
mitophagy and cell viability.
Mitochondrial Mg2+ transport processes
may play an important role in cellular Mg2+ homeostasis
and the regulation of cell and mitochondrial functions (26). MRS2 encodes a mitochondrial
membrane protein, that is an essential component of the major
electrophoretic Mg2+ influx system in mitochondria (26). Mutant alleles of MRS2 and its
overexpression increase intra-mitochondrial Mg2+
concentrations. Two yeast homolog genes of LETM1, MRS7 and YOL027,
are multicopy suppressors of MRS2D (disruptiing the open reading
frames of MRS2 and defective in mitochondrial Mg2+
influx, respectively), whereas Yo1027p overexpression may improve
Mg2+ influx in MRS2D cells (23,26).
Furthermore, Mdm38 mutation suppresses the growth deficiency caused
by mutation in the MRS2 gene (27,28).
Together, these findings suggest LETM1 acts as an Mg2+
transporter in the mitochondrion.
LETM1 and its partners cooperate to
influence mitochondria morphology, respiration and biogenesis
Yeast Mdm38, a homolog of LETM1 was identified in a
comprehensive genome-wide screen for mutants with mitochondrial
morphology defects (29). Within
LETM1-knockdown cells, mitochondrial swelling and the
disorganization of cristae structures were observed using electron
microscopy (11). LETM1 knockdown
caused mitochondria to become dot-like structures and lose their
tubular networks to a significantly greater extent than fragmented
mitochondria observed in OPA1-knockdown cells (11,30).
Images of mitochondria lacking LETM1 were reminiscent of
observations following overexpression of pro-fission proteins such
as Fis1 (31) or knockdown of
pro-fusion proteins sfiguch as OPA1 (32). In both cases, inhibition of the
dynamin-related protein Drp1-dependent fission machinery, by
silencing or using a dominant negative, caused the Drp1K38A mutant
to recover its mitochondrial morphology (33,34).
Nevertheless, Drp1 inhibition in LETM1 knockdown cells, did not
recover the fragmented phenotype, indicating that blockage of the
fission machinery is not induced by reduction of LETM1 and that
lack of LETM1 causes Drp1-independent mitochondrial fragmentation
(30). Moreover, following the
silencing of both Drp1 and LETM1, mitochondria remained partly
swollen, suggesting that mitochondrial membrane fusion is
unaffected by downregulation of LETM1 or co-silencing of Drp1
(11). These results suggest that
LETM1 is not directly implicated in mitochondrial membrane fission
and fusion. Thus, neither lack nor excess of LETM1 is beneficial to
cells, and fragmented mitochondria and swollen cristae were
observed in LETM1-overexpressed HeLa cells (14).
LETM1 downregulation caused reduced numbers, and
morphological changes in cristae, leading to a substantially lower
membrane potential (ΔΨm), which is consistent with reports that
isolated mitochondria from yeast Mdm38 mutants exhibited low
membrane potential (23,35); however, no significant changes in
ΔΨm were observed between controls and LETM1-transduced cells
(14). LETM1 knockdown caused the
disassembly of three different proton pumps complex I (NADH
dehydrogenase), III (cytochrome b complex), and IV (cytochrome c
oxidase) (36), suggesting that
LETM1 regulates the biogenesis of respiratory chains. Low membrane
potential appears to be a consequence of the failure to form
super-complexes: complexes I, III and IV of the respiratory chains
(11).
Therefore, LETM1 is critical for respiratory chain
biogenesis, being physically associated with mitochondrial
ribosomes to mediate membrane insertion of several proteins of
nuclear and mitochondrial origin, and facilitating their transport
across the inner membrane (24,35).
A ribosome-associated site has been identified on the LETM1
ribosome binding domain (RBD); it displays a matrix-exposed
14-3-3-like fold, which is critical for respiratory chain assembly
through the regulation of Cox1 and Cytb translation (37). Mitochondrial ribosomal protein L36
(MRPL36) has been reported to be associated with the inner-membrane
(38). The addition of puromycin
was used to access possible interactions between LETM1 and MRPL36;
LETM1 associated with MRPL36 both in vivo and in
vitro. MRPL36 siRNA significantly recovered LETM1-mediated ATP
reduction, suggesting that LETM1 may regulate the mitochondrial
translation system and reduce mitochondrial biogenesis through
association with MRPL36 (14). The
inhibition of mitochondrial biogenesis by LETM1 offers a possible
explanation of WHS phenotypes, especially neuromuscular defects and
seizures, which likely reflect oxidative phosphorylation defects
and thus resembles classical mitochondrial encephalomyopathies such
as mitochondrial encephalomyopathy, lactic acidosis, and stroke
like episodes (35).
Cells lacking mitochondrial DNA lose active
respiratory chains, but maintain mitochondrial tubular networks,
indicating that mitochondrial swelling caused by LETM1 knockdown is
not caused by the disassembly of respiratory chains (11). Human AAA-ATPase BCS1L, which is a
mitochondria inner membrane protein, is responsible for human
disorders and the assembly of respiratory chains (39–41).
BCS1L interacts specifically with LETM1, stimulating the formation
of the LETM1 major complex; thus, BCS1L and LETM1 function in
different process to maintain mitochondria and form tubular network
structures (11).
Role of LETM1 in mitochondrial quality
control
The term mitophagy was first introduced by Dr J.J.
Lemasters for the selective autophagic degradation of damaged and
dysfunctional mitochondria (42).
An accumulation of studies suggest that mitochondrial dysfunction
and morphological changes, which are interrelated factors, are
responsible for the induction of mitophagy (24,43,44).
Mitochondrial fission produces two subsets of daughter mitochondria
with either increased or decreased membrane potential. The
depolarized daughter mitochondrion, which is incapable of fusing
into the polarized network, is removed through the process of
mitophagy (44). Loss of
K+/H+ activity and subsequent decrease in
membrane potential due to Mdm38 depletion leads to mitochondrial
fragmentation into discrete spheres and their association with
vacuoles, suggestive of mitophagy induction. Furthermore, the
depletion of DNM1, a mitochondrial fission protein, and Mdm38 leads
to the continued fusion of swollen mitochondria, indicating the
blockage of selective removal by mitophagy (24). LETM1 overexpression has been
reported to result in a mitochondrial ATP decrease, mitochondrial
dysmorphology, swollen mitochondria cristae, and fragmentation in
HeLa cells in an OPA1-dependent manner (45). The mitochondria within
autophagosomes with reduced OPA1 levels (44) and fragmented discrete mitochondria
targeted by autophagic vacuoles in an Mdm38-depleted yeast strain
point toward a possible role for LETM1 in mitophagy induction.
Mitochondrial depolarization also leads to the stabilization and
accumulation of PINK1 in the outer membrane of the mitochondrion
(46), which can recruit parkin;
the depolarized, damaged mitochondria are subsequently removed by
mitophagy (47). Interestingly,
PINK1-phosphorylated LETM1 has been shown to modulate mitochondrial
Ca2+ transport and protect neurons against mitochondrial
stress (48). These studies paved
the way for further studies to reveal the role of LETM1 in the
quality control machinery of mitochondria.
New insights into tumorigenesis
Tumors have been reported in several WHS cases
(49–52). In particular, the discovery of
neuroblastoma in a child with WHS implicated the association of
these two phenotypes (51).
Because WHS is rare, the occurrence of these tumors raises concern
that cancer may be a component feature of WHS. LETM1 is highly
expressed in many human malignancies and is correlated with poor
prognosis (14,53,54).
LETM1 knockdown by siRNA repressed proliferation, migration, and
invasion in bladder cancer cells (53). Several oncogenic proteins in the
Wnt/β-catenin signaling pathway (β-catenin, cyclin D1 and c-Myc)
were significantly decreased by LETM1 siRNA. These finding clearly
indicate the roles of LETM1 in tumor progression. However, the
potential molecular mechanism of LETM1-mediated tumorigenesis
remains to be elucidated.
Because mitochondrial dysfunction has been
implicated in a wide variety of human diseases, including cancers
and age-related disorders, mitochondria have emerged as effective
targets for anticancer therapy (55–57).
Mitochondria function as central components of cell survival
through ATP production and govern cell fate by mitochondrial
membrane-dependent cell death signaling (58). Loss of Mdm38 results in a variety
of phenotypic effects, including defects of respiratory chain,
altered mitochondrial morphology, osmotic swelling, and mitophagy
(24). Downregulation of LETM1
causes Drp1-independent fragmentation of the mitochondrial network,
but is not associated with respiratory chain, whereas
overexpression of LETM1 leads to mitochondrial fragmentation
through OPA1 modulation (30).
Although the functions and mechanisms of LETM1 with respect to cell
viability and tumorigenesis remain controversial, accumulating data
suggest that LETM1 will be a crucial candidate to clarify how
mitochondria regulate the normal life of the cell.
Maintenance of mitochondrial
morphology by LETM1 is required for cell viability
The regulation of mitochondrial volume and
morphology has been associated with a wide range of important
biological functions and pathologies. Any disruption in the osmotic
balance between mitochondria and the cytosol, mainly as a result of
intracellular ion fluxes (particularly K+, due to its
higher concentration), induces water movement between these two
compartments, leading to loss of mitochondrial volume and
homeostasis, which then cause morphological changes (59). Fission-fusion events can also
result in mitochondrial morphological changes, which are modulated
by a complex network of cytosolic and mitochondrial proteins and
are coordinated to respond to specific cellular demands (60,61).
Osmotic swelling, which indicates pathological states in
mitochondria, has been found to activate downstream cascades,
notably determining the viability of the cell as a whole (61).
Mitochondrial swelling also plays an important role
in the release of cytochrome c, which is associated with apoptotic
cell death (59).
Adenovirus-meditated LETM1 overexpression leads to mitochondrial
dysmorphology, swollen mitochondria cristae, and an increase in
mitochondrial fragmentation, as shown by electron microscopy, and
sensitizes the cell to apoptosis in an OPA-1-dependent manner
(45).
However, long-term LETM1 overexpression leads to
necrotic cell death via decreases in intracellular ATP levels, in a
time-dependent manner, because it is independent of AIF nuclear
translocation and caspase activation. A previous study found that
the addition of extra glucose led to a reduction in total
mitochondrial mass and the amount of ATP per cell, and partial
recovery, supporting the notion that LETM1-mediated cell death is
influenced by energy deprivation (14). However, downregulation of LETM1 has
also been shown to result in caspase-independent and
Bcl-2-insensitive necrotic cell death. To date, it remains unknown
how both the gain and loss of LETM1 causes similar phenotypes in
cells.
Mitochondrial morphology is also strongly associated
with energy metabolism, as mitochondria with increased respiration
levels appear in morphologically interconnected networks with
enlarged cristae compartments, whereas those with low respiration
and therefore high glycolysis levels are fragmented, with smaller
inter-cristae space (62). LETM1
has been demonstrated to cause such fragmented morphology (24,45)
along with altered energy metabolism leading to tumorigenesis
(14). Taken together, these
findings demonstrate that the role of LETM1 in mitochondrial volume
and morphological homeostasis is critical to determining cell
viability and correlation with tumorigenesis.
LETM1 contributes to cancerous
metabolic alteration
In 1931, Otto Warburg was awarded the Nobel Prize
for oncology for his breakthrough hypothesis and research on
cancerous metabolism (63). He
observed that cancer cells are predominantly dependent on energy
produced by glycolysis, rather than by pyruvate oxidation in
mitochondria (57). Although the
exact mechanisms responsible for this metabolic alteration remain
to be elucidated, mitochondrial respiratory defects, due in part to
mitochondrial DNA mutations/deletions and hypoxia, are thought to
be important contributing factors. Mitochondrial respiration
deficiency and an increase in ATP production via glycolysis,
leading to activation of the PKB/Akt survival pathway through
NADH-mediated inactivation of PTEN, is a novel mechanism
contributing to altered cancerous metabolism (64). Consistent with these results, Piao
and colleagues demonstrated that LETM1 overexpression (45); led to increased ATP production via
glycolysis and increased lactate production, also causing PTEN
inactivation and PKB activation, suggesting that LETM1-induced
mitochondrial dysfunction resulted in PKB activation which
increased the ratio of NADH/NADPH to inactivate PTEN enzyme
activity. -Similar levels of LETM1 expression in cancerous tissue
and in overexpressed HeLa cells, along with the detection of LETM1
in two bands from six patients who had undergone surgery for
malignant cancer, strongly suggest that LETM1 overexpression is a
feature of altered cancerous metabolism accompanied by metastasis
(14). In contrast, AMPK
activation and consequent inhibition of G1/S cell cycle
progression, as well as decreased PKB and mTOR phosphorylation by
LETM1 overexpression can alter lung cancer cell growth (65). Using liquid chromatography tandem
mass spectrometry and a power law global error model for reliable
bio-signature mapping of hepatocellular carcinoma,, LETM1 was found
to have increased expression level, and its potential translocation
to the tumor nuclear fraction was inferred, especially to the
peripheral nuclear region and outer nucleus as confirmed by western
blot, immunohistochemical, and immunofluorescent analyses (66). Collectively, these finding
underscore the putative role of LETM1 in altered cancerous
metabolism and indicate the need for further study to explore the
roles of LETM1 in different tumors, and establish it as an
effective therapeutic target in cancer-treatment.
Association of LETM1 with
mitochondrial ribosomes and mitochondrial ATP regulation
Mutations in mitochondrial DNA have been shown to
play a key role in tumorigenesis within various organs, as these
mutations lead to the malformation or/and malfunctioning of the
mitochondrial respiratory chain, compelling cells to depend on
glycolysis to fulfill their ATP demand (67,68).
The biogenesis of respiratory chain complexes requires the
synthesis of proteins encoded by the mitochondrial genome and their
subsequent insertion into the inner membrane. Mdm38 has been found
to be associated with newly synthesized mitochondrial proteins via
the ribosome, and is specifically required for efficient membrane
insertion of cytochrome b and Atp6, and polytopic membrane proteins
of complexes III and V, respectively (35). The LETM1 ortholog Mdm38 plays a
role in respiratory chain function at the cellular level, as
demonstrated by growth defects and reduced ∆ψ observed in Mdm38∆
mitochondria. LETM1 partially rescues growth defects in Mdm38∆
cells, suggesting that both proteins fulfill similar cellular
functions (23).
An LETM1 RBD has been shown to be important to
respiratory chain assembly through regulation of Cox1 and Cytb
translation; this matrix-exposed domain displays a 14-3-3-like fold
(37). LETM1 overexpression has
been reported to decrease mitochondrial ATP production, oxygen
consumption, and MRPL36 depletion through the use of siRNA-MRPL36
to significantly revert the LETM1-mediated ATP decrease, suggesting
a functional association between LETM1 and mitochondrial ribosomes
(14). Thus, the association of
the LETM1 family with mitochondrial ribosomes and its consequent
role in the mitochondrial translation system and biogenesis
highlights the putative role of LETM1 in tumorigenesis.
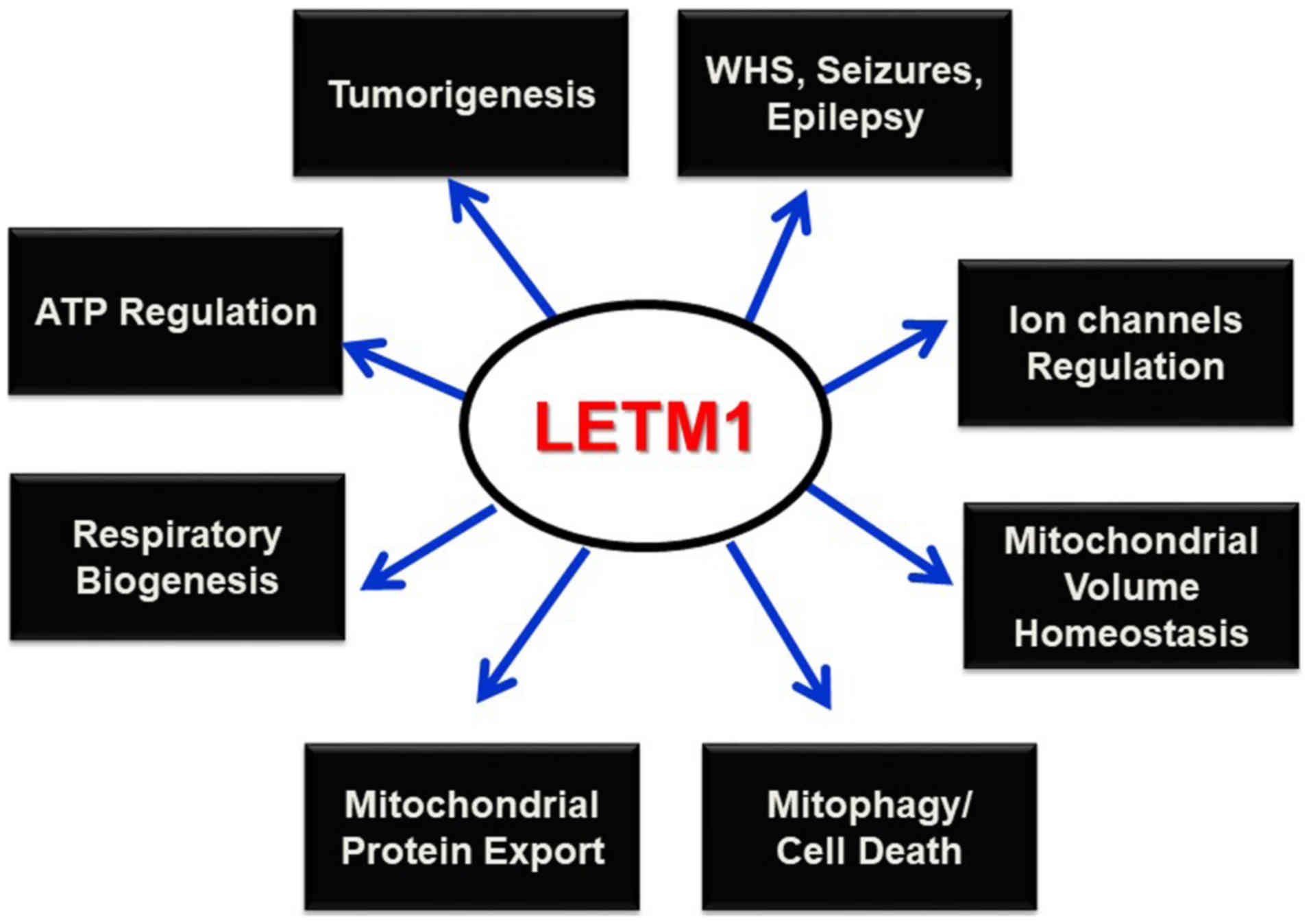
Conclusion
LETM1 has been cloned in an attempt to identify the
genes deleted in WHS (6), and was
found to be responsible for seizure development (4,10).
Since this discovery, LETM1 has been shown to play prominent roles
in mitochondrial K+ and Ca2+ homeostasis,
volume, and morphology maintenance, and respiratory chain
biogenesis, and to be indispensable in maintaining organelles and
cellular physiological integrity. Its potential interaction with
mitochondrial ribosomes and biogenesis, and regulation of
mitochondrial ATP and morphological homeostasis underscore its
putative candidacy in altered metabolism and subsequent
tumorigenesis. Mitophagy has been reported as the selective
degradation of depolarized mitochondria, as a result of lost
K+/H+ exchange activity due to Mdm38
depletion (24); however, this
process must be further explored to determine the contributing
roles of the PINK/PARKIN pathway and LETM1. Studies of the possible
interactions between LETM1 and mitochondrial ribosomes and its
functional relevance as a translocase in mitochondrial protein
synthesis and export machinery may help to establish this protein
as a potential therapeutic target for various diseases such as WHS,
epilepsy, cancer, and other pathophysiological conditions (Fig. 3).
Acknowledgements
Not applicable.
Funding
This work was financially supported by research fund
of Chungnam National University (Jongsun Park) and by the National
Research Foundation of Korea (NRF) grant funded by the Korea
Government (MEST) (grant nos. NRF-2012M3A9B6055302,
NRF-2015R1A2A2A01003597 and NRF-2016K1A3A1A08953546).
Availability of data and materials
All data used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YL conceived the present study. QT analyzed the
LETM1 literature and made the substantial contribution in the
finalization of this manuscript. LP assessed the experimental data.
RS and SP contributed to data interpretation and writing the
discussion. JiP contributed to data interpretation and discussion,
particularly regarding the mitochondria function of LETM1. JoP
contributed to designing the study and was involved in data
interpretation and writing the discussion. JoP also approved the
final manuscript for publication. All authors reviewed the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Schlickum S, Moghekar A, Simpson JC,
Steglich C, O'Brien RJ, Winterpacht A and Endele SU: LETM1, a gene
deleted in Wolf-Hirschhorn syndrome, encodes an evolutionarily
conserved mitochondrial protein. Genomics. 83:254–261. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McQuibban AG, Joza N, Megighian A,
Scorzeto M, Zanini D, Reipert S, Richter C, Schweyen RJ and
Nowikovsky K: A Drosophila mutant of LETM1, a candidate gene for
seizures in Wolf-Hirschhorn syndrome. Hum Mol Genet. 19:987–1000.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
South ST, Bleyl SB and Carey JC: Two
unique patients with novel microdeletions in 4p16.3 that exclude
the WHS critical regions: Implications for critical region
designation. Am J Med Genet A. 143A:2137–2142. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rauch A, Schellmoser S, Kraus C, Dörr HG,
Trautmann U, Altherr MR, Pfeiffer RA and Reis A: First known
microdeletion within the Wolf-Hirschhorn syndrome critical region
refines genotype-phenotype correlation. Am J Med Genet. 99:338–342.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zollino M, Lecce R, Fischetto R, Murdolo
M, Faravelli F, Selicorni A, Buttè C, Memo L, Capovilla G and Neri
G: Mapping the Wolf-Hirschhorn syndrome phenotype outside the
currently accepted WHS critical region and defining a new critical
region, WHSCR-2. Am J Hum Genet. 72:590–597. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Endele S, Fuhry M, Pak SJ, Zabel BU and
Winterpacht A: LETM1, a novel gene encoding a putative EF-hand
Ca(2+)-binding protein, flanks the Wolf-Hirschhorn syndrome (WHS)
critical region and is deleted in most WHS patients. Genomics.
60:218–225. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Simon R and Bergemann AD: Mouse models of
Wolf-Hirschhorn syndrome. Am J Med Genet C Semin Med Genet.
148C:275–280. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Piekorz RP, Hoffmeyer A, Duntsch CD, McKay
C, Nakajima H, Sexl V, Snyder L, Rehg J and Ihle JN: The
centrosomal protein TACC3 is essential for hematopoietic stem cell
function and genetically interfaces with p53-regulated apoptosis.
EMBO J. 21:653–664. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hasegawa A and van der Bliek AM: Inverse
correlation between expression of the Wolfs Hirschhorn candidate
gene Letm1 and mitochondrial volume in C. elegans and in mammalian
cells. Hum Mol Genet. 16:2061–2071. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang X, Chen G, Lu Y, Liu J, Fang M, Luo
J, Cao Q and Wang X: Association of mitochondrial letm1 with
epileptic seizures. Cereb Cortex. 24:2533–2540. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tamai S, Iida H, Yokota S, Sayano T,
Kiguchiya S, Ishihara N, Hayashi J, Mihara K and Oka T:
Characterization of the mitochondrial protein LETM1, which
maintains the mitochondrial tubular shapes and interacts with the
AAA-ATPase BCS1L. J Cell Sci. 121:2588–2600. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Froschauer E, Nowikovsky K and Schweyen
RJ: Electroneutral K+/H+ exchange in mitochondrial membrane
vesicles involves Yol027/Letm1 proteins. Biochim Biophys Acta.
1711:41–48. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Perkins SJ and Bonner A: Structure
determinations of human and chimaeric antibodies by solution
scattering and constrained molecular modelling. Biochem Soc Trans.
36:37–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Piao L, Li Y, Kim SJ, Byun HS, Huang SM,
Hwang SK, Yang KJ, Park KA, Won M, Hong J, et al: Association of
LETM1 and MRPL36 contributes to the regulation of mitochondrial ATP
production and necrotic cell death. Cancer Res. 69:3397–3404. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shao J, Fu Z, Ji Y, Guan X, Guo S, Ding Z,
Yang X, Cong Y and Shen Y: Leucine zipper-EF-hand containing
transmembrane protein 1 (LETM1) forms a
Ca2+/H+ antiporter. Sci Rep. 6:341742016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee SY, Kang MG, Shin S, Kwak C, Kwon T,
Seo JK, Kim JS and Rhee HW: Architecture mapping of the inner
mitochondrial membrane proteome by chemical tools in live cells. J
Am Chem Soc. 139:3651–3662. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stec I, van Ommen GJ and den Dunnen JT:
WHSC1L1, on human chromosome 8p11.2, closely resembles WHSC1 and
maps to a duplicated region shared with 4p16.3. Genomics. 76:5–8.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiang D, Zhao L and Clapham DE:
Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+
antiporter. Science. 326:144–147. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bernardi P: Mitochondrial transport of
cations: Channels, exchangers, and permeability transition. Physiol
Rev. 79:1127–1155. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Collins TJ, Lipp P, Berridge MJ and
Bootman MD: Mitochondrial Ca(2+) uptake depends on the spatial and
temporal profile of cytosolic Ca(2+) signals. J Biol Chem.
276:26411–26420. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Santo-Domingo J and Demaurex N: Calcium
uptake mechanisms of mitochondria. Biochim Biophys Acta.
1797:907–912. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Waldeck-Weiermair M, Jean-Quartier C, Rost
R, Khan MJ, Vishnu N, Bondarenko AI, Imamura H, Malli R and Graier
WF: Leucine zipper EF hand-containing transmembrane protein 1
(Letm1) and uncoupling proteins 2 and 3 (UCP2/3) contribute to two
distinct mitochondrial Ca2+ uptake pathways. J Biol Chem.
286:28444–28455. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nowikovsky K, Froschauer EM, Zsurka G,
Samaj J, Reipert S, Kolisek M, Wiesenberger G and Schweyen RJ: The
LETM1/YOL027 gene family encodes a factor of the mitochondrial K+
homeostasis with a potential role in the Wolf-Hirschhorn syndrome.
J Biol Chem. 279:30307–30315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nowikovsky K, Reipert S, Devenish RJ and
Schweyen RJ: Mdm38 protein depletion causes loss of mitochondrial
K+/H+ exchange activity, osmotic swelling and mitophagy. Cell Death
Differ. 14:1647–1656. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mijaljica D, Prescott M and Devenish RJ:
Different fates of mitochondria: Alternative ways for degradation?
Autophagy. 3:4–9. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kolisek M, Zsurka G, Samaj J, Weghuber J,
Schweyen RJ and Schweigel M: Mrs2p is an essential component of the
major electrophoretic Mg2+ influx system in mitochondria. EMBO J.
22:1235–1244. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Waldherr M, Ragnini A, Schweyer RJ and
Boguski MS: MRS6-yeast homologue of the choroideraemia gene. Nat
Genet. 3:193–194. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gregan J, Kolisek M and Schweyen RJ:
Mitochondrial Mg(2+) homeostasis is critical for group II intron
splicing in vivo. Genes Dev. 15:2229–2237. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dimmer KS, Fritz S, Fuchs F, Messerschmitt
M, Weinbach N, Neupert W and Westermann B: Genetic basis of
mitochondrial function and morphology in Saccharomyces cerevisiae.
Mol Biol Cell. 13:847–853. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dimmer KS, Navoni F, Casarin A, Trevisson
E, Endele S, Winterpacht A, Salviati L and Scorrano L: LETM1,
deleted in Wolf-Hirschhorn syndrome is required for normal
mitochondrial morphology and cellular viability. Hum Mol Genet.
17:201–214. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Alirol E, James D, Huber D, Marchetto A,
Vergani L, Martinou JC and Scorrano L: The mitochondrial fission
protein hFis1 requires the endoplasmic reticulum gateway to induce
apoptosis. Mol Biol Cell. 17:4593–4605. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cipolat S, Martins de Brito O, Dal Zilio B
and Scorrano L: OPA1 requires mitofusin 1 to promote mitochondrial
fusion. Proc Natl Acad Sci USA. 101:15927–15932. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
James DI, Parone PA, Mattenberger Y and
Martinou JC: hFis1, a novel component of the mammalian
mitochondrial fission machinery. J Biol Chem. 278:36373–36379.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sesaki H, Southard SM, Yaffe MP and Jensen
RE: Mgm1p, a dynamin-related GTPase, is essential for fusion of the
mitochondrial outer membrane. Mol Biol Cell. 14:2342–2356. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Frazier AE, Taylor RD, Mick DU, Warscheid
B, Stoepel N, Meyer HE, Ryan MT, Guiard B and Rehling P: Mdm38
interacts with ribosomes and is a component of the mitochondrial
protein export machinery. J Cell Biol. 172:553–564. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schägger H and Pfeiffer K: Supercomplexes
in the respiratory chains of yeast and mammalian mitochondria. EMBO
J. 19:1777–1783. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lupo D, Vollmer C, Deckers M, Mick DU,
Tews I, Sinning I and Rehling P: Mdm38 is a 14-3-3-like receptor
and associates with the protein synthesis machinery at the inner
mitochondrial membrane. Traffic. 12:1457–1466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ott M, Prestele M, Bauerschmitt H, Funes
S, Bonnefoy N and Herrmann JM: Mba1, a membrane-associated ribosome
receptor in mitochondria. EMBO J. 25:1603–1610. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
de Lonlay P, Valnot I, Barrientos A,
Gorbatyuk M, Tzagoloff A, Taanman JW, Benayoun E, Chrétien D,
Kadhom N, Lombès A, et al: A mutant mitochondrial respiratory chain
assembly protein causes complex III deficiency in patients with
tubulopathy, encephalopathy and liver failure. Nat Genet. 29:57–60.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Visapää I, Fellman V, Vesa J, Dasvarma A,
Hutton JL, Kumar V, Payne GS, Makarow M, Van Coster R, Taylor RW,
et al: GRACILE syndrome, a lethal metabolic disorder with iron
overload, is caused by a point mutation in BCS1L. Am J Hum Genet.
71:863–876. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hinson JT, Fantin VR, Schönberger J,
Breivik N, Siem G, McDonough B, Sharma P, Keogh I, Godinho R,
Santos F, et al: Missense mutations in the BCS1L gene as a cause of
the Björnstad syndrome. N Engl J Med. 356:809–819. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lemasters JJ: Selective mitochondrial
autophagy, or mitophagy, as a targeted defense against oxidative
stress, mitochondrial dysfunction, and aging. Rejuvenation Res.
8:3–5. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gomes LC and Scorrano L: High levels of
Fis1, a pro-fission mitochondrial protein, trigger autophagy.
Biochim Biophys Acta. 1777:860–866. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Twig G, Elorza A, Molina AJ, Mohamed H,
Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al:
Fission and selective fusion govern mitochondrial segregation and
elimination by autophagy. EMBO J. 27:433–446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Piao L, Li Y, Kim SJ, Sohn KC, Yang KJ,
Park KA, Byun HS, Won M, Hong J, Hur GM, et al: Regulation of
OPA1-mediated mitochondrial fusion by leucine
zipper/EF-hand-containing transmembrane protein-1 plays a role in
apoptosis. Cell Signal. 21:767–777. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jin SM, Lazarou M, Wang C, Kane LA,
Narendra DP and Youle RJ: Mitochondrial membrane potential
regulates PINK1 import and proteolytic destabilization by PARL. J
Cell Biol. 191:933–942. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Narendra D, Tanaka A, Suen DF and Youle
RJ: Parkin is recruited selectively to impaired mitochondria and
promotes their autophagy. J Cell Biol. 183:795–803. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Huang E, Qu D, Huang T, Rizzi N, Boonying
W, Krolak D, Ciana P, Woulfe J, Klein C, Slack RS, et al:
PINK1-mediated phosphorylation of LETM1 regulates mitochondrial
calcium transport and protects neurons against mitochondrial
stress. Nat Commun. 8:13992017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Battaglia A, Calhoun ARUL, Lortz A and
Carey JC: Risk of hepatic neoplasms in Wolf-Hirschhorn syndrome
(4p-): Four new cases and review of the literature. Am J Med Genet
A. 176:2389–2394. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bayhan T, Aydin B, Yalcin B, Orhan D and
Akyuz C: Hepatoblastoma and Wolf-Hirschhorn syndrome: Coincidence
or a new feature of a rare disease? Pediatr Int. 59:1028–1029.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ozcan A, Acer H, Ciraci S, Gumus H,
Karakukcu M, Patiroglu T, Ozdemir MA and Unal E: Neuroblastoma in a
child with Wolf-Hirschhorn syndrome. J Pediatr Hematol Oncol.
39:e224–e226. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Prunotto G, Cianci P, Cereda A, Scatigno
A, Fossati C, Maitz S, Biondi A and Selicorni A: Two cases of
hepatic adenomas in patients with Wolf-Hirschhorn syndrome: A new
rare complication? Am J Med Genet A. 161A:1759–1762. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Huang B, Zhang J, Zhang X, Huang C, Hu G,
Li S, Xie T, Liu M and Xu Y: Suppression of LETM1 by siRNA inhibits
cell proliferation and invasion of bladder cancer cells. Oncol Rep.
38:2935–2940. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li N, Zheng Y, Xuan C, Lin Z, Piao L and
Liu S: LETM1 overexpression is correlated with the clinical
features and survival outcome of breast cancer. Int J Clin Exp
Pathol. 8:12893–12900. 2015.PubMed/NCBI
|
|
55
|
Desagher S and Martinou JC: Mitochondria
as the central control point of apoptosis. Trends Cell Biol.
10:369–377. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wallace DC: Mitochondria and cancer:
Warburg addressed. Cold Spring Harb Symp Quant Biol. 70:363–374.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Don AS and Hogg PJ: Mitochondria as cancer
drug targets. Trends Mol Med. 10:372–378. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Karbowski M and Youle RJ: Dynamics of
mitochondrial morphology in healthy cells and during apoptosis.
Cell Death Differ. 10:870–880. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kaasik A, Safiulina D, Zharkovsky A and
Veksler V: Regulation of mitochondrial matrix volume. Am J Physiol
Cell Physiol. 292:C157–C163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cereghetti GM and Scorrano L: The many
shapes of mitochondrial death. Oncogene. 25:4717–4724. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Nowikovsky K, Schweyen RJ and Bernardi P:
Pathophysiology of mitochondrial volume homeostasis: Potassium
transport and permeability transition. Biochim Biophys Acta.
1787:345–350. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Alirol E and Martinou JC: Mitochondria and
cancer: Is there a morphological connection? Oncogene.
25:4706–4716. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Shaw RJ: Glucose metabolism and cancer.
Curr Opin Cell Biol. 18:598–608. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Pelicano H, Xu RH, Du M, Feng L, Sasaki R,
Carew JS, Hu Y, Ramdas L, Hu L, Keating MJ, et al: Mitochondrial
respiration defects in cancer cells cause activation of Akt
survival pathway through a redox-mediated mechanism. J Cell Biol.
175:913–923. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hwang SK, Piao L, Lim HT, Minai-Tehrani A,
Yu KN, Ha YC, Chae CH, Lee KH, Beck GR, Park J and Cho MH:
Suppression of lung tumorigenesis by leucine zipper/EF
hand-containing transmembrane-1. PLoS One. 5:e125352010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lee YY, McKinney KQ, Ghosh S, Iannitti DA,
Martinie JB, Caballes FR, Russo MW, Ahrens WA, Lundgren DH, Han DK,
et al: Subcellular tissue proteomics of hepatocellular carcinoma
for molecular signature discovery. J Proteome Res. 10:5070–5083.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chen Z, Lu W, Garcia-Prieto C and Huang P:
The Warburg effect and its cancer therapeutic implications. J
Bioenerg Biomembr. 39:267–274. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Tran Q, Park J, Lee H, Hong Y, Hong S,
Park S, Park J and Kim SH: TMEM39A and human diseases: A brief
review. Toxicol Res. 33:205–209. 2017. View Article : Google Scholar : PubMed/NCBI
|