Introduction
In the wide array of inherited metabolic disorders,
maple syrup urine disease (MSUD; Online Mendelian Inheritance in
Man no. 248600; http://www.omim.org/entry/248600) has attracted
increasing attention due to the potential neurological damage
caused by this disorder (1). MSUD
was first reported by Menkes et al (2) in 1954. MSUD is a rare autosomal
recessive disorder with an incidence of 1 in 185,000 children
worldwide (3). However, the
incidence in some regions is expected to be higher due to an
increased rate of consanguineous marriages (4).
MSUD is characterized by a deficiency of the
branched-chain α-ketoacid dehydrogenase (BCKD) complex, affecting
the metabolism of branched-chain amino acids (BCAAs), including
leucine, isoleucine and valine. Increased circulating levels of
BCAAs and their corresponding branched-chain keto acids (BCKAs) in
blood and cerebrospinal fluid are neurovirulent and may become
life-threatening (5). In addition,
increases in BCKAs levels result in a characteristic odor of the
urine of patients with MSUD, reminiscent of maple syrup (6).
The BCKD complex consists of three catalytic
components: E1, E2 and E3. E1 is a branched-chain α-ketoacid
decarboxylase, consisting of two E1α and two E1β subunits, encoded
by the branched chain keto acid dehydrogenase E1, α polypeptide
(BCKDHA) and by the branched chain keto acid dehydrogenase E1, β
polypeptide (BCKDHB) genes, respectively. E2 is a dihydrolipoyl
transacylase encoded by the dihydrolipoamide branched chain
transacylase E2 (DBT) gene. E3, a dihydrolipoamide dehydrogenase,
is encoded by the dihydrolipoamide dehydrogenase (DLD) gene
(7). Mutations in any of the genes
that encode components of the BCKD complex may result in MSUD. In
the Chinese population, numerous genetic variations have been
previously reported, primarily in the BCKDHA and BCKDHB genes, and
certain variations have been reported in the DBT gene (8).
The present study examined the clinical features and
genetic variations of two siblings with MSUD in a Chinese
family.
Materials and methods
Clinical data
The present study examined a Chinese family with
five members (Fig. 1). The
10-year-old daughter of the family was admitted to the County
Hospital of Juye, Heze in November 2007 with paroxysmal spasticity
of lower limbs, when she was 7 days old. Additional clinical
manifestations included poor feeding, vomiting and lethargy.
Initially, no definitive diagnosis was given, due to the limited
medical resources of the local hospital. At the age of 4 months,
the girl was misdiagnosed with developmental dysplasia of the hip
due to limited leg motor activity, and she was treated with a
Pavlik harness for 9 months. However, the clinical symptoms did not
improve. At the age of 1.5 years, the girl was diagnosed with MSUD
in April 2009 at Peking University First Hospital, Beijing
following tandem mass spectrometry (MS/MS) performed in blood
samples and gas chromatography combined with mass spectrometry
(GC/MS) performed using urine samples. MS/MS analysis in 2009
revealed that the concentrations of leucine and isoleucine, and
valine were 635.26 and 530.10 µmol/l, respectively. The BCAAs
levels in the blood were significantly higher than normal (9). GC/MS analysis identified elevated
levels of organic acids in the urine, including 2-OH-isovaleric,
2-keto-isovaleric and acetoacetic acids.
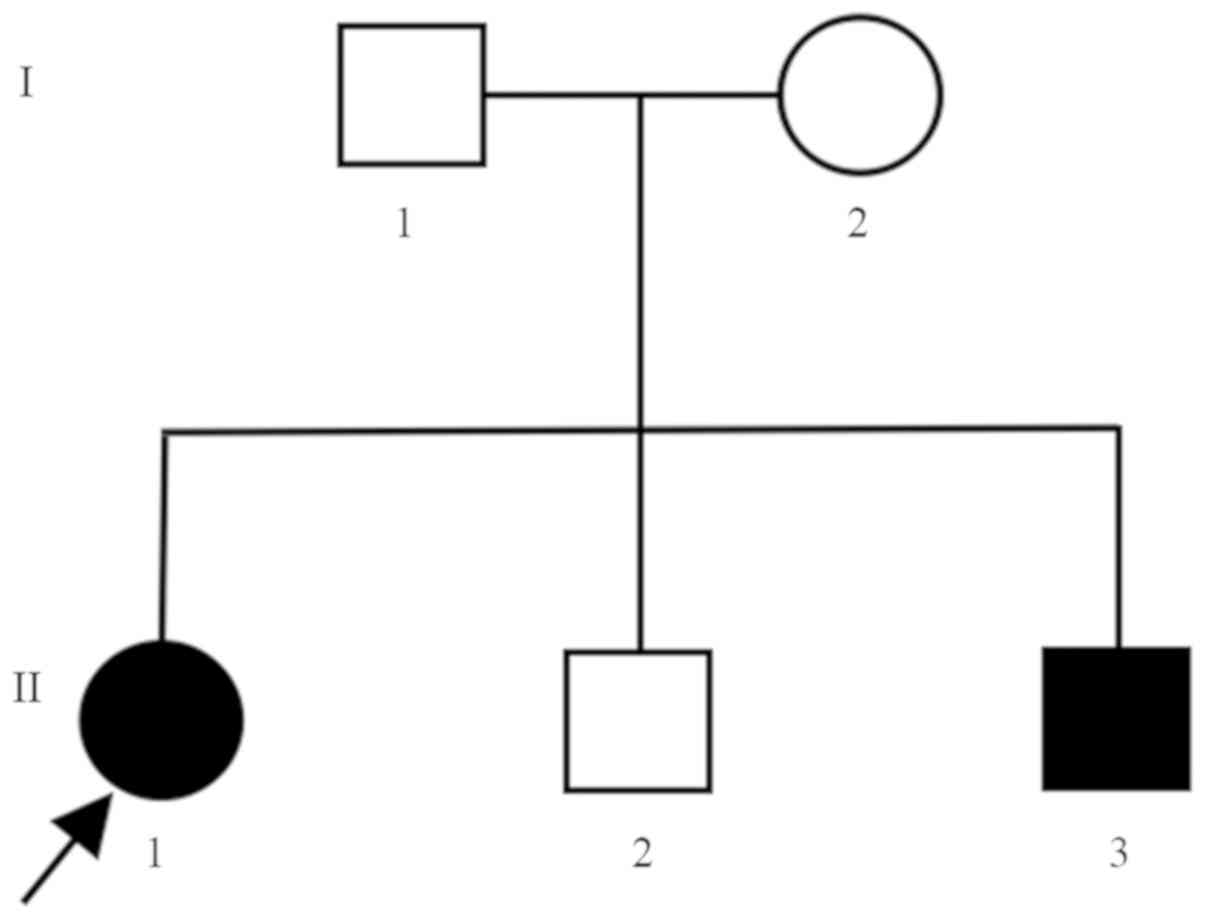 | Figure 1.Pedigree chart of the examined family.
The Chinese family investigated consisted of five members. I1, the
father, 34 years old, healthy. I2, the mother, 34 years old,
healthy. II1, the daughter, 10 years old, with MSUD. II2, the first
son, 9 years old, healthy. II3, the second son, 11 days old, with
MSUD. MSUD, maple syrup urine disease. |
The parents of the patient were healthy and
non-consanguineous, without significant disorders (cardiovascular,
neurological, endocrine, digestive, respiratory or genitourinary)
in their family history. The first son of the family was a year
younger than the daughter, and of good health from birth to the
present day.
Their third child was born when the eldest daughter
was 10 years old. The 11-day-old male was admitted to the
Affiliated Hospital of Jining Medical University, Jining in October
2017, due to paroxysmal spasticity of lower extremities, poor
feeding, vomiting and lethargy manifested for 3 days, with a urine
odor reminiscent of maple syrup. The symptoms were similar to the
symptoms of his sister during the neonatal period. Physical
examination identified hypermyotonia, poor sucking and rooting
reflexes, and incomplete clasping and grasping reflexes. MSUD was a
possible diagnosis, and the patient was admitted to the intensive
care unit. Convulsions occurred occasionally during
hospitalization, characterized by circular movements of the upper
limbs and spasticity of lower limbs.
Medical examinations
Metabolic analyses were performed when the youngest
son was 11 days old. Peripheral blood (3 drops) was collected from
the boy, permeating the filter paper to form dried blood spots, and
sent to Shanghai ADICON Clinical Laboratories, Inc. The levels of
blood amino acids, including BCAAs (valine and leucine), were
measured via MS/MS. Dried blood spot samples with a diameter of 3
mm were placed in microporous plates. The extraction and
derivatization procedures were performed using an Amino Acid and
Carnitine Tandem Mass Spectrometry kit (Fenghua Biotech Holding
Co., Ltd.) according to the manufacturer's protocol. Samples were
analyzed by Shanghai ADICON Clinical Laboratories, Inc. using a TQD
LC-MS/MS system (Waters Corporation) and ChemoView software
(SCIEX). The following day, fresh morning urine was collected from
the patient, permeating the filter paper; the sample was left to
dry naturally and then sent to Shanghai ADICON Clinical
Laboratories, Inc. (Shanghai, China) for analysis of the levels of
organic acids in urine via gas chromatography-MS, using the
GC-MS-QP2010plus analyzer (Shimadzu Corporation, Kyoto, Japan) and
the Inborn Errors of Metabolism Screening System software (Shimadzu
Corporation). When the boy was 1 month old, these metabolic
analyses were repeated.
At the age of 13 days, the boy underwent cerebral
magnetic resonance imaging (MRI) and video-electroencephalogram
(EEG). For his sister, MRI and video-EEG were also performed when
she was 10 years old. The revised Wechsler intelligence scale for
children was used to assess intelligence (10).
Genetic screening
To identify the genetic etiology of the disease,
genetic screening was performed when the youngest son was 2.5
months old. Peripheral blood samples (2 ml) were collected from
each member of the family and were sent to Beijing Kangso Medical
Inspection, Co., Ltd. (Beijing, China) for gene panel testing.
The FlexiGene DNA kit (Qiagen GmbH, Hilden, Germany)
was used to extract genomic DNA from the blood samples, and the
procedures were performed according to the manufacturer's protocol.
To construct the DNA library, genomic DNA samples were fragmented
into 150–300 bp DNA fragments by an ultrasonic processor (20 cycles
of 30 sec on, 30 sec off; Bioruptor® Plus Water Cooler;
Diagenode). Adaptors were ligated to both ends, and the cohesive
ends were removed. Then, the DNA library was amplified using a
PrimeSTAR HS DNA Polymerase (New England BioLabs, Inc., Ipswich,
MA, USA) under the following thermocycling conditions: Initial
denaturation at 95°C for 3 min, followed by 6 cycles of 20 sec at
98°C, 15 sec at 62°C and 30 sec at 72°C, with a final extension at
72°C for 5 min (forward primer,
5′-AATGATACGGCGACCACCGAGATCTACACACACTCTTTCCCTACACGACGCTCTTCCGATC-s-T-3′
and reverse primer,
5′-CAAGCAGAAGACGGCATACGAGAT[i7]GTGACTGGAGTTCAGACGTGTGCTCTTCCGATC-s-T-3′).
PCR products were purified using a nucleic acid purification kit
(Agencourt AMPure XP; Beckman Coulter, Inc., Brea, CA, USA)
according to the manufacturer's protocol.
A customized gene panel for inherited metabolic
diseases was designed, which comprised a total of 324 genes. The
DNA fragments were hybridized, isolated and then amplified using
the SureSelect Target Enrichment System (Herculase II Fusion Enzyme
dNTP combo; Agilent Technologies, Inc., Santa Clara, CA, USA) under
the following thermocycling conditions: Initial denaturation at
98°C for 2 min, followed by 15 cycles of 30 sec at 98°C, 30 sec at
62°C and 1 min at 72°C, with a final extension at 72°C for 10 min
(forward primer, 5′-AATGATACGGCGACCACCGA-3′ and reverse primer,
5′-CAAGCAGAAGACGGCATACGA-3′). Then, the products were purified as
aforementioned and quantified. Single-read sequencing was performed
using the NextSeq500 (Illumina, Inc., San Diego, CA, USA). Raw data
were obtained in FASTQ format and were transformed into
identifiable base sequences using CASAVA software (version 1.8.2;
Illumina, Inc.). Sequences were aligned to GRCh37 (as known as
hg19) using Burrow-Wheeler Aligner Version 0.7.15-r1140 (11), and single nucleotide and
deletion/insertion polymorphisms analyses were performed to obtain
mutation information within the targeted regions using GATK Version
3.6 (12).
The candidate mutated regions in BCKDHB gene were
selected for further validation. The primers were designed using
the online tool PrimerZ (Release 105; ncbi36.genepipe.ncgm.sinica.edu.tw/primerz/beginDesign.do)
and were subsequently synthesized (Tianyi Huiyuan Biotech Co.,
Ltd.). The sequences of the primers used are listed in Table I. The candidate mutation sites were
amplified by PCR using a EasyTaq PCR SuperMix (Beijing TransGen
Biotech Co., Ltd., Beijing, China), under the following
thermocycling conditions: Initial denaturation at 95°C for 10 min,
followed by 35 cycles of 30 sec at 95°C, 30 sec at 60°C and 45 sec
at 72°C, with a final extension at 72°C for 5 min. PCR products
were subsequently sequenced by Sanger sequencing.
 | Table I.BCKDHB (NM_183050) primer
sequences. |
Table I.
BCKDHB (NM_183050) primer
sequences.
| Genomic
coordinates | Nucleotide
substitution | Primer sequence
(5′-3′) |
|---|
| chr6:80881070 | c.705delT | F:
CAGCCCTTCTTAGCAGCGAGT |
|
|
| R:
CAGCACCTCCTTCACAGTCAAA |
| chr6:81053418 | c.1,076G>A | F:
TGGGGCATGGAGGAATTACA |
|
|
| R:
TCATTTTTCGAAGGGCATCA |
Bioinformatics analysis
To investigate the effects of the detected variants,
bioinformatics analyses were performed. RaptorX (http://raptorx.uchicago.edu) was used to predict the
tertiary structures of the mutated and wild-type BCKDHB proteins
(13). Then, the impact of a
certain mutation on the biological function of BCKDHB protein was
investigated using SIFT (Version 2; http://sift.bii.a-star.edu.sg/), PolyPhen2 (Version 2;
http://genetics.bwh.harvard.edu/pph2/) and
MutationTaster (NCBI 37/Ensembl 69; http://www.mutationtaster.org/).
Results
MS/MS revealed that the 11-day-old boy's blood
levels of leucine and valine were 2,027.40 and 736.86 µmol/l,
respectively, notably increased compared with usual levels
(9). GC/MS also revealed abnormal
increases in the urine levels of organic acids, including
2-OH-isovaleric, 3-OH-isovaleric, 2-keto-isovaleric and
2-keto-3-methylvaleric acids (data not shown). The results of the
metabolic analyses were consistent with a diagnosis of MSUD
(14).
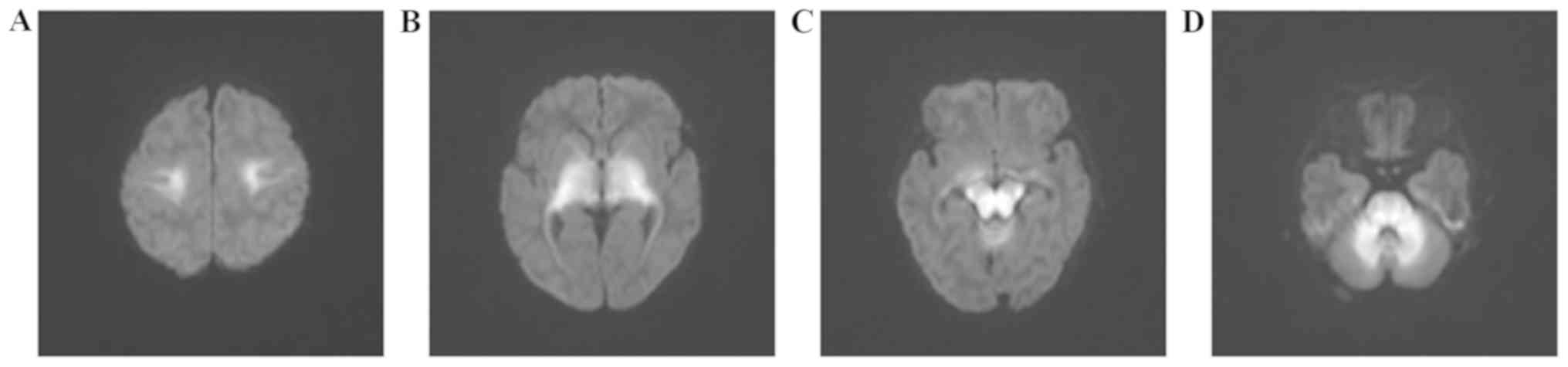
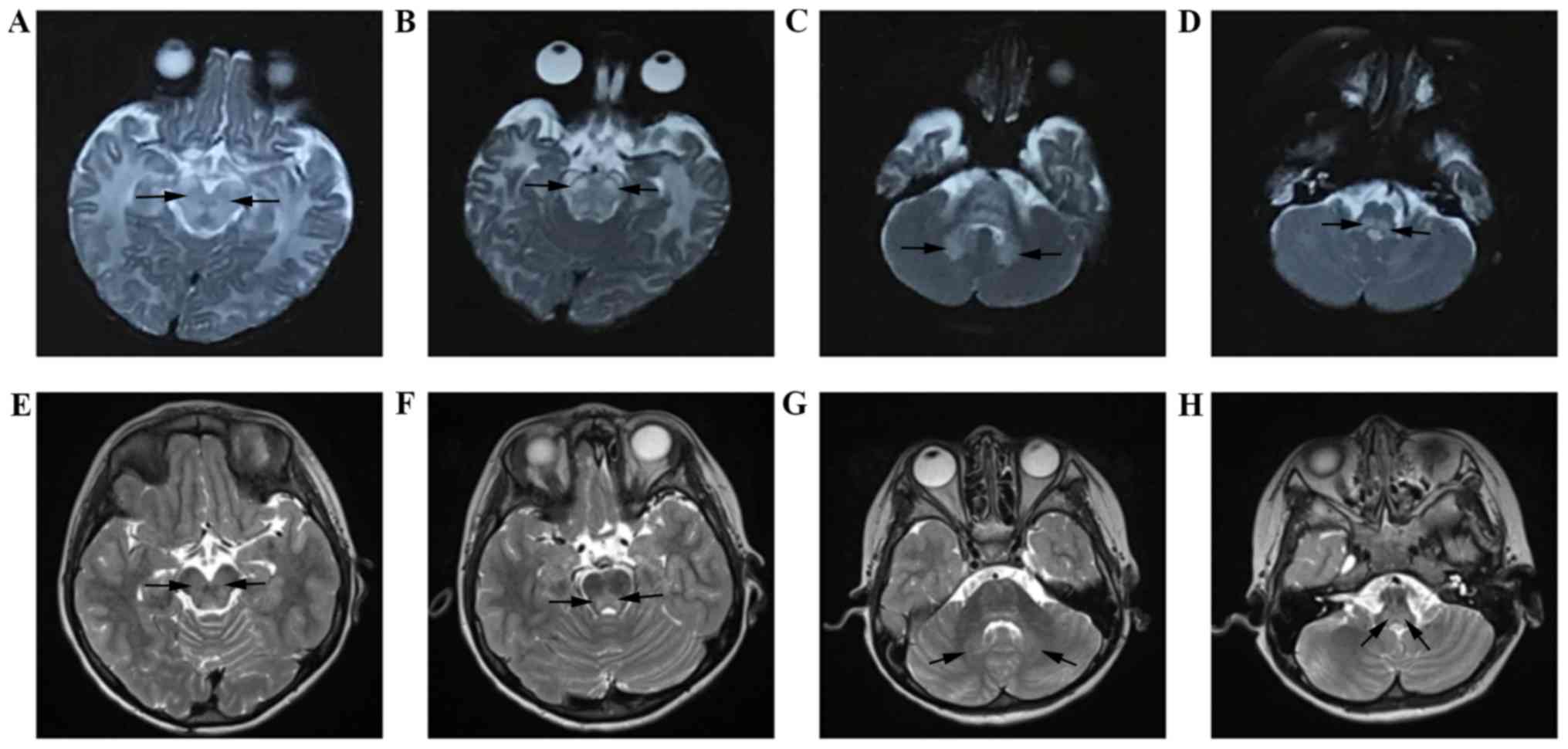
At the age of 13 days, the cranial MRI of the boy
demonstrated large areas of eudipleural long T2 signals with
limited diffusion in the frontoparietal white matter, basal
ganglia-thalamus, brainstem and cerebellum (Fig. 2). For his sister, cranial MRI
revealed eudipleural plaque-like long T2 signals in bilateral
cerebral peduncles, pons, dentate nucleus and medulla (Fig. 3).
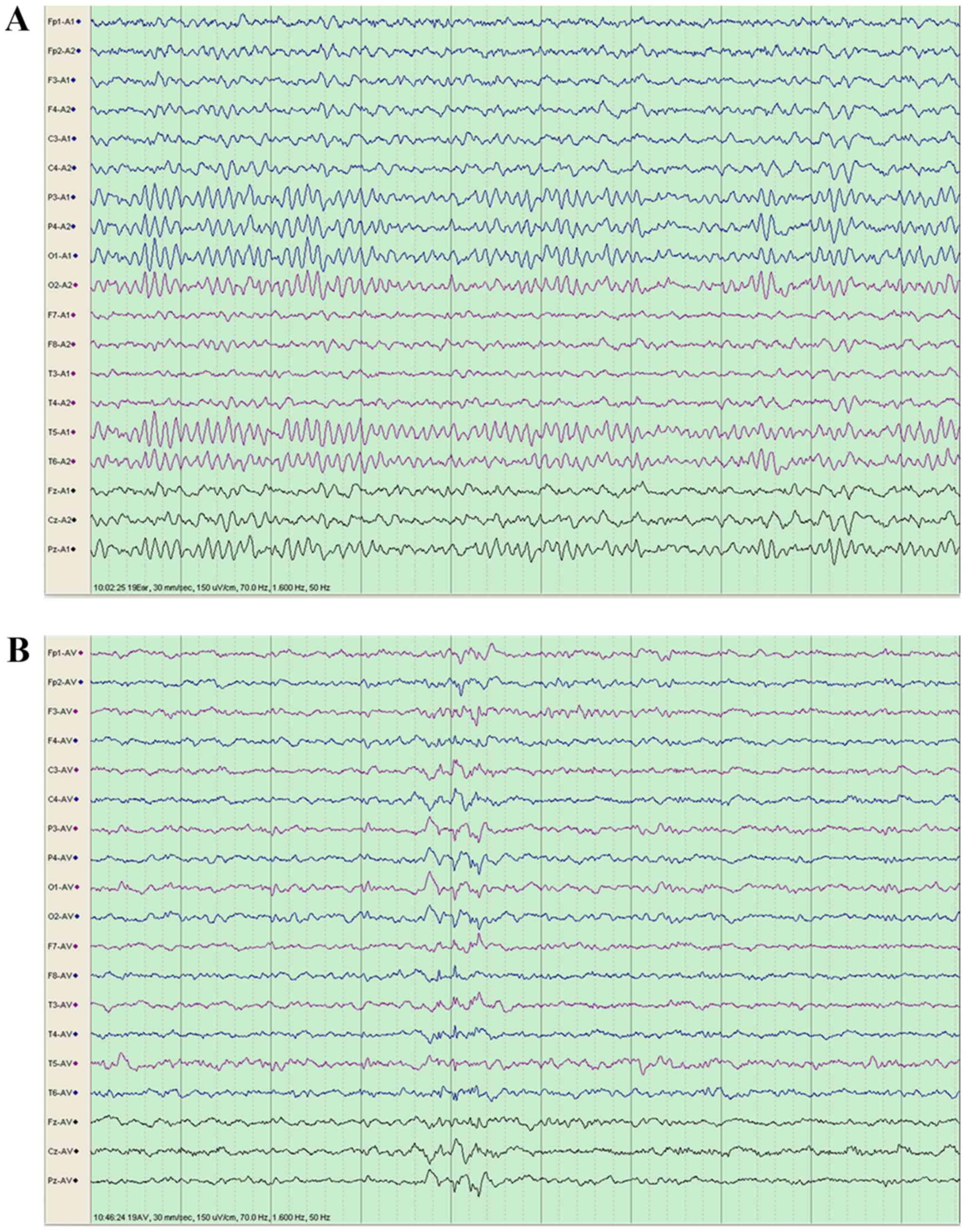
Video-EEG was performed when awake and when the
10-year-old girl was sleeping. During sleep, a small number of
spikes and slow wave complexes of medium-high amplitude were
identified (Fig. 4A and B,
respectively). For the boy, no abnormal waves were identified via
video-EEG analysis (data not shown).
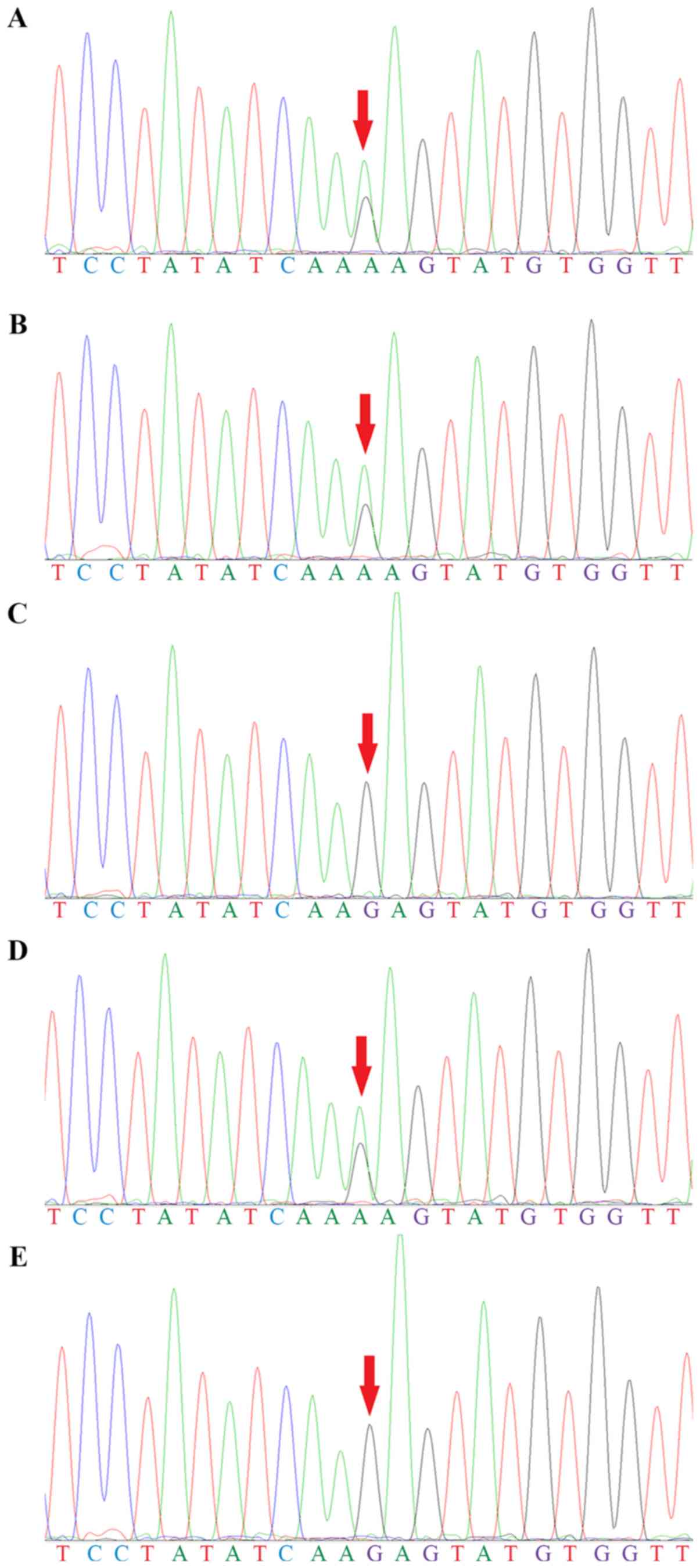
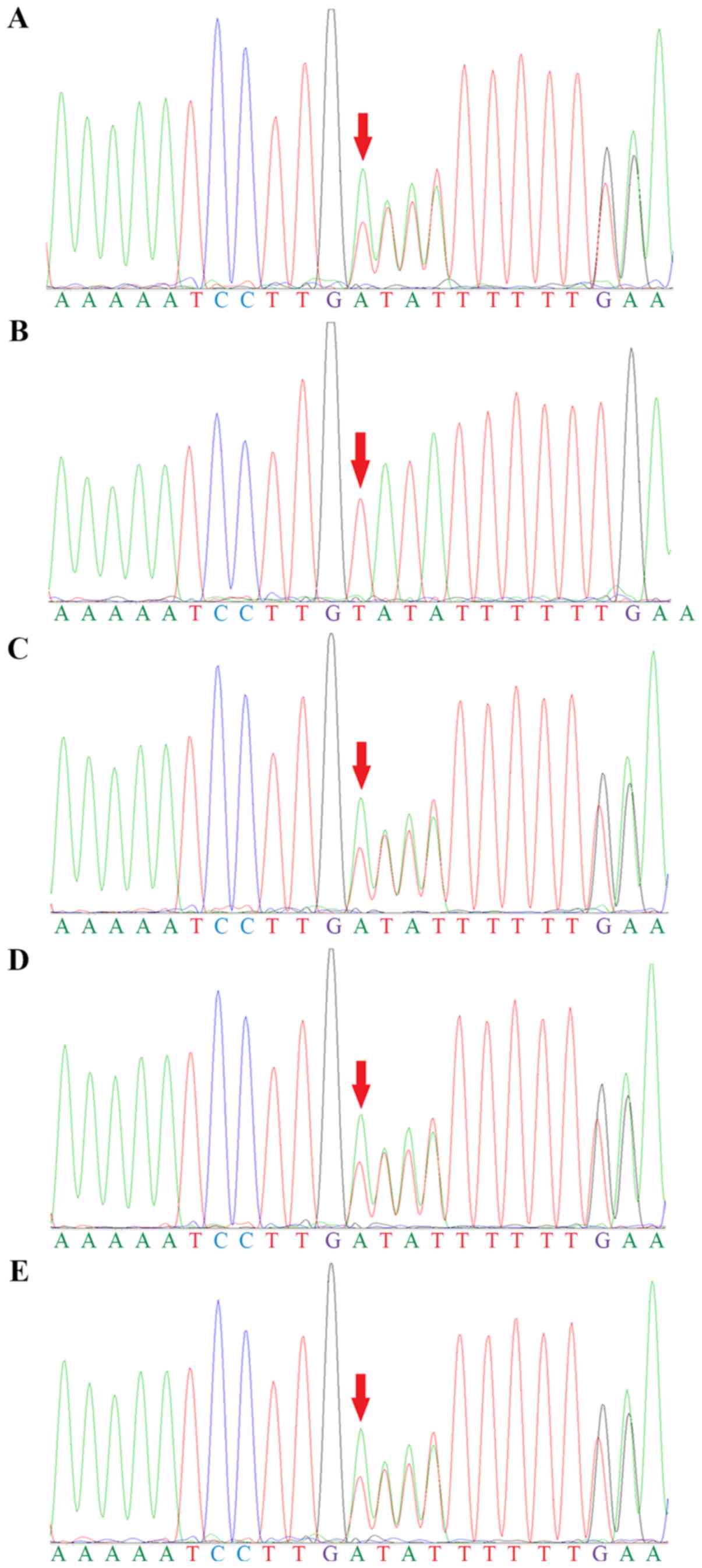
Genetic screening identified two compound
heterozygous mutations in BCKDHB; a substitution from guanine to
adenine in the coding region at position 1,076 (c.1,076G>A) in
exon 10 (Fig. 5) and a deletion of
a thymine at position 705 (c.705delT) in exon 6 (Fig. 6). The two siblings had both
mutations. Their father, mother and healthy brother had only one of
the two heterozygous mutations. The genotypic profiles of the
members of the family are presented in Table II.
| Table II.Genotype of the five family
members. |
Table II.
Genotype of the five family
members.
|
| Family member |
|---|
|
|
|
|---|
| Type of mutation in
BCKDHB | Second
sona | Fatherb | Motherb |
Daughtera | First
sonb |
|---|
| c.1,076G>A | Heterozygosis | Heterozygosis | WT | Heterozygosis | WT |
| c.705delT | Heterozygosis | WT | Heterozygosis | Heterozygosis | Heterozygosis |
The mutation c.1,076G>A results in an amino acid
substitution from arginine to lysine at position 359 (p.Arg359Lys).
SIFT and Polyphen2 identified that the Arg359Lys mutation may be
‘deleterious’ and ‘damaging’, respectively. MutationTaster software
indicated that the Arg359Lys mutation could be a ‘disease-causing’
mutation. The mutation c.705delT results in the replacement of a
cysteine at position 235 with a stop codon (p.Cys235Ter). According
to the American College of Medical Genetics and Genomics (ACMG)
criteria and guidelines (15), the
Cys235Ter mutation is classified as ‘likely pathogenic’. To
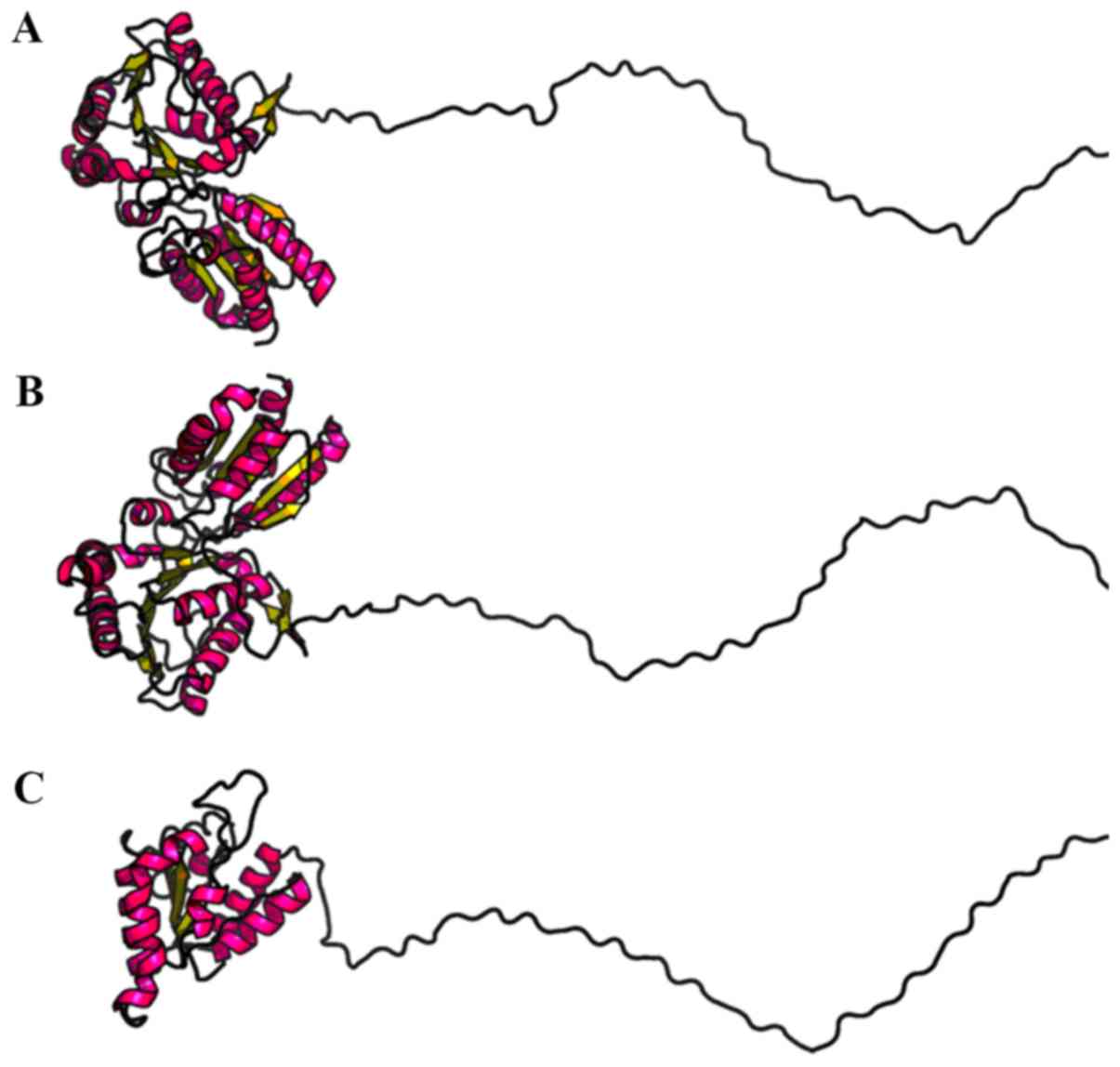
investigate the structural differences between the mutated and
wild-type BCKDHB proteins, RaptorX was used to predict the tertiary
structures (Fig. 7). Compared with
the wild-type BCKDHB protein, there were noticeable differences in
the predicted structure of the BCKDHB-mutated protein. Gene
mutations were predicted to cause changes in the three-dimensional
structure, which could affect the function of the protein.
The 11-day-old boy was promptly treated with a
combined therapy. Levocarnitine (500 mg/day) was used to decrease
the circulating levels of malondialdehyde in order to prevent
neurological damage. Thiamine (190 mg three times a day) and
BCAA-free medical formula (40 ml every 3 h) were administered to
decrease the circulating levels of BCAAs. After the treatment for
MSUD, the circulating levels of BCAAs were normal at 1 month of
age. At the time of writing, the 6-month-old boy had developed
normally without intellectual disability. However, due to a lack of
early diagnosis and prompt treatment in the neonatal period, his
sister developed an irreversible intellectual disability.
Specifically, the 10-year-old girl exhibited the intelligence level
of a 3-year-old child and an intelligence quotient <40, based on
the revised Wechsler intelligence scale for children (10).
Discussion
In the present study, the two novel heterozygous
mutations in the BCKDHB gene found in the family were hypothesized
to be responsible for the phenotype of the two children with MSUD.
The BCKDHB gene encodes the E1β subunit of the BCKD complex that
contains 392 amino acids. There are two Pfam conserved domains in
BCKDHB, including a transketolase, pyrimidine binding domain
between the amino acid 69 and 247, and a transketolase, C-terminal
domain between the amino acid 261 and 382 [(UniProt Knowledgebase;
https://www.uniprot.org/ (human BCKDHB)].
The mutation c.1,076G>A results in an amino acid
substitution: Lysine replaces arginine at position 359
(p.Arg359Lys). c.1,076G>A is a missense mutation in the
C-terminal domain of transketolase, which was identified to be a
regulatory domain with a ligand binding site (16). The C-terminal domain has a major
role in E1-E2 subunit association, which may be destabilized by the
mutation in the C-terminal domain (17). In addition, the Arg359 residue is
located in the homodimer interface of β-β' subunits, which may
influence the dimerization of E1β (18), and mutations in this site may
affect the interaction of the E1β subunit with other subunits,
altering the assembly of a functional heterotetramer (19). These effects may lead to changes in
the structure of the BCKD complex, impairing its BCAAs-degradation
activity, thus causing MSUD. SIFT analysis identified that the
Arg359Lys mutation may be deleterious. PolyPhen2 predicted that the
mutation was possibly damaging. In addition, MutationTaster
indicated that Arg359Lys may be a disease-causing mutation.
Therefore, Arg359Lys may lead to an unfavorable alteration in the
BCKD structure.
The mutation c.705delT results in a replacement of a
cysteine to a termination codon at position 235 (p.Cys235Ter).
Therefore, the synthesis of the peptide chain is terminated 158
amino acids earlier than the wild-type protein. The truncating
mutation affects the pyrimidine binding domain, thus inhibiting the
transketolase activity (20).
Additionally, the protein may lack the C-terminal domain including
the E1β interface segment, thus affecting the E1α-E1β interaction
(18). According to the ACMG
criteria and guidelines, this variant is classified as ‘likely
pathogenic’. Collectively, the present results suggested that the
c.705delT mutation, which results in the premature termination of
the peptide at the amino acid number 235, may influence the
function of the BCKD complex.
Neither of the BCKDHB alleles may be able to encode
functional E1β subunits in the compound-heterozygote patients,
resulting in an impaired activity of BCKD, which may lead to
increased circulating levels of BCAAs and BCKAs, thus causing MSUD.
Impairment of BCKD function results in accumulation of BCAAs,
leucine in particular, which may compete with other essential amino
acids for transport through the blood-brain barrier, leading to
decreased protein synthesis and demyelination in the brain
(21). Furthermore, accumulation
of BCKAs may inhibit pyruvate dehydrogenase and α-ketogutarate
dehydrogenase resulting in Krebs cycle dysfunction, leading to cell
swelling and cerebral edema (21).
In addition, oxidative stress is involved in neurological damage in
MSUD, and leucine may be the principal metabolite causing oxidative
brain damage (22).
Although MSUD is an autosomal recessive inherited
disorder, compound heterozygous mutations in BCKDHB alleles can
induce clinical manifestation of MSUD (23). In the present study, two children
with compound heterozygous mutations presented with MSUD, whereas
their parents and their brother with only one mutation in one of
the two BCKDHB alleles were healthy. In the present study, it was
hypothesized that the two mutations, including c.1076G>A and
c.705delT, are responsible for the phenotype of the two children
with MSUD.
In total, 181 BCKDHB mutations are listed in ClinVar
(update in April 2018, ncbi.nlm.nih.gov/clinvar/). Notably, neither of the
two mutations identified in the present study have been previously
reported in other databases, including the Human Gene Mutation
Database (update in January 2018, hgmd.cf.ac.uk/) and PubMed (update in January 2018,
ncbi.nlm.nih.gov/pubmed/). MSUD can be
classified as type IA, IB, II and III based on mutations in BCKDHA,
BCKHB, DBT and DLD, respectively (20). According to the MSUD
classification, the two siblings in this Chinese family presented
MSUD type IB. The clinical phenotypes of MSUD include classic,
intermediate, thiamine-responsive and intermittent forms. Mutations
in the genes that encode components of the BCKD complex can result
in MSUD. The IA and IB types are associated with the classic form
of MSUD, whereas the II and III types are associated with other
forms (23).
In addition to the common genetic background, the
two siblings had similar clinical manifestations; however, their
outcomes were markedly different. The male child had a favorable
prognosis because of the early diagnosis and treatment. By
contrast, the 10-year-old female exhibited intellectual disability
and impaired cognitive functions, due to inadequate treatment
during the acute phase of MSUD and poor long-term metabolic
management (24).
Early diagnosis of MSUD is important to prevent
irreversible brain injury. A maple syrup-like odor in urine and
cerumen may indicate MSUD (25).
MSUD may be suspected if early signs of metabolic decompensation,
including vomiting, poor feeding, diarrhea, fever and lethargy, or
neurological features, such as altered level of consciousness,
headaches, ataxia, dystonia and seizures, are observed (5). In the two siblings, paroxysmal
spasticity of lower extremities was the initial symptom. In
addition, cerebral MRI results showed restricted diffusion in the
myelinated areas on the diffusion-weighted imaging, and this
phenotype was identified to be an indicator of MSUD in newborns
(26). Additionally, adolescents
and young adults with MSUD exhibit increased signals on T2-weighted
imaging (27). Notably, the
definitive diagnosis of MSUD requires MS/MS and GC/MS analyses, and
genetic testing.
Appropriate treatment is crucial in patients with
MSUD. Patients with MSUD exhibit defects in the function of the
BCKD complexes that may reduce the interactions between the BCKD
complex and its cofactor, thiamine. The decreased affinity for
thiamine can be treated by administering high concentrations of
thiamine; therefore, thiamine should be used as a therapeutic agent
to treat MSUD (28). Another
beneficial therapy is L-carnitine supplementation (29). L-carnitine is a potent antioxidant
that prevents neurological damage, as molecule is able to cross the
blood-brain barrier (30). To
maintain normal circulating levels of BCAAs, long-term
administration of BCAAs-free medical formula and dietary protein
restriction are required (31).
Moreover, liver transplantation is an effective treatment, which
may reduce the progression of brain damage; however, liver
transplantation is not sufficient to reverse pre-existing
neurological damages (32).
Therefore, early treatment of MSUD is essential to allow normal
neuronal development (33).
In summary, the present study investigated clinical
and genetic features of two children with MSUD in a Chinese family.
In the present study, two novel mutations were identified in
BCKDHB, increasing the number of mutations known to be associated
with MSUD. Distinct neurocognitive outcomes of the two siblings
indicate the importance of early diagnosis and treatment of
MSUD.
Acknowledgements
The authors would like to thank Dr Ying-Ying Su, Dr
Sai-Sai Ren, Dr Lin Wang, Dr Xi-Bin Hu and Dr Ting-Ting Bian of the
Affiliated Hospital of Jining Medical University for their
assistance in collecting the clinical data; Dr Wei Wei of Beijing
Kangso Medical Inspection for technical support; and Dr Nan-Nan
Jiang of Beijing Children's Hospital, Dr Wei-Yang Li of Jining
Medical University and Dr Zhong-Yi Liu of the University of Hong
Kong for their assistance in drafting the manuscript.
Funding
The present study was supported by The Training
Program of National Natural Science Foundation of Jining Medical
University (grant no. JYP201740) and The Medicine, Health, Science
and Technology Development Project of Shandong Province (grant no.
2018WS450).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
QXK and QBL designed the present study. RHL, YS and
YDL collected the clinical data. YDL and XC analyzed the data and
wrote the paper. All authors read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
The present study was approved by The Ethics
Committee of The Affiliated Hospital of Jining Medical University.
The parents of the patients provided written informed consent.
Patient consent for publication
The parents of the patients provided written
informed consent for the publication of any associated data and
accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sperringer JE, Addington A and Hutson SM:
Branched-chain amino acids and brain metabolism. Neurochem Res.
42:1697–1709. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Menkes JH, Hurst PL and Craig JM: A new
syndrome: progressive familial infantile cerebral dysfunction
associated with an unusual urinary substance. Pediatrics.
14:462–467. 1954.PubMed/NCBI
|
|
3
|
Wasim M, Awan FR, Khan HN, Tawab A, Iqbal
M and Ayesha H: Aminoacidopathies: Prevalence, etiology, screening,
and treatment options. Biochem Genet. 56:7–21. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tabbouche O, Saker A and Mountain H:
Identification of three novel mutations by studying the molecular
genetics of maple syrup urine disease (MSUD) in the lebanese
population. Mol Genet Metab Rep. 1:273–279. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rodan LH, Aldubayan SH, Berry GT and Levy
HL: Acute illness protocol for maple syrup urine disease. Pediatr
Emerg Care. 34:64–67. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Puckett RL, Lorey F, Rinaldo P, Lipson MH,
Matern D, Sowa ME, Levine S, Chang R, Wang RY and Abdenur JE: Maple
syrup urine disease: Further evidence that newborn screening may
fail to identify variant forms. Mol Genet Metab. 100:136–142. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Su L, Lu Z, Li F, Shao Y, Sheng H, Cai Y
and Liu L: Two homozygous mutations in the exon 5 of BCKDHB gene
that may cause the classic form of maple syrup urine disease. Metab
Brain Dis. 32:765–772. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li X, Ding Y, Liu Y, Ma Y, Song J, Wang Q,
Li M, Qin Y and Yang Y: Eleven novel mutations of the BCKDHA,
BCKDHB and DBT genes associated with maple syrup urine disease in
the Chinese population: Report on eight cases. Eur J Med Genet.
58:617–623. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sowell J, Pollard L and Wood T:
Quantification of branched-chain amino acids in blood spots and
plasma by liquid chromatography tandem mass spectrometry for the
diagnosis of maple syrup urine disease. J Sep Sci. 34:631–639.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wechsler D: Wechsler Intelligence Scale
for Children. 5th. NCS Pearson; San Antonio, TX: 2014
|
|
11
|
Li H: Aligning sequence reads, clone
sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv.
1303.3997v2. 2013.
|
|
12
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
D'Angelo R, Donato L, Venza I, Scimone C,
Aragona P and Sidoti A: Possible protective role of the ABCA4 gene
c.1268A>G missense variant in Stargardt disease and syndromic
retinitis pigmentosa in a Sicilian family: Preliminary data. Int J
Mol Med. 39:1011–1020. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hoffmann GF and Kölker S: Defects in amino
acid catabolism and the urea cycle. Handb Clin Neurol.
113:1755–1773. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lindqvist Y, Schneider G, Ermler U and
Sundström M: Three-dimensional structure of transketolase, a
thiamine diphosphate dependent enzyme, at 2.5 A resolution. EMBO J.
11:2373–2379. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
AEvarsson A, Chuang JL, Wynn RM, Turley S,
Chuang DT and Hol WG: Crystal structure of human branched-chain
alpha-ketoacid dehydrogenase and the molecular basis of multienzyme
complex deficiency in maple syrup urine disease. Structure.
8:277–291. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bashyam MD, Chaudhary AK, Sinha M,
Nagarajaram HA, Devi AR, Bashyam L, Reddy EC and Dalal A: Molecular
genetic analysis of MSUD from India reveals mutations causing
altered protein truncation affecting the C-termini of E1α and E1β.
J Cell Biochem. 113:3122–3132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Edelmann L, Wasserstein MP, Kornreich R,
Sansaricq C, Snyderman SE and Diaz GA: Maple syrup urine disease:
Identification and carrier-frequency determination of a novel
founder mutation in the Ashkenazi Jewish population. Am J Hum
Genet. 69:863–868. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo Y, Liming L and Jiang L: Two novel
compound heterozygous mutations in the BCKDHB gene that cause the
intermittent form of maple syrup urine disease. Metab Brain Dis.
30:1395–1400. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zinnanti WJ, Lazovic J, Griffin K, Skvorak
KJ, Paul HS, Homanics GE, Bewley MC, Cheng KC, Lanoue KF and
Flanagan JM: Dual mechanism of brain injury and novel treatment
strategy in maple syrup urine disease. Brain. 132:903–918. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sitta A, Ribas GS, Mescka CP, Barschak AG,
Wajner M and Vargas CR: Neurological damage in MSUD: The role of
oxidative stress. Cell Mol Neurobiol. 34:157–165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang YP, Qi ML, Li TT and Zhao YJ: Two
novel mutations in the BCKDHB gene (R170H, Q346R) cause the classic
form of maple syrup urine disease (MSUD). Gene. 498:112–115. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Simon E, Schwarz M and Wendel U: Social
outcome in adults with maple syrup urine disease (MSUD). J Inherit
Metab Dis. 30:2642007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Blackburn PR, Gass JM, Vairo FPE, Farnham
KM, Atwal HK, Macklin S, Klee EW and Atwal PS: Maple syrup urine
disease: Mechanisms and management. Appl Clin Genet. 10:57–66.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kilicarslan R, Alkan A, Demirkol D, Toprak
H and Sharifov R: Maple syrup urine disease: Diffusion-weighted MRI
findings during acute metabolic encephalopathic crisis. Jpn J
Radiol. 30:522–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schönberger S, Schweiger B, Schwahn B,
Schwarz M and Wendel U: Dysmyelination in the brain of adolescents
and young adults with maple syrup urine disease. Mol Genet Metab.
82:69–75. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brown G: Defects of thiamine transport and
metabolism. J Inherit Metab Dis. 37:577–585. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mescka CP, Guerreiro G, Donida B,
Marchetti D, Wayhs CA, Ribas GS, Coitinho AS, Wajner M, Dutra-Filho
CS and Vargas CR: Investigation of inflammatory profile in MSUD
patients: Benefit of L-carnitine supplementation. Metab Brain Dis.
30:1167–1174. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ribas GS, Vargas CR and Wajner M:
L-carnitine supplementation as a potential antioxidant therapy for
inherited neurometabolic disorders. Gene. 533:469–476. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li X, Yang Y, Gao Q, Gao M, Lv Y, Dong R,
Liu Y, Zhang K and Gai Z: Clinical characteristics and mutation
analysis of five Chinese patients with maple syrup urine disease.
Metab Brain Dis. 33:741–751. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Díaz VM, Camarena C, de la Vega Á,
Martínez-Pardo M, Díaz C, López M, Hernández F, Andrés A and Jara
P: Liver transplantation for classical maple syrup urine disease:
Long-term follow-up. J Pediatr Gastroenterol Nutr. 59:636–639.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bouchereau J, Leduc-Leballeur J, Pichard
S, Imbard A, Benoist JF, Abi Warde MT, Arnoux JB, Barbier V,
Brassier A, Broué P, et al: Neurocognitive profiles in MSUD
school-age patients. J Inherit Metab Dis. 40:377–383. 2017.
View Article : Google Scholar : PubMed/NCBI
|