Introduction
Acute myocardial infarction (AMI) is a serious
cardiovascular disease caused by coronary occlusion, leading to
acute and persistent ischemia and hypoxia in the myocardium.
Revascularization by thrombolysis or percutaneous coronary
intervention (PCI) is the main treatment for AMI, and effectively
reduces AMI injury and limits the size of the myocardial infarction
(1); however, myocardial
reperfusion injury may be induced by the reestablished blood flow
in the ischemic myocardium (2).
Therefore, ischemia/reperfusion (I/R) injury is a major cause of
AMI damage. Hypoxia/reoxygenation (H/R) is an important factor in
the pathogenesis of tissue I/R injury (3).
Previous studies have reported that endoplasmic
reticulum (ER) stress and autophagy are associated with I/R
(4,5). The ER is an indispensable eukaryotic
organelle that is primarily responsible for the translocation and
post-transcriptional modification of proteins, and the maintenance
of cellular calcium (Ca2+) homeostasis and lipid
synthesis (6); however, under
extreme conditions, such as I/R, the homeostasis is disrupted,
inducing ER stress, which leads to the apoptosis of cells (7). Autophagy is a highly conserved
catabolic process involving the degradation and recycling of
excess/impaired proteins and organelles via autophagosome formation
and lysosome degradation, serving an important role in maintaining
cell homeostasis (8); however,
under stressful conditions, such as hypoxia or starvation,
autophagy is activated as a pathway to promote cell survival by
conserving energy and reducing toxic substances (9).
A previous study reported that the activation of ER
stress induces cardiomyocyte apoptosis during H/R (10), and a separate study reported that
autophagy serves a protective role against H/R injury (11). In addition, an association between
ER stress and autophagy was reported in ovarian injury and HCT116
cell apoptosis (12,13); however, whether there is potential
crosstalk between autophagy and ER stress during cardiomyocyte H/R
injury is yet to be determined. Thus, the present study aimed to
investigate the roles and mechanisms of ER stress and autophagy in
H9c2 cell apoptosis during H/R injury.
Materials and methods
Materials
Rapamycin (Rap, autophagy agonist), 3-methyladenine
(3-MA, autophagy inhibitor), thapsigargin (Tg, ER stress agonist)
and 4-phenylbutyrate (4-PBA, ER stress inhibitor) were purchased
from Sigma-Aldrich (Merck KGaA). Fetal bovine serum (FBS) was
purchased from Gibco (Thermo Fisher Scientific, Inc.). High-glucose
Dulbecco's Modified Eagle's medium (DMEM) was obtained from HyClone
(GE Healthcare Life Sciences). The rat H9c2 cell line was purchased
from the Chinese Academy of Sciences. Caspase-3 Activity Assay kit
(cat. no. G015-1-3), Lactate Dehydrogenase (LDH) Assay kit (cat.
no. A020-2-2), radio immunoprecipitation assay (RIPA) lysis buffer
and Bicinchoninic Acid (BCA) Protein Assay kits were purchased from
Nanjing Jiancheng Bio-Engineering Institute Co., Ltd. A Cell
Counting Kit-8 (CCK-8) and an Annexin V-fluorescein isothiocyante
(FITC)/propidium iodide (PI) Apoptosis Analysis kit were purchased
from Beijing Zoman Biotechnology Co., Ltd. TRIzol®
Reagent was purchased from Invitrogen (Thermo Fisher Scientific,
Inc.) and a First Strand cDNA Synthesis kit was purchased from
Tiangen Biotech Co., Ltd. A SYBR Green Master Mix kit was purchased
from Takara Bio, Inc. Adenovirus expressing mCherry-green
fluorescent protein (GFP)-microtubule-associated proteins 1A/1B
light chain 3B (LC3B) was obtained from Beyotime Institute of
Biotechnology. Control small interfering RNA (siRNA), specific
siRNA for Beclin1 and specific siRNA for C/EBP homologous protein
(CHOP) were obtained from Shanghai GeneChem Co., Ltd.
Lipofectamine® 2000 was obtained from Invitrogen (Thermo
Fisher Scientific, Inc.). The primary antibodies rabbit anti-BAX
(cat. no. sc-6236), rabbit anti-Bcl2 (cat. no. sc-23960) and rabbit
anti-GAPDH (cat. no. sc-32233) were obtained from Santa Cruz
Biotechnology, Inc. The primary antibodies rabbit anti-activating
transcription factor 6 (ATF6; cat. no. ab37149), rabbit anti-CHOP
(cat. no. ab10444), rabbit anti-glucose-regulated protein 78
(GRP78; cat. no. ab32618), rabbit anti-Beclin1 (cat. no. ab62557),
rabbit anti-P62 (cat. no. ab91526) and rabbit anti-LC3 (cat. no.
ab48394) were purchased from Abcam. The goat anti-rabbit secondary
antibodies (cat. no. SA00001-2) were obtained from ProteinTech
Group, Inc.
Cell culture and treatment
H9c2 cells were cultured in DMEM containing 10% FBS,
50 U/ml penicillin and 50 µg/ml streptomycin, and incubated at 37°C
in a humidified atmosphere containing 5% CO2/95% air.
For hypoxia treatment, the H9c2 cell media were replaced with
serum-free and glucose-free DMEM prior to incubation in an
anaerobic chamber with a humidified atmosphere consisting of 5%
CO2/95% N2 for 3, 6, 12 and 18 h. The cells
were then subjected to reperfusion by replacing the media with
high-glucose DMEM containing 10% FBS followed by incubation under
normoxic conditions for 6 h and 12/6 h H/R was used for all H/R
treatment following the initial viability, LDH, and apoptosis
experiments. In the experimental group, the cells were pretreated
with Rap (5 µM), 3-MA (5 mM) or 4-PBA (5 µM) at 37°C for 4 h prior
to H/R, and the positive control cells were treated with Rap (5
µM), 3-MA (5 mM), 4-PBA (5 µM) or Tg (2 µM) in normoxic conditions
at 37°C for 4 h before the cells were cultured in normal medium for
14 h.
Cell viability and lactate
dehydrogenase (LDH) assays
A CCK-8 assay was performed to determine cell
viability. Briefly, cells were seeded at a density of
1×104 cells/well in 96-well plates. Following the
induction of H/R, cells were incubated with 10 µl CCK-8 solution
for an additional 2 h at 37°C, and the absorbance value was
measured at a wavelength of 450 nm using a microplate reader. Cell
injury was verified using an LDH assay. After treatment, 0.2 ml
culture medium was used to measure LDH activity using an LDH assay
kit according to the manufacturer's protocol. The CCK-8 and LDH
results were presented as a percentage of the values measured for
control cells that were incubated under normoxic conditions for the
same time.
Measurement of apoptosis by flow
cytometry
The apoptotic rates of H9c2 cells were determined
using an Annexin V-FITC Apoptosis Detection kit. Briefly,
1×105/ml H9c2 cells were inoculated into 6-well culture
plates at 37°C for 24 h. Following the different treatments, the
cells were collected and resuspended in 500 µl binding buffer.
After incubation with 10 µl Annexin V-FITC and 5 µl PI for 15 min
at room temperature in the dark, the apoptotic rates of the cells
were measured using a flow cytometer (FACSCalibur™; BD Biosciences)
and analyzed using CellQuest Pro software (version 3.3, BD
Biosciences).
siRNA transfection
The siRNA sequences used were as follows: Control,
5′-UUCUCCGAACGUGUCACGUTT-3′; Beclin1,
5′-GAUGGUGUCUCUCGAAGAUdTdT-3′; CHOP, 5′-GGUCCUGUCCUCAGAUGAAdTdT-3′.
1×105 cells/ml H9c2 cells were seeded into 6-well plates
and cultured to 70–80% confluence. H9c2 cells were transfected with
10 nM control siRNA (Con siRNA), Beclin1 siRNA or CHOP siRNA using
Lipofectamine® 2000 according to the manufacturer's
protocols. H/R or other treatments were performed at 6 h
post-transfection.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from H9c2 cells using
TRIzol® Reagent. The total RNA of each sample was
quantified using a spectrophotometer (NanoDrop Technologies; Thermo
Fisher Scientific, Inc.). RNA (1 µg) was reverse-transcribed in a
20 µl reaction volume with oligo dT primers using a First Strand
cDNA Synthesis kit (Tiangen Biotech Co., Ltd). qPCR was performed
using an Applied Biosystems 7500 Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) with a SYBR Green
Master Mix kit. The PCR cycling conditions consisted of initial
denaturation at 95°C for 1 min, followed by 40 cycles at 95°C for
15 sec and at 60°C for 34 sec. The expression levels of Beclin1 and
CHOP were normalized to GAPDH. Relative quantification of gene
expression was performed using the 2−ΔΔCq method
(14). The mRNA primers used were:
Beclin1, forward 5′-GCCTCTGAAACTGGACACG-3′, reverse,
5′-CCTCTTCCTCCTGGCTCTCT-3′; CHOP, forward
5′-CTGGAAGCCTGGTATGAGGAT-3′, reverse, 5′-CAGGGTCAAGAGTAGTGAAGGT-3′;
and GAPDH, forward 5′-CTCGTCTCATAGACAAGATGGT-3′ and reverse,
5′-GGGTAGAGTCATACTGGAACATG-3′.
Western blotting
After treatment, the cells were collected, washed
with ice-cold PBS and lysed with RIPA buffer; the total protein
concentration was determined via a BCA assay. Subsequently, 30
µg/lane total protein was separated via 15% SDS-PAGE (with the
exception of experiments involving CHOP siRNA, for which the
concentration of total protein used was 50 µg), after which they
were transferred to PVDF membranes. After blocking in TBS-0.1%
Tween-20 with 10% non-fat milk for 2 h at room temperature, the
samples were incubated overnight at 4°C with primary antibodies
against Bcl2 (1:1,000), ATF6 (1:1,000), BAX (1:500), GAPDH
(1:2,000), CHOP (1:1,000), GRP78 (1:1,000) Beclin1 (1:1,000), P62
(1:1,000) and LC3 (1:1,000). After washing, the membranes were
incubated with secondary antibody (1:4,000) conjugated to
horseradish peroxidase at 37°C for 30 min. The immunoreactive bands
were visualized using a Super Signal West Pico kit (Beijing
Solarbio Science & Technology Co., Ltd.) according to the
manufacturer's protocol, and the protein band densities were
semi-quantified by densitometric analysis using ImageJ software
(version 1.48, National Institutes of Health).
mCherry-GFP-LC3 adenovirus
transduction and autophagy assay
H9c2 cells cultured in 24-well plates
(1×105 cells/well) were transduced with mCherry-GFP-LC3
adenovirus at 40 MOI (multiplicity of infection) for 24 h at 37°C
in a humidified atmosphere containing 5% CO2/95% air.
Following transduction, the cells were incubated with fresh culture
medium for 24 h at 37°C. The numbers of GFP and mCherry dots per
cell were counted in three randomly selected fields under a
fluorescence microscope (Olympus Corporation).
Caspase-3 activity measurement
Caspase-3 activity was measured using a Caspase-3
Activity Colorimetric Assay kit. Briefly, following the various
aforementioned treatments, cells were harvested by scraping,
collected by centrifugation at 800 × g and 4°C for 5 min, and lysed
with RIPA buffer on ice for 15 min. Subsequently, the lysate was
centrifuged at 12,000 × g and 4°C for 15 min, and the protein
content was determined, following which the caspase-3 substrate was
measured at 405 nm using a microplate reader (Model 680, Bio-Rad
Laboratories, Inc.).
Statistical analyses
All experiments were repeated at least three times
for each group, and the data are presented as the means ± standard
error of the mean. The data were analyzed by one-way analysis of
variance followed by Fisher's least significant difference test
using SPSS version 13.0 software (SPSS, Inc.).
Results
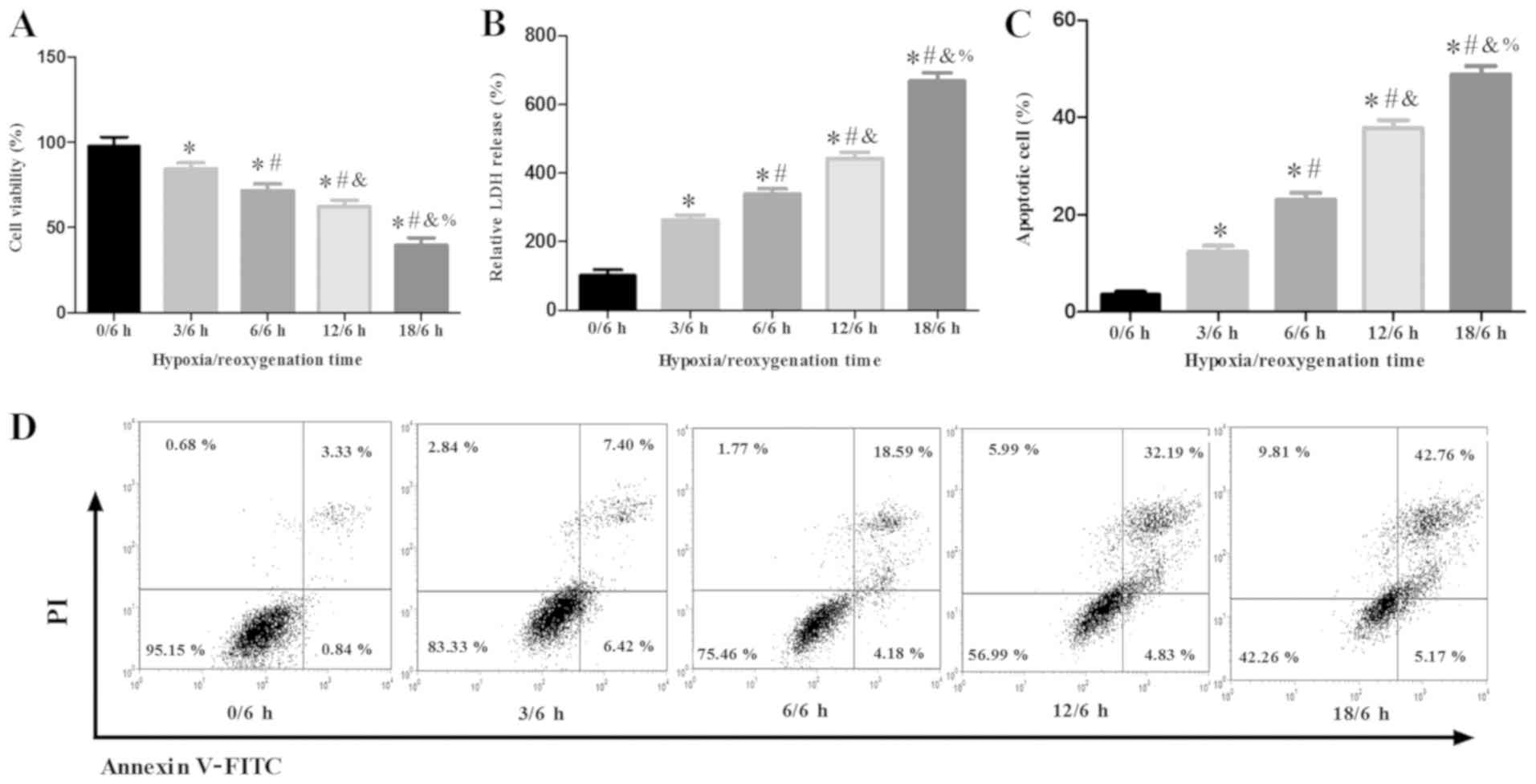
Effects of H/R treatment on damage and
apoptosis in H9c2 cells
To investigate the injurious effects of H/R on H9c2
cells, cells were subjected to hypoxia for 0, 3, 6, 12 and 18 h
prior to reperfusion for 6 h. H9c2 cell viability was significantly
decreased in a time-dependent manner under hypoxic conditions for
0, 3, 6, 12 and 18 h followed by reperfusion for 6 h, with a ~40%
reduction in viability following 12 h of hypoxia (Fig. 1A). LDH results indicated that H/R
induced a significant increase in the release of LDH in H9c2 cells
in a time-dependent manner (Fig.
1B). Consistent with these findings, as the duration of H/R
increased, the number of apoptotic H9c2 cells increased (Fig. 1C and D). It was revealed that H/R
treatment of H9c2 cells for 12/6 h resulted in the apoptosis of
>30% of cells. Therefore, 12/6 h H/R was used for all H/R
treatments following the viability, LDH and apoptosis
experiments.
Effects of H/R-treatment on autophagy
and ER stress in H9c2 cells
Western blot analysis suggested that the expression
patterns of Beclin1 and LC3-II/LC3-I were similar. Beclin1
expression and LC3II/LC3I ratio were increased in a time-dependent
manner following H/R, whereas the expression of P62 protein was
downregulated (Fig. 2B and F-H).
These results indicated that the expression of Beclin1 and ratio of
LC3II/LC3I were promoted but the expression of P62 was inhibited by
H/R. In addition, fluorescent microscopy revealed that H/R promoted
the formation of autophagosomes and autolysosomes compared with the
control (Fig. 3C and D). These
results indicated that autophagy was activated in H/R-treated H9c2
cells. Additionally, the expression levels of ER stress proteins
(ATF6, GRP78 and CHOP) were upregulated following H/R in a
time-dependent manner (Fig. 2A and
C-E).
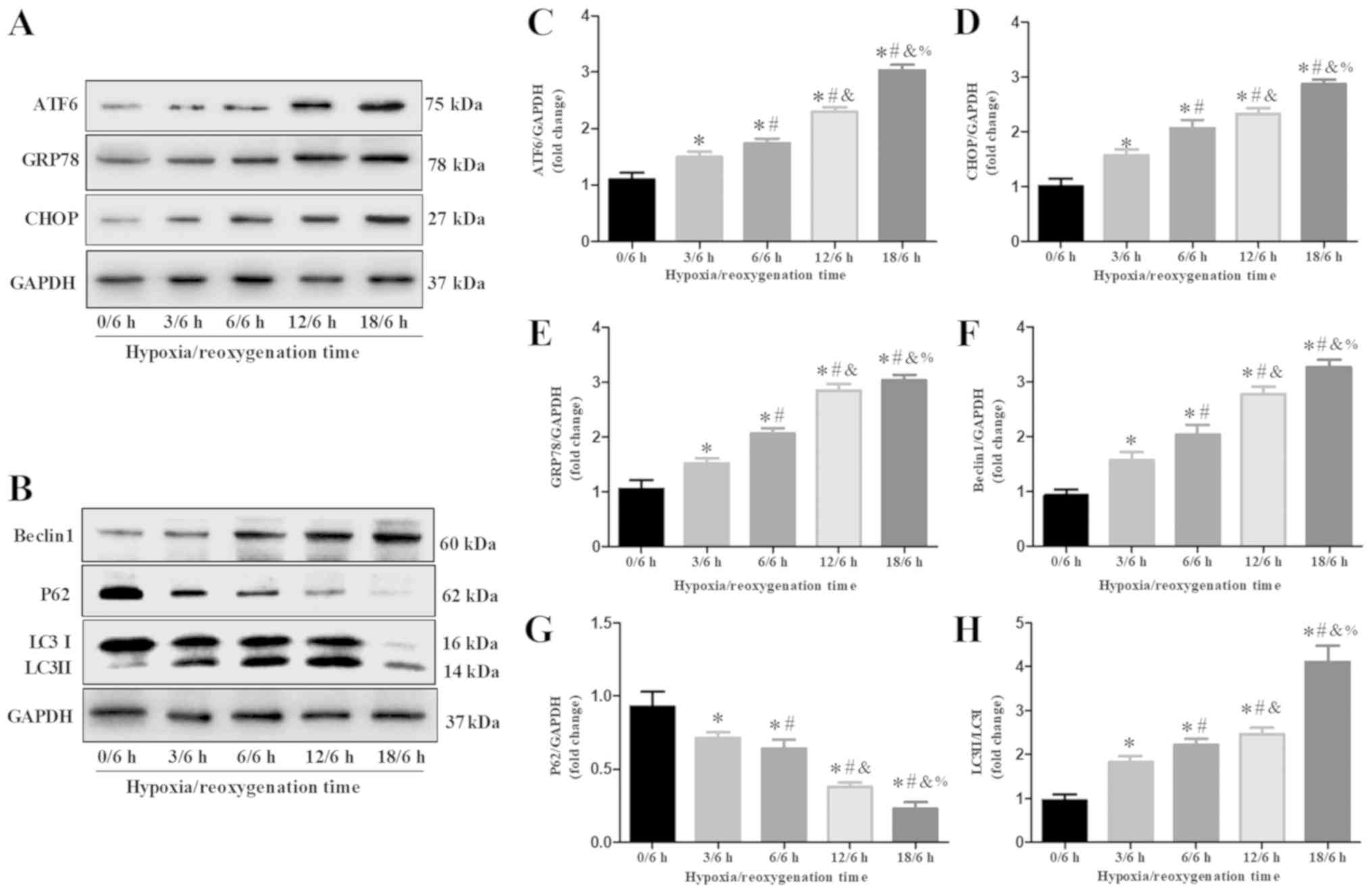 | Figure 2.Endoplasmic reticulum stress and
autophagy in H9c2 cells following H/R. (A) Expression of ATF6,
GRP78 and CHOP following various periods of hypoxia and 6 h of
reoxygenation in H9c2 cells. (B) Expression of Beclin1, P62, and
LC3 following various periods of hypoxia and 6 h of reoxygenation
in H9c2 cells. (C-H) Semi-quantification of the relative ratio of
ATF6, GRP78, CHOP, Beclin1, P62 and LC3II/LC3I following H/R;
protein expression levels were normalized to GAPDH. Data are
presented as the means ± standard error of the mean of three
independent experiments. *P<0.05 vs. 0/6 h;
#P<0.05 vs. 3/6 h; &P<0.05 vs. 6/6
h; %P<0.05 vs. 12/6 h. ATF6, activating transcription
factor 6; CHOP, C/EBP homologous protein; GRP78, glucose-regulated
protein 78; H/R, hypoxia/reoxygenation; LC3, microtubule-associated
proteins 1A/1B light chain 3B. |
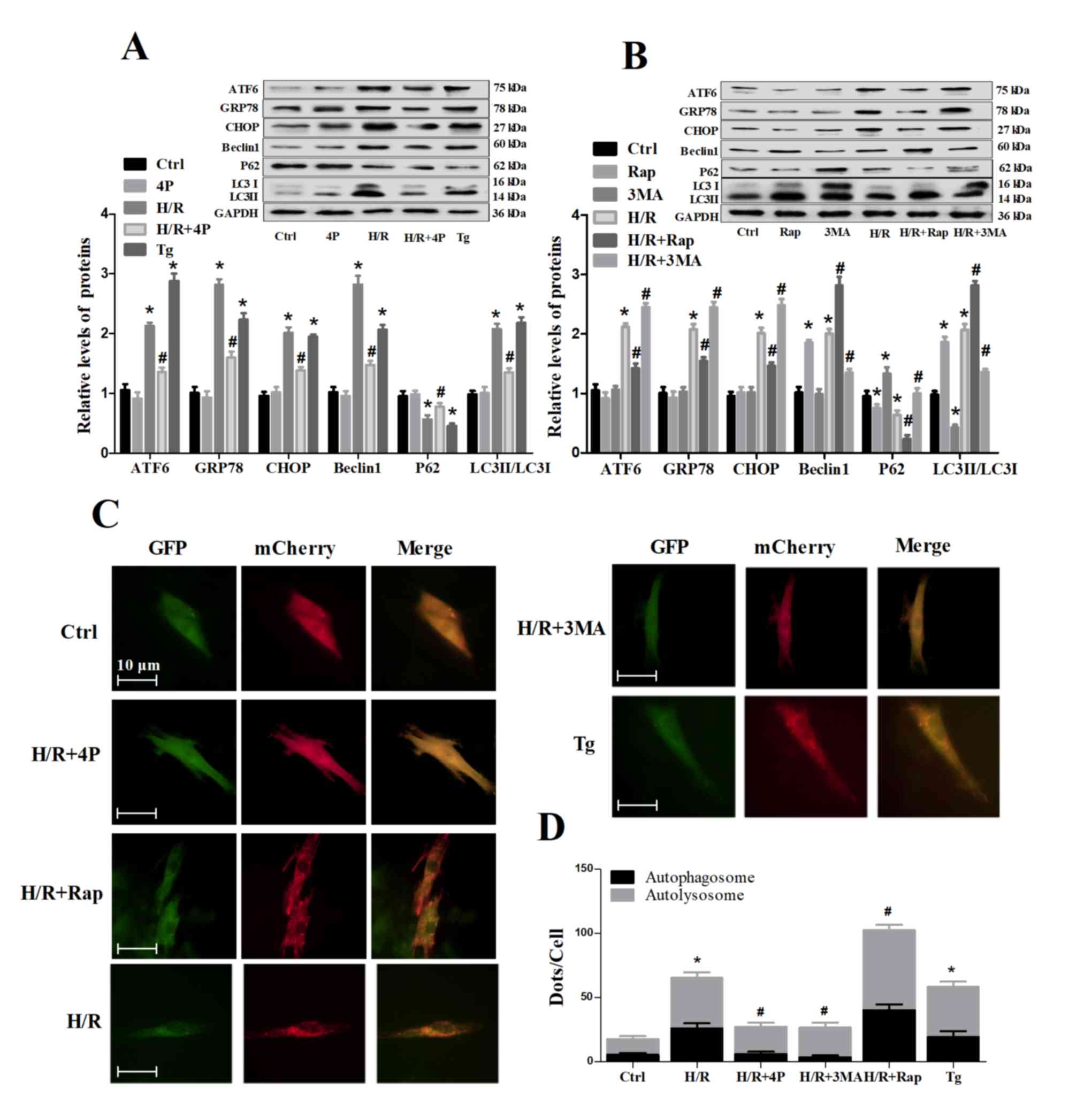 | Figure 3.Effects of Rap, 3-MA, Tg and 4-PBA on
the autophagy and endoplasmic reticulum stress of H/R-treated H9c2
cells. Untreated H9c2 cells, or cells pretreated with Rap, 3-MA or
4-PBA for 4 h were subjected to H/R injury, as were H9c2 cells
treated with Tg alone. (A and B) Western blot analysis of Beclin1,
P62, LC3, ATF6, GRP78 and CHOP expression. Beclin1, P62, ATF6,
GRP78, CHOP and LC3II/LC3I were normalized to GAPDH. (C)
Fluorescence microscopy analysis of H9c2 cells transfected with
mCherry-GFP-LC3. (D) Statistical analysis of fluorescent dots in
H9c2 cells. Yellow spots indicate autophagosomes and red spots
indicate autolysosomes in the merged images. Data are presented as
the means ± standard error of the mean of three independent
experiments. *P<0.05 vs. Ctrl; #P<0.05 vs. H/R.
3-MA/3MA, 3-methyladenine; 4P/4-PBA, 4-phenylbutyrate; ATF6,
activating transcription factor 6; CHOP, C/EBP homologous protein;
Ctrl, Control; GFP, green fluorescent protein; GRP78,
glucose-regulated protein 78; H/R, hypoxia/reoxygenation; LC3,
microtubule-associated proteins 1A/1B light chain 3B; Rap,
rapamycin; Tg, thapsigargin. |
Autophagy is altered by ER stress
levels in H/R-treated H9c2 cells
To determine whether autophagy was influenced by ER
stress in cells during H/R injury, 4-PBA was used to inhibit ER
stress during treatment (Fig. 3A).
4-PBA pretreatment significantly decreased the expression levels of
ATF6, GRP78 and CHOP compared with in the H/R treatment group.
Additionally, 4-PBA pretreatment effectively decreased Beclin1
expression and LC3-II/LC3-I ratio, but increased P62 expression,
compared with the H/R treatment group, and decreased the formation
of H/R-induced autophagosomes and autolysosomes (Fig. 3C and D). Conversely, Tg (an ER
stress agonist) significantly increased the expression levels of
ATF6, GRP78 and CHOP in H9c2 cells, and the expression of the
autophagy-associated proteins Beclin1 and LC3, but not P62, was
increased by Tg compared with control group in H9c2 cells (Fig. 3A). The numbers of autophagosomes
and autolysosomes were also significantly increased following the
treatment of H9c2 cells with Tg (Fig.
3C and D).
ER stress is altered by autophagy
activity in H/R-treated H9c2 cells
To determine whether ER stress was influenced by
autophagy activity in HR injury, Rap or 3-MA were used to activate
or inhibit autophagy prior to treatment, respectively (Fig. 3B). The results revealed that Rap
pretreatment effectively increased Beclin1 expression and
LC3-II/LC3-I ratio, but decreased P62 expression, during H/R
treatment, and in the absence of H/R treatment. Conversely, 3-MA
pretreatment successfully decreased Beclin1 expression and
LC3-II/LC3-I ratio, but increased P62 expression, in H9c2 cells
during H/R treatment. Additionally, Rap pretreatment increased the
formation of H/R-induced autophagosomes and autolysosomes, whereas
3-MA pretreatment induced opposing effects (Fig. 3C and D). Rap pretreatment also
resulted in a significant decrease in the protein expression of
ATF6, GRP78 and CHOP during H/R, whereas 3-MA pretreatment
significantly increased their protein expression (Fig. 3B).
Effects of autophagy and ER stress on
cell injury and apoptosis in H/R-treated H9c2 cells
CCK-8 and LDH assays were performed to analyze the
extent of cell injury (Fig. 4A and
B). It was revealed that cell injury was significantly
decreased following pretreatment with Rap or 4-PBA compared with
the H/R treatment group. In contrast, 3-MA pretreatment increased
the injury of H/R-treated H9c2 cells. Subsequently, the apoptotic
rate was measured by flow cytometry; pretreatment with Rap or 4-PBA
significantly decreased the relative number of apoptotic
H/R-treated H9c2 cells, whereas 3-MA pretreatment significantly
increased the number of apoptotic cells (Fig. 4C and D). Furthermore, BAX and
caspase-3 levels were downregulated, whereas those of Bcl2 were
markedly upregulated by the pretreatment of H/R-treated H9c2 cells
with Rap or 4-PBA; however, 3-MA pretreatment significantly
increased the levels of BAX and caspase-3, but decreased those of
Bcl2 in H/R-treated H9c2 cells. In addition, cell injury and
apoptosis were increased by Tg compared with the control group in
H9c2 cells (Fig. 4E-I).
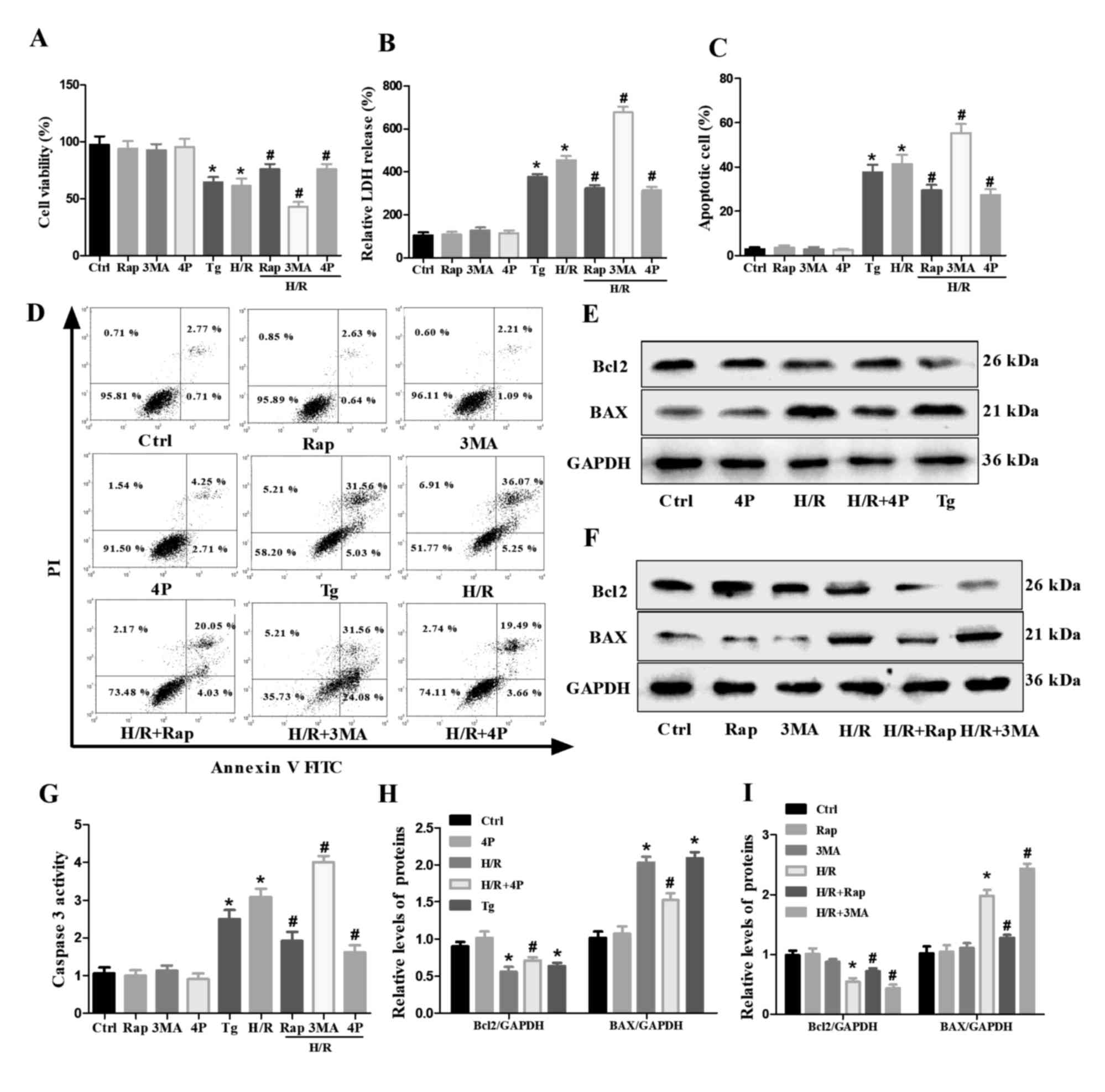 | Figure 4.Effects of Rap, 3-MA, Tg and 4-PBA on
the H/R-induced damage and apoptosis of H9c2 cells. Untreated H9c2
cells, or cells pretreated with Rap, 3-MA or 4-PBA for 4 h were
subjected to H/R injury as were H9c2 cells treated with Tg alone.
(A) Cell viability and (B) LDH release were determined via Cell
Counting Kit-8 and LDH assays. (C and D) Apoptosis of H9c2 cells as
determined via flow cytometry. (E and F) Western blot analysis of
Bcl2 and BAX in H9c2 cells. (G) Caspase-3 activity as determined
via a caspase-3 activity assay. (H and I) Semi-quantified
expression of Bcl2 and BAX normalized to GAPDH. Data are presented
as the means ± standard error of the mean of three independent
experiments. *P<0.05 vs. Ctrl; #P<0.05 vs. H/R.
3-MA/3MA, 3-methyladenine; 4P/4-PBA, 4-phenylbutyrate; Ctrl,
Control; H/R, hypoxia/reoxygenation; PI, propidium iodide; Rap,
Rapamycin; Tg, thapsigargin. |
Effects of CHOP and Beclin1 on the
interaction between autophagy and ER stress in H/R-treated H9c2
cells
To further investigate the molecular mechanisms
underlying the crosstalk between autophagy and ER stress,
endogenous CHOP and Beclin1 levels were downregulated using siRNA.
siRNA transfection significantly decreased the expression levels of
CHOP and Beclin1 (Fig. S1).
Western blot analysis revealed that CHOP siRNA significantly
decreased the protein expression levels of CHOP in H/R-induced H9c2
cells. Notably, it also decreased Beclin1 levels and LC3-II/LC3-I
ratio, and increased levels of P62 during H/R treatment (Fig. 5A). In addition, Beclin1 siRNA
significantly decreased Beclin1 expression and LC3-II/LC3-I ratio,
but increased P62 expression, in control and H/R-treated H9c2
cells; it also upregulated the expression of ATF6, GRP78 and CHOP
in H/R-induced H9c2 cells (Fig.
5B). The apoptotic rate was measured by flow cytometry, and it
was demonstrated that CHOP siRNA decreased the number of apoptotic
H/R-treated H9c2 cells, whereas Beclin1 siRNA transfection induced
opposing effects (Fig. 5C and D).
Additionally, CHOP siRNA decreased the levels of BAX, but increased
those of Bcl2 in H/R-treated H9c2 cells, with Beclin1 siRNA
increased the levels of BAX but decreased the levels of Bcl2 during
H/R treatment as well as without H/R treatment (Fig. 5E).
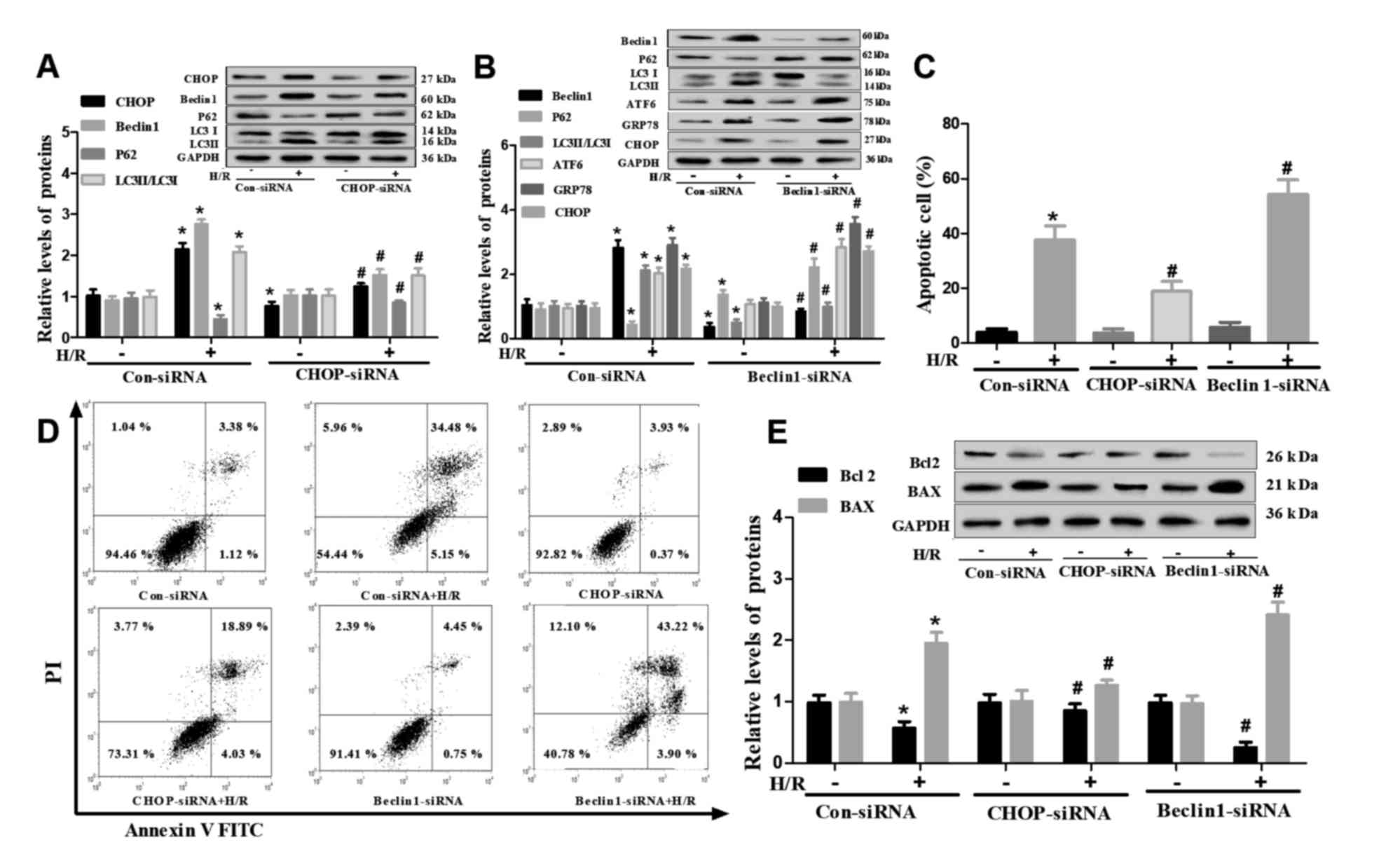 | Figure 5.Effects of siRNA-mediated knockdown
of Beclin1 and CHOP on the autophagy, endoplasmic reticulum stress
and apoptosis of H/R-injured H9c2 cells. H9c2 cells were subjected
to H/R injury following transfection with CHOP siRNA, Beclin1 siRNA
or Con siRNA for 6 h. (A and B) Western blot analysis of Beclin1,
P62, LC3, ATF6, GRP78 and CHOP expression. Beclin1, P62, ATF6,
GRP78, CHOP and LC3II/LC3I were normalized to GAPDH. (C and D)
Apoptosis of H9c2 as determined via flow cytometry. (E) Western
blot analysis of Bcl2 and BAX in H9c2 cells. Data are presented as
the means ± standard error of the mean of three independent
experiments. *P<0.05 vs. Con siRNA; #P<0.05 vs.
Con siRNA + H/R. ATF6, activating transcription factor 6; CHOP,
C/EBP homologous protein; Con, Control; GRP78, glucose-regulated
protein 78; H/R, hypoxia/reoxygenation; LC3, microtubule-associated
proteins 1A/1B light chain 3B; PI, propidium iodide; siRNA, small
interfering RNA. |
Discussion
The present study investigated the effects of H/R on
the viability, damage and apoptosis of H9c2 cells. Consistent with
a previous study (15), the data
revealed that H/R decreased cell viability, increased cell damage
and induced cell apoptosis in a time-dependent manner. Cell
viability was reduced by ~40% following a 12/6 h
hypoxia/reperfusion period. Therefore, 12/6 h hypoxia/reperfusion
treatment was selected to induce H/R injury in subsequent
experiments.
ER stress is an adaptive or protective response that
decreases the accumulation of unfolded or misfolded proteins via
the unfolded protein response (UPR) (7); however, under excessive stress, ER
stress results in cell apoptosis (16). GRP78, a main indicator of ER
stress, is a chaperone protein localized in the ER whose expression
is typically increased in response to ER stress (17). ATF6 is activated by the
accumulation of misfolded proteins and initiation of the UPR,
inhibiting general protein translation (18). The UPR is a defense mechanism
directed toward cellular adaptation to alleviate the unfolded
protein load; however, prolonged stress is associated with the
activation of proapoptotic proteins, such as CHOP (19). A previous study reported that ER
stress is increased following H/R treatment (15). Consistent with this study, the
present findings indicated that GRP78, ATF6 and CHOP were
upregulated in a time-dependent manner following H/R injury.
Autophagy is an important intracellular bulk
degradation process involving the lysosome-dependent turnover of
damaged cytosolic proteins and organelles, which is critical for
the maintenance of normal cell phenotypes and functions (20); however, the autophagic machinery
may also contribute to cell death when pathological stress induces
autophagy dysfunction (2). Beclin1
and LC3 are important proteins in autophagy. LC3 is a frequently
used biomarker of the autophagosome membrane and reflects cellular
autophagy activity. The P62 protein is an important substrate for
autophagy degradation and has been defined as a marker of
autophagic flux (7). Therefore,
Beclin1, P62 and LC3 were selected as biomarkers of autophagy in
the present study. Autophagy has been reported to be a regulatory
target involved in H/R injury in cardiomyocytes (21). In the present study, it was
observed that Beclin1 expression, LC3II lipidation and P62
degradation were increased in a time-dependent manner following H/R
injury. These results suggested that ER stress and autophagy were
increased following H/R injury, consistent with the results of a
previous study reporting that autophagy and ER stress are increased
during myocardial H/R injury (21).
Subsequently, the association between autophagy and
ER stress was investigated in H9c2 cells during H/R injury. It was
demonstrated that autophagy was decreased in H/R-treated H9c2 cells
following 4-PBA treatment, whereas autophagy was increased
following the treatment of H9c2 cells with Tg. These results
suggested that autophagy was attenuated following the inhibition of
ER stress in H/R-injured H9c2 cells, and that increased ER stress
following Tg treatment promoted autophagic flux. Therefore, these
results indicated that autophagy was activated by ER stress during
H/R injury, which is consistent with a previous study reporting
that ER stress induces autophagy in mammalian cells (22,23).
A possible explanation for these results is that the UPR also leads
to the transcriptional upregulation of a number of
autophagy-associated genes required for the induction and
construction of the autophagy machinery during ER stress (24).
To determine the effects of autophagy on ER stress
during H/R-induced injury, ER stress was analyzed following the
regulation of autophagy activity. The results indicated that
promoting autophagy by pretreating H9c2 cells with Rap decreased ER
stress during H/R injury. In contrast, inhibiting autophagy by
pretreating H9c2 cells with 3-MA increased ER stress during H/R
injury. Therefore, the levels of ER stress during H/R injury were
influenced by alterations in autophagy activity. This may be a
result of the activation of autophagy relieving stress and
reinstating homeostasis in the ER by removing unfolded proteins,
protein aggregates and damaged organelles (25). Therefore, the present findings
indicated the presence of a negative-feedback loop between
autophagy and ER stress in H9c2 cells during H/R injury, with
autophagy inhibiting ER stress but increased ER stress promoting
autophagy.
Cardiomyocyte damage and apoptosis are prominent
features of H/R injury. Therefore, the mechanisms of ER stress and
autophagy that underlie cell damage and apoptosis in cardiomyocytes
during H/R injury were investigated. It was observed that cell
damage and apoptosis were alleviated in H9c2 cells during H/R
injury after inhibiting ER stress using 4-PBA. Conversely,
promoting autophagy alleviated the apoptosis and damage of
H/R-treated H9c2 cells, whereas inhibiting autophagy further
promoted apoptosis and damage. These findings indicated that
increasing ER stress increased apoptosis and damage, but that
inducing autophagy served a protective role in H9c2 cells during
H/R injury. Thus, combined with the conclusions that ER stress
activated autophagy and increased apoptosis, the further activation
of autophagy alleviated ER stress and apoptosis in H9c2 cells
during H/R injury. Therefore, it was hypothesized that apoptosis is
induced depending on the levels of ER stress, and that autophagy
serves a protective role by alleviating ER stress in H9c2 cells
during H/R injury. These findings are consistent with previous
studies that reported that ER stress serves an important role in
the H/R-induced apoptosis of cardiomyocytes (15,26).
In addition, these results supported the previous finding that
increased autophagy alleviates H/R injury (11). Conversely, it has been reported
elsewhere that inhibition of autophagy alleviates H/R injury
(27). One possible explanation
for this discrepancy may be that the distinct effects of autophagy
are dependent on the duration or method of H/R treatment.
To further determine the effects of H/R injury on
cell apoptosis, the activity (caspase-3) or expression (BAX and
Bcl2) of apoptosis-associated proteins was investigated. Caspase-3
is processed into cleaved-caspase-3 during the early stages of
apoptosis, and the expression of caspase-3 is positively associated
with the rate of apoptosis in cells (28). Bcl2 is an important protein in
promoting cellular survival and inhibiting the actions of
proapoptotic proteins (29).
Conversely, BAX, a member of the Bcl2 family, induces proapoptotic
effects (30). The present results
indicated that 4-PBA and Rap pretreatment decreased the activity of
caspase-3 and expression of BAX, but increased the levels of Bcl2
in H9c2 cells during H/R injury; however, 3-MA pretreatment induced
opposing effects. These findings further supported the data from
the flow cytometry assays.
CHOP is a major point of convergence for the three
major upstream ER stress transducers, and is also the most
extensively characterized factor in the transition from ER stress
to apoptosis (31,32). Beclin1 is required for the
initiation of autophagy (33). To
investigate the molecular mechanisms underlying the roles of ER
stress and autophagy in the apoptosis of H/R-induced H9c2 cells,
the levels of CHOP and Beclin1 were downregulated with siRNA. It
was observed that CHOP siRNA decreased autophagy and attenuated the
apoptosis of H9c2 cells during H/R injury, indicating that the
activation of autophagy and the promotion of apoptosis depended on
the increased expression of CHOP in H/R injury. Conversely, Beclin1
siRNA inhibited autophagy, but aggravated ER stress and cell
apoptosis. These results further indicated that autophagy serves a
protective role against ER stress and apoptosis in H9c2 cells
during H/R injury.
In summary, the results of the present study
provided further evidence that H/R induces apoptosis and autophagy
via ER stress in cardiomyocytes. The findings suggested that CHOP
serves an important role in ER stress-induced autophagy and
apoptosis. Additionally, autophagy, as an adaptive response,
alleviated ER stress and ER stress-induced apoptosis in H9c2 cells
during H/R injury. The novel findings that ER stress-induced
autophagy serves a protective role in H9c2 cells during H/R injury
may aid future research.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant no. 81660152) and the Natural
Science Foundation of Jiangxi Province (grant no.
20181BAB205004).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LY and XG were involved in the conception and design
of the experiments. LY supervised the work. GPG, WYH, PHZ and JZ
performed the experiments. GPG, HY and SYL analyzed data. GPG
contributed to drafting and revising the manuscript. XG approved
the final version to be published.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kalkman DN, Aquino M, Claessen BE, Baber
U, Guedeney P, Sorrentino S, Vogel B, de Winter RJ, Sweeny J,
Kovacic JC, et al: Residual inflammatory risk and the impact on
clinical outcomes in patients after percutaneous coronary
interventions. Eur Heart J. 39:4101–4108. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ma X, Liu H, Foyil SR, Godar RJ,
Weinheimer CJ, Hill JA and Diwan A: Impaired autophagosome
clearance contributes to cardiomyocyte death in
ischemia/reperfusion injury. Circulation. 125:3170–3181. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang Y, Ding S, Xu G, Chen F and Ding F:
MicroRNA-15a inhibition protects against
hypoxia/reoxygenation-induced apoptosis of cardiomyocytes by
targeting mothers against decapentaplegic homolog 7. Mol Med Rep.
15:3699–3705. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liao F, Zheng Y, Cai J, Fan J, Wang J,
Yang J, Cui Q, Xu G, Tang C and Geng B: Catestatin attenuates
endoplasmic reticulum induced cell apoptosis by activation type 2
muscarinic acetylcholine receptor in cardiac ischemia/reperfusion.
Sci Rep. 5:165902015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yao T, Ying X, Zhao Y, Yuan A, He Q, Tong
H, Ding S, Liu J, Peng X, Gao E, et al: Vitamin D receptor
activation protects against myocardial reperfusion injury through
inhibition of apoptosis and modulation of autophagy. Antioxid Redox
Signal. 22:633–650. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ma Y, Shimizu Y, Mann MJ, Jin Y and
Hendershot LM: Plasma cell differentiation initiates a limited ER
stress response by specifically suppressing the PERK-dependent
branch of the unfolded protein response. Cell Stress Chaperones.
15:281–293. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rashid HO, Yadav RK, Kim HR and Chae HJ:
ER stress: Autophagy induction, inhibition and selection.
Autophagy. 11:1956–1977. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gatica D, Chiong M, Lavandero S and
Klionsky DJ: Molecular mechanisms of autophagy in the
cardiovascular system. Circ Res. 116:456–467. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Salabei JK and Conklin DJ: Cardiovascular
autophagy: Crossroads of pathology, pharmacology and toxicology.
Cardiovasc Toxicol. 13:220–229. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guo JJ, Xu FQ, Li YH, Li J, Liu X, Wang
XF, Hu LG and An Y: Alginate oligosaccharide alleviates myocardial
reperfusion injury by inhibiting nitrative and oxidative stress and
endoplasmic reticulum stress-mediated apoptosis. Drug Des Dev Ther.
11:2387–2397. 2017. View Article : Google Scholar
|
|
11
|
Li H, Zhang X, Tan J, Sun L, Xu LH, Jiang
YG, Lou JS, Shi XY and Mi WD: Propofol postconditioning protects
H9c2 cells from hypoxia/reoxygenation injury by inducing autophagy
via the SAPK/JNK pathway. Mol Med Rep. 17:4573–4580.
2018.PubMed/NCBI
|
|
12
|
Ding WX, Ni HM, Gao W, Hou YF, Melan MA,
Chen X, Stolz DB, Shao ZM and Yin XM: Differential effects of
endoplasmic reticulum stress-induced autophagy on cell survival. J
Biol Chem. 282:4702–4710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang C, Zhang S, Ma R, Zhang X, Zhang C,
Li B, Niu Q, Chen J, Xia T, Li P, et al: Roles of endoplasmic
reticulum stress, apoptosis and autophagy in
2,2′,4,4′-tetrabromodiphenyl ether-induced rat ovarian injury.
Reprod Toxicol. 65:187–193. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Du Y, Wang M, Liu X, Zhang J, Xu X, Xu H,
Sun G and Sun X: Araloside C prevents hypoxia/reoxygenation-induced
endoplasmic reticulum stress via increasing heat shock protein 90
in H9c2 cardiomyocytes. Front Pharmacol. 9:1802018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sano R and Reed JC: ER stress-induced cell
death mechanisms. Biochim Biophys Acta. 1833:3460–3470. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee AS: The ER chaperone and signaling
regulator GRP78/BiP as a monitor of endoplasmic reticulum stress.
Methods. 35:373–381. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harding HP, Novoa I, Zhang Y, Zeng H, Wek
R, Schapira M and Ron D: Regulated translation initiation controls
stress-induced gene expression in mammalian cells. Mol Cell.
6:1099–1108. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gorman AM, Healy SJ, Jäger R and Samali A:
Stress management at the ER: Regulators of ER stress-induced
apoptosis. Pharmacol Ther. 134:306–316. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Przyklenk K, Dong Y, Undyala VV and
Whittaker P: Autophagy as a therapeutic target for
ischaemia/reperfusion injury? Concepts, controversies, and
challenges. Cardiovasc Res. 94:197–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Matsui Y, Takagi H, Qu X, Abdellatif M,
Sakoda H, Asano T, Levine B and Sadoshima J: Distinct roles of
autophagy in the heart during ischemia and reperfusion: Roles of
AMP-activated protein kinase and Beclin 1 in mediating autophagy.
Circ Res. 100:914–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Høyer-Hansen M and Jäättelä M: Connecting
endoplasmic reticulum stress to autophagy by unfolded protein
response and calcium. Cell Death Differ. 14:1576–1582. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kawakami T, Inagi R, Takano H, Sato S,
Ingelfinger JR, Fujita T and Nangaku M: Endoplasmic reticulum
stress induces autophagy in renal proximal tubular cells. Nephrol
Dial Transplant. 24:2665–2672. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rouschop KM, van den Beucken T, Dubois L,
Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W,
Voncken JW, et al: The unfolded protein response protects human
tumor cells during hypoxia through regulation of the autophagy
genes MAP1LC3B and ATG5. J Clin Invest. 120:127–141. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Doyle KM, Kennedy D, Gorman AM, Gupta S,
Healy SJ and Samali A: Unfolded proteins and endoplasmic reticulum
stress in neurodegenerative disorders. J Cell Mol Med.
15:2025–2039. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao Y, Yang H, Chi J, Xu Q, Zhao L and
Yang W, Liu W and Yang W: Hydrogen gas attenuates myocardial
ischemia reperfusion injury independent of postconditioning in rats
by attenuating endoplasmic reticulum stress-induced Autophagy. Cell
Physiol Biochem. 43:1503–1514. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Y, Wang Q, Zhang L, Ke Z, Zhao Y,
Wang D, Chen H, Jiang X, Gu M, Fan S and Huang C: Coptisine
protects cardiomyocyte against hypoxia/reoxygenation-induced damage
via inhibition of autophagy. Biochem Biophys Res Commun.
490:231–238. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mirzayans R, Andrais B, Kumar P and Murray
D: The growing complexity of cancer cell response to DNA-damaging
agents: Caspase 3 mediates cell death or survival? Int J Mol Sci.
17(pii): E7082016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hardwick JM and Soane L: Multiple
functions of BCL-2 family proteins. Cold Spring Harb Perspect Biol.
5(pii): a0087222013.PubMed/NCBI
|
|
30
|
Sobhan PK, Seervi M, Deb L, Varghese S,
Soman A, Joseph J, Mathew KA, Raghu G, Thomas G, E S, et al:
Calpain and reactive oxygen species targets Bax for mitochondrial
permeabilisation and caspase activation in zerumbone induced
apoptosis. PLoS One. 8:e593502013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Su J, Zhou L, Kong X, Yang X, Xiang X,
Zhang Y, Li X and Sun L: Endoplasmic reticulum is at the crossroads
of autophagy, inflammation, and apoptosis signaling pathways and
participates in the pathogenesis of diabetes mellitus. J Diabetes
Res. 2013:1934612013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Marciniak SJ, Yun CY, Oyadomari S, Novoa
I, Zhang Y, Jungreis R, Nagata K, Harding HP and Ron D: CHOP
induces death by promoting protein synthesis and oxidation in the
stressed endoplasmic reticulum. Genes Dev. 18:3066–3077. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|