Introduction
Due to the widespread use of percutaneous coronary
intervention (PCI) in clinical practice, balloon angioplasty is one
of the most common medical procedures in the world (1). Although balloon angioplasty is
effective in the treatment of coronary stenosis, complications have
restricted its use. Previous studies have indicated a high rate of
restenosis in patients undergoing successful coronary angioplasty,
because of constrictive remodeling and neointimal hyperplasia
(2). Neointimal hyperplasia
causing vessel renarrowing is one of the key limitations for
long-term outcomes. However, the mechanisms of balloon
injury-induced neointimal hyperplasia are complex and remain to a
large extent unexplored.
Through largely unknown mechanisms, calcium
signaling plays an important role in neointimal hyperplasia
(3,4). Store operated calcium entry (SOCE)
represents a key mechanism, by which cells convey Ca2+
signals and maintain Ca2+ homeostasis (5), and is critical for the proliferation
and migration of vascular smooth muscle cells (VSMCs) in neointima.
Previous studies found that transmembrane protein 66 (TMEM66), also
named hereafter as SARAF (for SOCE-associated regulatory factor),
plays a key role in shaping cytosolic Ca2+ signals and
determining the content of the major intracellular Ca2+
stores, as a negative regulator of SOCE (6). In addition, TMEM66 is a protein with
a putative transmembrane domain, a N-terminal region that is
required for the activation of the protein, and a C-terminal domain
that has been shown to be involved in the interaction with the
C-terminal inhibitory domain of Stim1, downstream of the Stim1 and
Orai1 activation region (7). Thus,
we speculated that TMEM66 may be critical for the proliferation and
migration of VSMCs in neointima.
Materials and methods
Rat balloon-induced carotid artery
injury model
A total of 16 male Sprague-Dawley (SD) rats, 8 month
old and weighing 310 to 380 g, were obtained from the Experimental
Animal Centre of The General Hospital of Western Theater Command at
Chengdu, China. Rats were housed in a room with a 12 light-dark
cycle at 23°C and given free access to standard rodent food and tap
water. Rats randomized to the sham-operated group, balloon injury
group and TMEM66-overexpression group. Balloon denudation was
performed as previously described, with a 2-French catheter in the
left common carotid artery (8,9).
After balloon injury, a 100 µl solution of lenti-TMEM66-GFP
(108 IU per rat) or PBS were infused into the injured
artery segment and incubated for 15 min. The efficiency of
transfection was identified by fluorescence microscopy (Fig. S1). After 4 week balloon injury and
lentiviral infection, the rats were euthanized using an overdose of
sodium pentobarbital. The vessel tissues were harvested for the
subsequent experiments. This study was approved by the Research
Council and Animal Care and Use Committee of The General Hospital
of Western Theater Command. All experiments conformed to the
guidelines of the American Association for the Accreditation of
Laboratory Animal Care and conformed to the guidelines of the
ethical use of animals, and all efforts were made to minimize
animal suffering and to reduce the number of animals used.
Immunoblotting
Carotid artery tissues were lysed in lysis buffer
(Beyotime Institute of Biotechnology) and centrifuged at 15,000 ×
g. The homogenates were separated by SDS-polyacrylamide gel (4%
stacking gel with an 8% separating gel) and transferred to
nitrocellulose filter membranes. The transblots were probed with
the rabbit anti-TMEM66 antibody (1:500; cat. no. ab80890; Abcam),
rabbit anti-α-SMA antibody (1:500; cat. no. ab32575; Abcam) and
rabbit anti-calponin antibody (1:500; cat. no. ab46794; Abcam) at
4°C overnight, after washing with TBST and blocking with 1% BSA.
The membranes were then washed and incubated with goat
anti-rabbit-IgG (1:4,000; cat. no. BA1039; Boster Biological
Technology) conjugated to horseradish peroxidase at room
temperature for 1 h, and the bands were visualized with enhanced
chemiluminescence (EMD Millipore). The amount of protein
transferred onto the membranes was verified by immunoblotting for
GAPDH (rabbit anti-GAPDH; 1:1,000; cat. no. ab181602; Abcam).
RNA extraction and PCR
RNA from the carotid artery was extracted using
Trizol (Thermo Fisher Scientific, Inc.), synthesized to cDNA and
served as a template for amplification of TMEM66 using an RNA
reverse transcriptase kit (Takara Biotechnology Co., Ltd.) under
the following conditions: 37°C for 15 min and 85°C for 5 sec. The
cDNA was then stored at 4°C. Primer information is described in a
previous study (10).
Amplification was performed using quantitative PCR (qPCR) using a
SYBR Green Premix (SYBR Real-Time PCR Kit; Takara Biotechnology
Co., Ltd.), according to the manufacturer's instructions using the
following conditions: 50°C for 2 min, 95°C for 2 min and 35 cycles
of 58°C for 30 sec and 72°C for 40 sec. The relative expression of
target genes was standardized to GAPDH, evaluated using the
2−ΔΔCq method and expressed as a ratio to control the
experiments (11).
Histological analysis
The injured artery tissues from SD rats were cleared
of blood with ice-cold PBS and kept in 4% paraformaldehyde for 24 h
at 4°C. After embedding in paraffin, sectioning to a thickness of 4
µm and mounting on slides, tissues were deparaffinized and
rehydrated by successive incubations in xylene, 100% ethanol, 95%
ethanol, 75% ethanol, and PBS. Sections were then stained with
hematoxylin and eosin using a standard protocol, or treated with
rabbit anti-TMEM66 antibody (1:100, Abcam) for
immunohistochemistry.
Co-immunoprecipitation
The artery tissue lysates (400 µg protein/ml
supernatant) were incubated with affinity-purified anti-Orai1
receptor antibody (5 µl/ml; cat. no. ab59330; Abcam) for 2 h at
room temperature and protein-G agarose at 4°C for 12 h. The
immunoprecipitates were subjected to immunoblotting with the Stim1
antibody (1:300; cat. no. ab57834; Abcam).
Cell culture and treatment
Embryonic thoracic aortic smooth muscle A10 cells
(ATCC, American Type Culture Collection) were cultured at 37°C in a
95% air/5% CO2 atmosphere in DMEM/F-12. TMEMT66
transduction was performed using the lentivirus-based
pLenti6.3-TMEM66 plasmid (Invitrogen Life Technologies; Thermo
Fisher Scientific, Inc.). Next, the A10 cells were cultured in 3 ml
DMEM containing 3% FBS, 10 µg/ml polybrene and virus (MOI=100). The
medium was replaced 48 h after transfection, and then 5 µg/ml
blasticidin was incubated in the medium for the next 48 h. The
cells were incubated with platelet-derived growth factor-BB
(PDGF-BB (30 ng/ml) (R&D Systems) for 24 h.
Cell proliferation and migration
assays
Cell proliferation was assessed using a
5-bromo-2′-deoxyuridine (BrdU) staining kit (BioVision, Inc.) and
the Cell Counting Kit-8 assay (CCK-8; Beyotime Institute of
Biotechnology), according to the manufacturer's instructions.
Briefly, for BrdU staining, cells were pulsed with BrdU for 2 h
after 24 h PDGF-BB stimulation. BrdU incorporation was visualized
by immunocytochemical staining with the anti-BrdU monoclonal
antibody (BioVision, Inc.). The cells were then fixed with 4%
paraformaldehyde and stained with 4′,6-diamidino-2-phenylindole
(DAPI). The number of BrdU-positive cells were counted using
fluorescent microscopy from three randomly chosen high power fields
(magnification, ×20). Cells were also seeded into 96-well plates
and incubated with CCK-8 for 2 h at 37°C. Absorbance was determined
using a spectrophotometer (Model 680; Bio-Rad Laboratories, Inc.)
at 450 nm. Moreover, a wound scratch assay was used to determine
cell migration. A10 cells were cultured in a 6-well dish until
>90% confluent was achieved. The scratching was performed with a
200-µl sterile pipette tip by creating a line. The cells were then
incubated with PDGF-BB for 24 h. Images were obtained by phase
contrast microscopy at ×40, and the number of migrated cells were
counted under a light microscope.
Intracellular calcium measurement
Intracellular calcium was measured using a Thermo
Varioskan flash instrument (Thermo Fisher Scientific, Inc.) as
previously described (12) with
various modifications. Briefly, A10 cells were seeded in 96-well
cell culture plates together with 5 µM calcium-dependent
fluorescent indicator Fura-2 in DMEM at 37°C. The cells were then
washed twice with Ca2+-free Krebs buffer (140 mM NaCl,
5.4 mM KCl, 1.2 mM MgSO4, 0.3 mM
NaH2PO4, 10 mM HEPES, 5 mM glucose, pH 7.4
with Tris base) and measured by the flash instrument with every 5
sec alternating between 340 and 380 nm excitation (2 nm slit size)
at 510 nm emission (5-nm slit size). The free Ca2+
concentration [Ca2+]free was calculated from
the equation (13):
[Ca2+]free=Kd[(R-Rmin)/(Rmax-R)](F380max/F380min);
The Kd is the dissociation constant of Fura-2 to
calcium. R is the ratio of each 340/380 nm. For the positive
control, the cells were incubated with calcium channel blocker,
verapamil, for 2 h, to reduce the Ca2+ concentration.
Minimum and maximum are the fluorescence values of cells treated by
Triton X-100 (saturating Ca2+ concentration) or by EGTA
(zero Ca2+ concentration).
Statistical analysis
Statistical analysis was performed using SPSS 22.0
software (IBM Corp.). Comparisons between two groups was carried
out using dependent paired t-test, and comparisons among more than
two groups was carried out by repeated measures one-way ANOVA. Data
are expressed as mean ± SEM. A probability value P<0.05 was
considered as indicative of statistical significance.
Results
TMEM66 mRNA and protein expression
after carotid artery injury
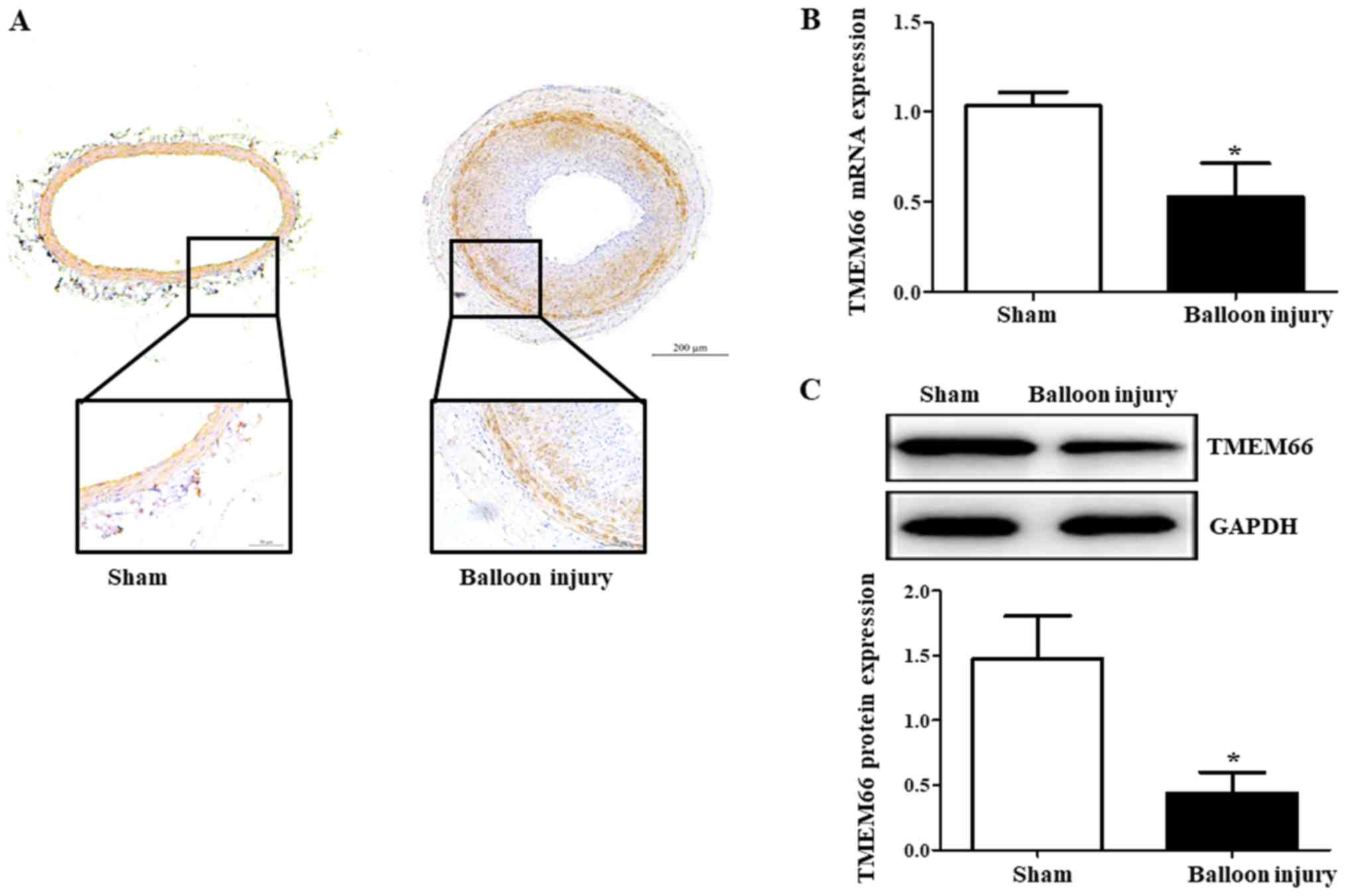
We first determined the expression of TMEM66 after
carotid artery injury by immunostaining, immunoblotting and qPCR.
Immunostaining of the arterial sections revealed expression of
TMEM66 in the medial and neointimal layers of the carotid artery
after balloon injury (Fig. 1A).
TMEM66 mRNA from the carotid artery tissue was significantly
decreased after balloon injury (Fig.
1B). Moreover, TMEM66 protein was lower in the balloon-injured
artery than that noted in the sham control (Fig. 1C), indicating that the impairment
of TMEM66 expression by balloon injury occurred at both the
post-translational and transcriptional levels.
TMEM66 overexpression attenuates
neointimal hyperplasia
To investigate whether or not TMEM66 is involved in
protection against neointimal hyperplasia, the carotid artery
thickness in TMEM66-overexpression mice was determined after
balloon injury. The histological analysis showed that balloon
injury significantly increased the ratio of the intima to media
thickness in the carotid artery, whereas the ratio of intima to
media thickness was reversed in the TMEM66-overexpression mice
(Fig. 2A).
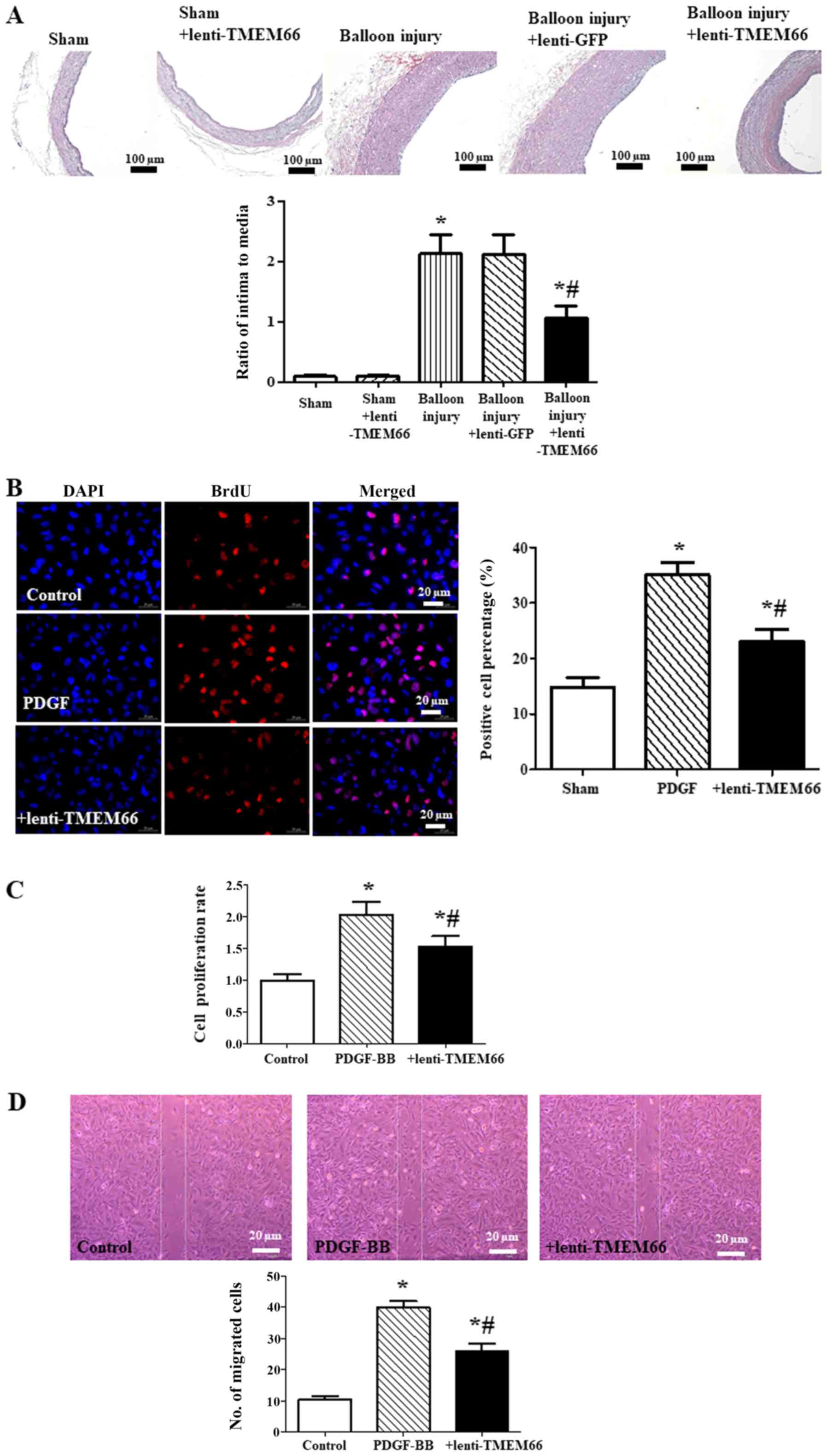 | Figure 2.Effect of TMEM66 overexpression on
neointimal hyperplasia. (A) Hematoxylin and eosin staining of
carotid artery showed the thickness of the neointima (*P<0.05,
vs. the sham control; #P<0.05, vs. the balloon injury
group, n=4). +lenti-TMEM66 means TMEM66 overexpression before
balloon injury. Scale bar, 100 µm. (B and C) BrdU labeling (B) and
CCK-8 assay (C) were used to determine A10 cell proliferation
(*P<0.05, vs. the control; #P<0.05, vs. PDGF-BB
treatment, n=6). +lenti-TMEM66 means TMEM66 overexpression before
PDGF-BB treatment. Scale bar, 20 µm. (D) Migration ability was
assessed by cell wound scratch assay. The number of migrated cells
was counted 24 h after scratching (*P<0.05, vs. the control;
#P<0.05 vs. PDGF-BB treatment, n=6). +lenti-TMEM66
means TMEM66 overexpression before PDGF-BB treatment. Scale bar, 20
µm. TMEM66, transmembrane protein 66; PDGF-BB, platelet-derived
growth factor-BB. |
Moreover, A10 cell proliferation was determined by
BrdU labeling (Fig. 2B) and Cell
Counting Kit-8 (CCK-8, Fig. 2C).
A10 cells with TMEM66 overexpression exhibited a significant
decrease in cell proliferation in response to PDGF-BB. The effect
of lentivirus overexpression of TMEM66 on PDGF-BB-induced A10 cell
migration was also assessed by scratch assay. It was demonstrated
that TMEM66 also reduced the cell migration induced by the PDGF-BB
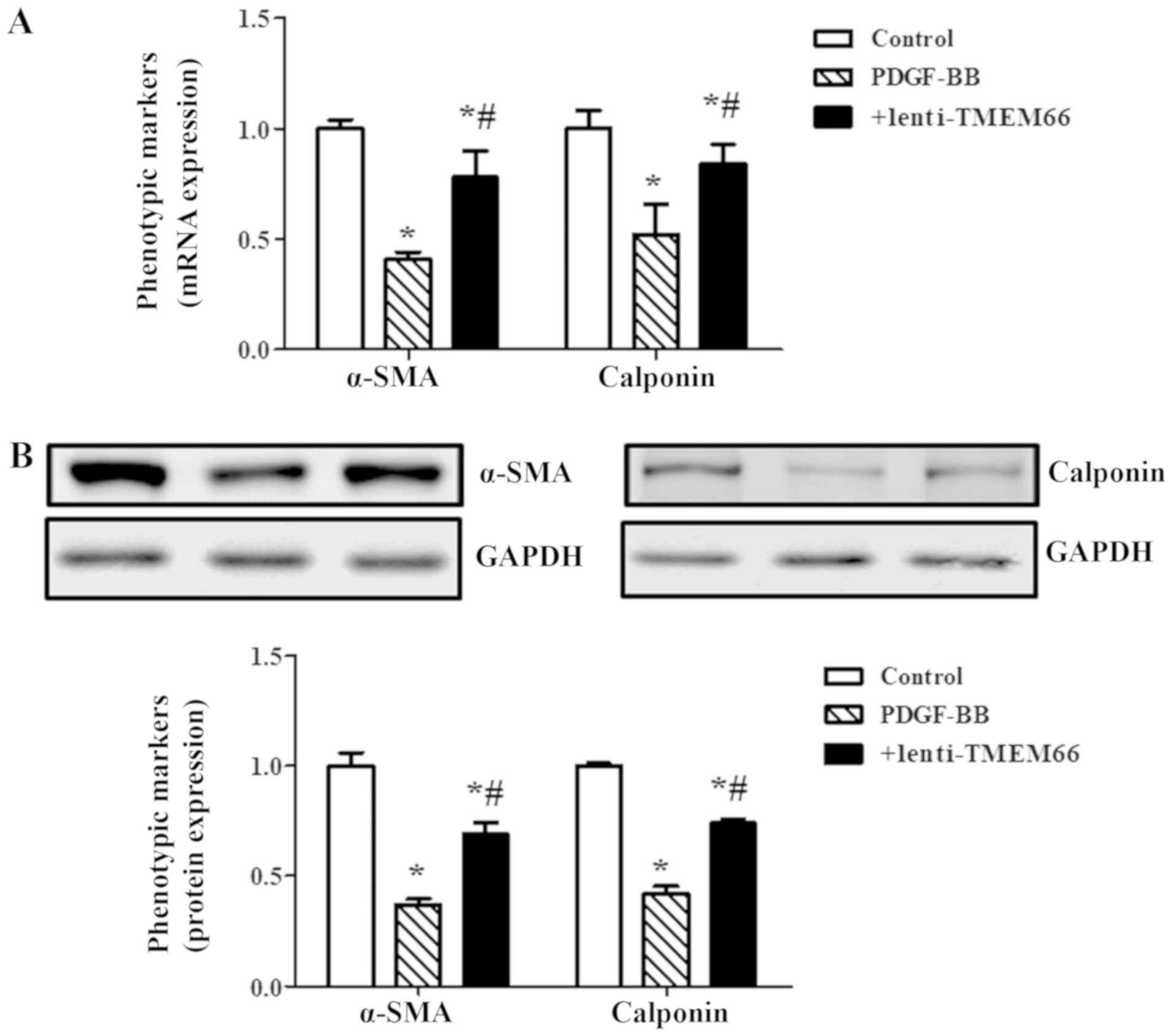
(Fig. 2D). In addition, expression
levels of α-smooth muscle actin (α-SMA) and calponin were decreased
following PDGF-BB treatment, while TMEM66 overexpression
significantly restored these VSMC phenotypic markers at the mRNA
(Fig. 3A) and protein (Fig. 3B) expression levels.
TMEM66 overexpression downregulates
modulators of SOCE
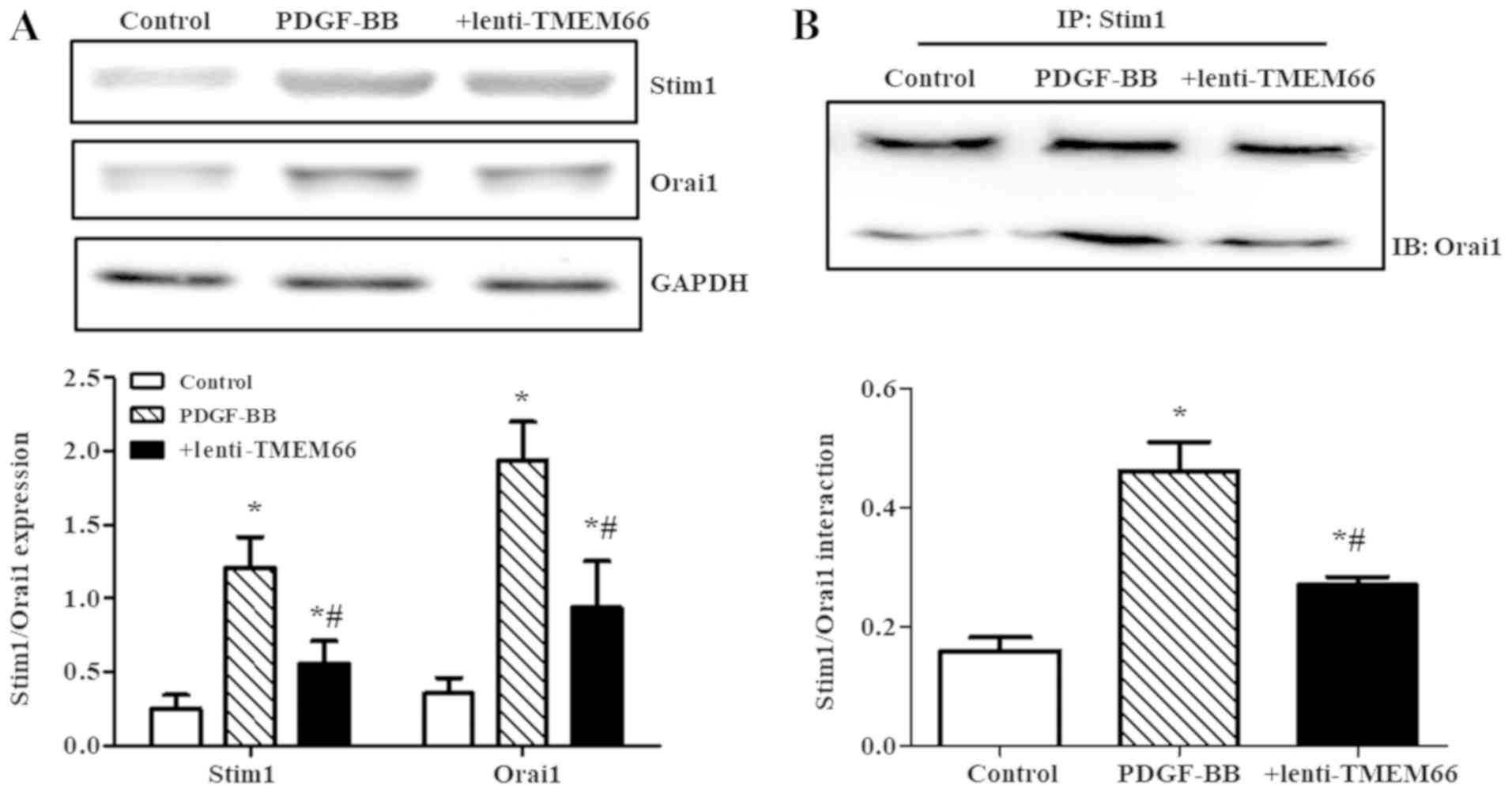
Stim1 and Orai1 are critical molecules involved in
the modulation of SOCE activation in VSMCs (14). Therefore, our further experiments
assessed the expression of Stim1 and Orai1 in balloon injured
carotid artery with or without TMEM66 transduction. The data
revealed that TMEM66 overexpression reduced the increased
expression of Stim1 and Orai1 after balloon injury (Fig. 4A). In addition, TMEM66 destabilized
the SOCE signalplexes: Interaction of Stim1 and Orai1 was increased
in the PDGF-BB-treated A10 cells, while it was deceased by TMEM66
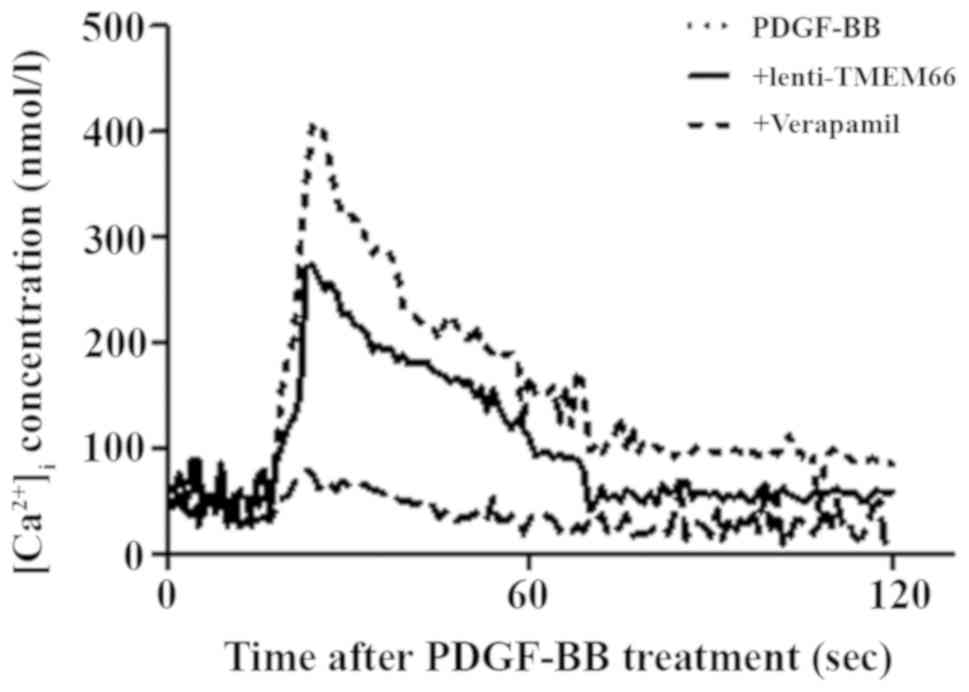
overexpression (Fig. 4B). Due to
the role of TMEM66 in SOCE modulators, the intracellular free
[Ca2+] was assessed. It was demonstrated that TMEM66
overexpression reduced the PDGF-BB treatment-enhanced
[Ca2+]i (Fig.
5).
Discussion
The calcium ion, as an important secondary
messenger, is a master regulatory molecule and an established
signal for promoting diseases. Neointimal hyperplasia and relevant
vascular smooth muscle cell (VSMC) phenotypes are dependent on
Ca2+ (4), especially
store operated Ca2+ entry (SOCE) (15). Transmembrane protein 66 (TMEM66),
known as the store-operated Ca2+ entry-associated
regulatory factor (SARAF), is a 339-amino acid protein that has
been presented as a novel regulator of SOCE and modulates
intracellular Ca2+ homeostasis (6,16).
The data presented in this study are consistent with the hypothesis
that TMEM66 was expressed in the medial and neointimal layers of
injured artery and the expression of TMEM66 was markedly decreased.
The results revealed that TMEM66 was impaired by balloon injury at
both the post-translational and transcriptional levels, and the
decreased expression of TMEM66 could be a key factor in the
pathological process of neointimal hyperplasia.
Due to the impairment of TMEM66 expression after
balloon injury, lenti-TMEM66 plasmids were transduced into the
carotid artery to induce TMEM66 protein overexpression. TMEM66
attenuated neointimal hyperplasia by suppressing the proliferation
and migration of the VSMCs, which is ascribed to the inhibitory
effect of TMEM66 on VSMC phenotypic changes. VSMCs have been
divided into contractile and synthetic/proliferative. The
proliferation and migration of proliferative VSMCs were found to be
significantly increased, and contribute to the formation of
neointimal hyperplasia and the development of restenosis after
vascular injury (17,18). In VSMCs, increased
[Ca2+]i initiates VSMC contraction as
excitation-contraction coupling, while Ca2+, as a
secondary messenger, affects gene expression as
excitation-transcription coupling (19). Excessive Ca2+ influx,
which has been thought as toxic [Ca2+]i,
leads to phenotypic marker expression and phenotypic modulation in
VSMCs, whereby the cells start to proliferate, migrate, and
synthesize excessive extracellular matrix (20), and finally causes neointimal
hyperplasia. Thus, decreasing [Ca2+]i may be
involved in the regulation of TMEM66 leading to VMSC proliferation
and migration.
The data presented in this study are consistent with
the hypothesis that TMEM66 reduces intracellular calcium
concentrations. The Ca2+ concentration is determined by
ion channels and pumps. Although the transient receptor potential
cation channel (TRPC) family members are involved in SOCE (21), the essential components of
Ca2+ release-activated Ca2+ (CRAC) channels,
Stim1 and Orai1, are key Ca2+ influx channels (22) and critical molecules for SOCE
activation in VSMCs (14). Stim1
is a Ca2+ sensor and detects the depletion of
Ca2+ concentration in the endoplasmic reticulum by a
conserved Ca2+ binding domain, which leads to Stim1
oligomerization and redistribution to binding to Orai1 directly
(23,24). The interaction of Stim1 and Orai1
causes channel opening and Ca2+ entry (25,26).
In the present study, it was shown that TMEM66 overexpression
reduced the increased expression of Stim1 and Orai1 after balloon
injury. Moreover, the stabilization of Stim1-Orai1 binding was
decreased by TMEM66 transduction as previously reported (6). TMEM66 responds to an elevated
intracellular Ca2+ concentration and promotes an
inactivation process of Stim1-mediated SOCE activity. Moreover, the
destabilization effect of TMEM66 on the Stim1/Orai1 complex is
dependent on the similar topology and overall structure with Stim1.
TMEM66 also contains a serine-proline rich domain at the C-terminal
tail, which may aid its interaction with plasma membrane
phospholipids (27,28).
In conclusion, our data indicated that TMEMT66
protects against the balloon injury-induced neointimal hyperplasia
by suppressing the Stim1/Orai1 complex expression and interaction.
Thus, TMEMT66 may be a pharmacological target for the treatment of
neointimal hyperplasia and restenosis after percutaneous coronary
intervention.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported in part by grants
from the National Natural Science Foundation of China (grant nos.
81770299 and 81470396) to DY and the National Natural Science
Foundation of China (grant no. 81500224) to QW.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JY, QW and DY conceived and designed the study. JY,
SL and DY performed the experiments. SL and DY wrote the paper. QW
and DY reviewed and edited the manuscript. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
This study was approved by the Research Council and
Animal Care and Use Committee of The General Hospital of Western
Theater Command. All experiments were conformed to the guidelines
of the American Association for the Accreditation of Laboratory
Animal Care and conformed to the guidelines of the ethical use of
animals, and all efforts were made to minimize animal suffering and
to reduce the number of animals used.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Byrne RA, Stone GW, Ormiston J and
Kastrati A: Coronary balloon angioplasty, stents, and scaffolds.
Lancet. 390:781–792. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Serruys PW, Luijten HE, Beatt KJ, Geuskens
R, de Feyter PJ, van den Brand M, Reiber JH, ten Katen HJ, van Es
GA and Hugenholtz PG: Incidence of restenosis after successful
coronary angioplasty: A time-related phenomenon. A quantitative
angiographic study in 342 consecutive patients at 1, 2, 3, and 4
months. Circulation. 77:361–371. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shimada Y, Kataoka T, Courtney BK, Morino
Y, Bonneau HN, Yock PG, Grube E, Honda Y and Fitzgerald PJ:
Influence of plaque calcium on neointimal hyperplasia following
bare metal and drug-eluting stent implantation. Catheter Cardiovasc
Interv. 67:866–869. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang W, Zhang X, Gonzalez-Cobos JC,
Stolwijk JA, Matrougui K and Trebak M: Leukotriene-C4 synthase, a
critical enzyme in the activation of store-independent Orai1/Orai3
channels, is required for neointimal hyperplasia. J Biol Chem.
290:5015–5027. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Parekh AB and Putney JW Jr: Store-operated
calcium channels. Physiol Rev. 85:757–810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Palty R, Raveh A, Kaminsky I, Meller R and
Reuveny E: Saraf inactivates the store operated calcium entry
machinery to prevent excess calcium refilling. Cell. 149:425–438.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jha A, Ahuja M, Maleth J, Moreno CM, Yuan
JP, Kim MS and Muallem S: The STIM1 CTID domain determines access
of SARAF to SOAR to regulate Orai1 channel function. J Cell Biol.
202:71–79. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grassia G, Maddaluno M, Guglielmotti A,
Mangano G, Biondi G, Maffia P and Ialenti A: The anti-inflammatory
agent bindarit inhibits neointima formation in both rats and
hyperlipidaemic mice. Cardiovasc Res. 84:485–493. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matsumae H, Yoshida Y, Ono K, Togi K,
Inoue K, Furukawa Y, Nakashima Y, Kojima Y, Nobuyoshi M, Kita T and
Tanaka M: CCN1 knockdown suppresses neointimal hyperplasia in a rat
artery balloon injury model. Arterioscler Thromb Vasc Biol.
28:1077–1083. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Romanuik TL, Wang G, Holt RA, Jones SJ,
Marra MA and Sadar MD: Identification of novel androgen-responsive
genes by sequencing of LongSAGE libraries. BMC Genomics.
10:4762009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Monck JR, Reynolds EE, Thomas AP and
Williamson JR: Novel kinetics of single cell Ca2+ transients in
stimulated hepatocytes and A10 cells measured using fura-2 and
fluorescent videomicroscopy. J Biol Chem. 263:4569–4575.
1988.PubMed/NCBI
|
|
13
|
Grynkiewicz G, Poenie M and Tsien RY: A
new generation of Ca2+ indicators with greatly improved
fluorescence properties. J Biol Chem. 260:3440–3450.
1985.PubMed/NCBI
|
|
14
|
Guo RW, Yang LX, Li MQ, Pan XH, Liu B and
Deng YL: Stim1- and Orai1-mediated store-operated calcium entry is
critical for angiotensin II-induced vascular smooth muscle cell
proliferation. Cardiovasc Res. 93:360–370. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Motiani RK, Stolwijk JA, Newton RL, Zhang
X and Trebak M: Emerging roles of Orai3 in pathophysiology.
Channels (Austin). 7:392–401. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Albarran L, Regodón S, Salido GM, Lopez JJ
and Rosado JA: Role of STIM1 in the surface expression of SARAF.
Channels (Austin). 11:84–88. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin G, Chow S, Lin J, Wang G, Lue TF and
Lin CS: Effect of cell passage and density on protein kinase G
expression and activation in vascular smooth muscle cells. J Cell
Biochem. 92:104–112. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yoshida T and Owens GK: Molecular
determinants of vascular smooth muscle cell diversity. Circ Res.
96:280–291. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wamhoff BR, Bowles DK and Owens GK:
Excitation-transcription coupling in arterial smooth muscle. Circ
Res. 98:868–878. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kudryavtseva O, Aalkjaer C and Matchkov
VV: Vascular smooth muscle cell phenotype is defined by
Ca2+-dependent transcription factors. FEBS J. 280:5488–5499. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eder P: Cardiac remodeling and disease:
SOCE and TRPC signaling in cardiac pathology. Adv Exp Med Biol.
993:505–521. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee KP, Yuan JP, Hong JH, So I, Worley PF
and Muallem S: An endoplasmic reticulum/plasma membrane junction:
STIM1/Orai1/TRPCs. FEBS Lett. 584:2022–2027. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stathopulos PB, Zheng L and Ikura M:
Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing
regions exhibit distinct unfolding and oligomerization kinetics. J
Biol Chem. 284:728–732. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hogan PG, Lewis RS and Rao A: Molecular
basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev
Immunol. 28:491–533. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Baba Y, Hayashi K, Fujii Y, Mizushima A,
Watarai H, Wakamori M, Numaga T, Mori Y, Iino M, Hikida M and
Kurosaki T: Coupling of STIM1 to store-operated Ca2+ entry through
its constitutive and inducible movement in the endoplasmic
reticulum. Proc Natl Acad Sci USA. 103:16704–16709. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou Y, Meraner P, Kwon HT, Machnes D,
Oh-hora M, Zimmer J, Huang Y, Stura A, Rao A and Hogan PG: STIM1
gates the store-operated calcium channel ORAI1 in vitro. Nat Struct
Mol Biol. 17:112–116. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kiselyov K, Xu X, Mozhayeva G, Kuo T,
Pessah I, Mignery G, Zhu X, Birnbaumer L and Muallem S: Functional
interaction between InsP3 receptors and store-operated HtrP3
channels. Nature. 396:478–482. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Korzeniowski MK, Popovic MA, Szentpetery
Z, Varnai P, Stojilkovic SS and Balla T: Dependence of
STIM1/Orai1-mediated calcium entry on plasma membrane
phosphoinositides. J Biol Chem. 284:21027–21035. 2009. View Article : Google Scholar : PubMed/NCBI
|