Introduction
Diabetic kidney disease (DKD) is increasingly
diagnosed and has become a leading cause of end-stage renal disease
(ESRD) throughout the world during the past few decades (1,2). DKD
is characterized by mesangial expansion, a thicker glomerular
basement membrane, progressive glomerulosclerosis, and finally the
development of progressive fibrosing kidney disease. Previous
studies have demonstrated that several pathological factors are
involved in the progression of diabetic nephropathy (DN), including
the downregulation of autophagy (3), podocyte apoptosis, detachment
(4) and epithelial-mesenchymal
transition (EMT) (5); however, the
precise mechanism remains elusive. Therefore, exploring the precise
mechanism and identifying effective strategies for the treatment of
DN remains crucial.
Recently, Krüppel-like factor 4 (KLF4), a zinc
finger-containing transcription factor, has aroused attention due
to its potential effect on podocyte and kidney disease. KLF4
belongs to the family of SP/KLF factors and plays a crucial role in
regulating various cellular processes (e.g., cell growth,
proliferation, differentiation, and inflammation) (6). A recent study revealed that
endothelial KLF4 is renoprotective and mediates statin-induced
protection against ischemic AKI by regulating the expression of
cell adhesion molecules and the concomitant recruitment of
inflammatory cells (7). In
addition, Hayashi et al (8)
confirmed that KLF4 is expressed in podocytes and found that KLF4
overexpression resulted in a sustained increase in nephrin
expression and a decrease in albuminuria, both in animal models and
humans exhibiting proteinuria. These findings suggest that KLF4 has
a renoprotective effect on chronic renal disease; however, little
is known about the specific mechanism.
In the present study, a KLF4 plasmid vector was
constructed, and a db/db spontaneous DKD mouse model, as well as
DKD mouse serum-induced podocyte injury, were utilized to explore
the potential role of KLF4 in DN and its underlying mechanism.
Materials and methods
Cell culture and grouping
Conditionally immortalized differentiated mouse
podocytes (MPC5) were purchased from the Institute of Basic
Medicine at the Chinese Academy of Medical Sciences. The cells were
cultured as described previously (9). Briefly, to induce proliferation, the
cells were cultured at 33°C in RPMI 1640 medium (cat. no.
SH30809.01B; HyClone; GE Healthcare Life Sciences) supplemented
with 10% fetal bovine serum (FBS), 100 U/ml penicillin G, 100 mg/ml
streptomycin and 100 U/ml interferon (IFN)-γ. To promote
differentiation, the cells were cultured in medium without IFN-γ at
37°C. The differentiated podocytes were diluted for all subsequent
experiments. Podocytes were divided into four groups: i) Control
serum group (treated with 10% serum from C57BL/KsJ db/m mice for 24
h); ii) DKD serum group (treated with 10% serum from C57BL/KsJ
db/db mice for 24 h); iii) DKD serum + Vector-negative control (NC)
group (transfected with an empty plasmid for 72 h following
treatment with DKD mouse serum); and iv) DKD serum + Vector-KLF4
group [transfected with the pcDNA3.1(+)-KLF4 plasmid for 72 h
following treatment with DKD mouse serum].
Animals and grouping
Male C57BL/KsJ db/db spontaneous DKD model mice
(n=10; 8 weeks old; weight, 22.3–26.5 g) and C57BL/KsJ db/m control
mice (n=5; 8 weeks old; weight, 20.8–24.1 g) were obtained from
Cavens Experimental Animal Co., Ltd. As previously described
(10), mice were bred in a 12-h
light/dark cycle in a pathogen-free facility at a constant
temperature (20±2°C) and humidity (50–60%) with free access to food
(a standard diet) and water for 5 weeks. C57BLKS/J db/db mice at 13
weeks old were randomly assigned to one of two groups: i) db/db +
vector-NC group (n=5; receiving an injection of 5×108 IU
lentiviral vector-NC every 5 days for 30 days); and ii) db/db +
vector-KLF4 group (n=5; receiving an injection of 5×108
IU lentiviral vector-KLF4 every 5 days for 30 days). Age-matched
db/m mice (n=5) were used as the control group. No mice succumbed
during the experiment. All mice were sacrificed and used for
subsequent experiments.
KLF4 vector construction and
transfection
The synthetic gene fragments of KLF4 (synthesized by
Shanghai Jierui Biological Engineering Co., Ltd.) were incorporated
into a plasmid (pcDNA3.1; cat. no. V79020; Invitrogen; Thermo
Fisher Scientific, Inc.) following double enzyme digestion with
NheI-BamHI (cat. nos. D1162A and D1010A; Takara
Biotechnology Co., Ltd.), and termed pcDNA3.1(+)-KLF4. The products
were transformed into DH5α-competent cells (cat. no. CD201-01;
Beijing TransGen Biotech Co., Ltd.), smeared on a lysogeny broth
(LB) ampicillin (AMP) plate, and cultured at 37°C overnight. An LB
culture solution containing AMP was used to screen for the
objective colonies. MPC5 cells were transfected with 800–1,500
ng/µl pcDNA3.1(+)-KLF4 using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) for 72 h, according to
the manufacturer's instructions.
The synthetic gene fragments of KLF4 were cloned
into a pCDH-CMV-MCS-EF1-GFP-Puro lentiviral vector (cat. no.
CD511B-1; System Biosciences). The constructs (1.5 µg) were then
transfected into 1×106/ml 293T cells (cat. no. CL-0005;
Procell Life Science & Technology Co., Ltd.) using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. After 72 h, the
lentivirus was collected. Mice were injected with 5×108
IU lentiviral vector-KLF4 every 5 days for 30 days through the tail
vein after the DKD model had been successfully established.
Biochemical measurements
Urine collection was performed in metabolic cages at
13 weeks before treatment and 30 days after vector-KLF4 injection.
Urine albumin was detected using an automatic biochemical analyzer
(Hitachi 7020; Hitachi, Ltd.). The concentration of serum
creatinine (Scr) and blood urea nitrogen (BUN) was detected using a
creatinine assay kit (cat. no. C011-1; Nanjing Jiancheng
Bioengineering Institute) and a urea nitrogen assay kit (cat. no.
C013-2; Nanjing Jiancheng Bioengineering Institute),
respectively.
Periodic acid-Schiff (PAS)
staining
Kidney samples were fixed in 10% neutral-buffered
formalin at room temperature for 12 h, embedded in paraffin wax and
cut into 4-µm thick sections. Kidney sections from the mice were
oxidized in 1% periodic acid for 10 min and washed with distilled
water after being dewaxed. The tissues were then stained with
Schiff's reagent (cat. no. G1281; Beijing Solarbio Science &
Technology Co., Ltd.) for 10–20 min and washed again. Afterwards,
the sections were counterstained in Harris's hematoxylin for 5–10
min. All steps were performed at room temperature. The images were
observed under a light microscope (magnification, ×400; DM2500;
Leica Microsystems GmbH).
Immunofluorescence staining
MPC5 cells from the different treatment groups
groups were seeded onto glass cover slips and fixed in 4%
paraformaldehyde for 30 min at room temperature, then treated with
phosphate buffered saline (PBS) supplemented with 0.1% Triton X-100
for 10 min at room temperature. Tissue sections were blocked with
PBS containing 5% BSA (cat. no. SH30574.03; HyClone; GE Healthcare
Life Sciences) for 1 h at room temperature after deparaffinization
using xylene (cat. no. 1330-20-7; Shanghai Macklin Biochemical Co.,
Ltd.). The podocytes and tissue sections were incubated with the
following primary antibodies at 4°C overnight: Anti-LC3 (Abcam;
cat. no. ab192890; 1:500), anti-nephrin (cat. no. sc-377246; 1:50;
Santa Cruz Biotechnology, Inc.), anti-filamentous (F-)actin (cat.
no. ab130935; 1:100; Abcam), and anti-vimentin (cat. no. ab92547;
1:100; Abcam). After washing, the samples were incubated with Alexa
Fluor 594-conjugated goat anti-rabbit IgG (cat. no. 111585003;
1:100; Jackson ImmunoResearch Inc), FITC-conjugated goat anti-mouse
IgG (cat. no. A0568; 1:100; Beyotime Institute of Biotechnology),
and Cy3-conjugated goat anti-rabbit IgG at 37°C for 1 h, followed
by counterstaining with DAPI (1:500; Beyotime Institute of
Biotechnology) at room temperature for 5 min. Images were observed
under a laser scanning confocal fluorescence microscope
(magnifications, ×100 and ×400; UltraVIEW VoX; PerkinElmer,
Inc.).
Western blot analysis
The cells were lysed with cell lysis buffer (cat.
no. P0013B; Beyotime Institute of Biotechnology) supplemented with
protease cocktail (cat. no. 89806; Invitrogen; Thermo Fisher
Scientific, Inc.) after washing three times. Animal tissues were
homogenized in RIPA lysis buffer and centrifuged for 5 min at
12,000 × g at 4°C. The protein concentration was quantified using a
bicinchoninic acid assay. The proteins (30 µg/lane) were resolved
by 8–12% SDS-PAGE and transferred onto PVDF membranes (EMD
Millipore). The membranes were blocked at room temperature with 5%
milk in TBS with Tween-20 for 1 h, and then probed with the
following primary antibodies: Anti-KLF4 (cat. no. ab129473,
1:2,000; Abcam), anti-caspase-3 (cat. no. 9662; 1:1,000; Cell
Signaling Technology, Inc.), anti-phosphorylated (p)-mTOR (cat. no.
AF3308; 1:1,000; Affinity Biosciences), anti-mTOR (cat. no. AF6308;
1:1,000; Affinity Biosciences), anti-S6K (cat. no. AF6226; 1:500;
Affinity Biosciences), anti-p-S6K (cat. no. AF3228; 1:500; Affinity
Biosciences), anti-E-cadherin (cat. no. AF013; 1:1,000; Affinity
Biosciences), anti-vimentin (cat. no. AF7013; 1:1,000; Affinity
Biosciences), anti-α-smooth muscle actin (SMA; cat. no. AF1032;
1:500; Affinity Biosciences), anti-microtubule associated protein 1
light chain 3α (LC3; cat. no. 4108S; 1:1,000; Cell Signaling
Technology, Inc.), anti-p62 (cat. no. 18420-1-AP; 1:500;
ProteinTech Group, Inc.) and anti-β-actin (cat. no. 4970; 1:1,000;
Cell Signaling Technology, Inc.) overnight at 4°C. The membranes
were then incubated with HRP-conjugated goat anti-rabbit IgG
antibody (cat. no. A0208; 1:1,000; Beyotime Institute of
Biotechnology) or HRP-conjugated goat anti-mouse IgG antibody (cat.
no. A0216; 1:1,000; Beyotime Institute of Biotechnology) for 1 h at
room temperature after being washed with TBS. The blots were then
visualized using an enhanced chemiluminescence reagent (cat. no.
WBKLS0100; EMD Millipore) and analyzed using ImageJ software,
version 1.8.0–112 (National Institutes of Health).
Analysis of apoptosis by flow
cytometry and TUNEL assay
The ratio of apoptotic cells was determined using a
fluorescein isothiocyanate-Annexin V Apoptosis Detection kit (BD
Biosciences). The analysis was performed as previously described
(9). Briefly, the cells were
stained with FITC-Annexin V and propidium iodide, according to the
manufacturer's instructions. Podocyte apoptosis was determined by
flow cytometry (FACSCalibur; BD Biosciences) and analyzed using
Accuri C6 software (version 1.0.264.21; BD Biosciences). The
presence of apoptotic cells in the tissue sections was determined
using a TUNEL Apoptosis kit (cat. no. BA27A; Nanjing Biobox Biotech
Co., Ltd.), according to the manufacturer's instructions. After
deparaffinization, sections of kidney tissue were decomposed with
protease K and successively incubated in terminal transferase
buffer with streptavidin-FITC-conjugated antibodies and a
POD-conjugated anti-FITC solution, followed by staining with a
diaminobenzidine solution (all in the TUNEL kit). Apoptotic cells
were observed under a light microscope in five fields of view
(magnification, ×400; DM2500; Leica Microsystems GmbH).
Statistical analysis
All statistical analyses were performed using SPSS
21.0 software (IBM Corp.). Data are presented as the mean ±
standard deviation. A multi-way ANOVA and one-way ANOVA followed by
a least significant difference test were used to analyze the
differences between multiple groups in the in vitro and
in vivo experiments, respectively. P<0.05 was considered
to indicate a statistically significant difference.
Results
Protective effect of KLF4 on podocytes
treated with DKD mouse serum
To determine the role of KLF4 in podocytes growing
in a high glucose environment, a plasmid encoding KLF4 (pcDNA3.1 +
KLF4) and an empty vector were constructed and transfected into
podocytes treated with DKD mouse serum. Western blotting revealed
that the level of KLF4 expression was significantly increased when
the podocytes were transfected with the KLF4 plasmid. As shown in
Fig. 1B, C, F and G, KLF4
overexpression significantly decreased high glucose-induced
podocyte apoptosis in line with the attenuation of cleaved
caspase-3 expression. Moreover, KLF4 overexpression significantly
reversed the disarrangement of F-actin, downregulated the
expression of vimentin and α-SMA, and upregulated E-cadherin
(Fig. 1D, F and G). Furthermore,
KLF4 overexpression inhibited the decline in nephrin induced by DKD
serum (Fig. 1E). These data
demonstrate a crucial role for KLF4 in protecting against podocyte
injury in a high glucose environment.
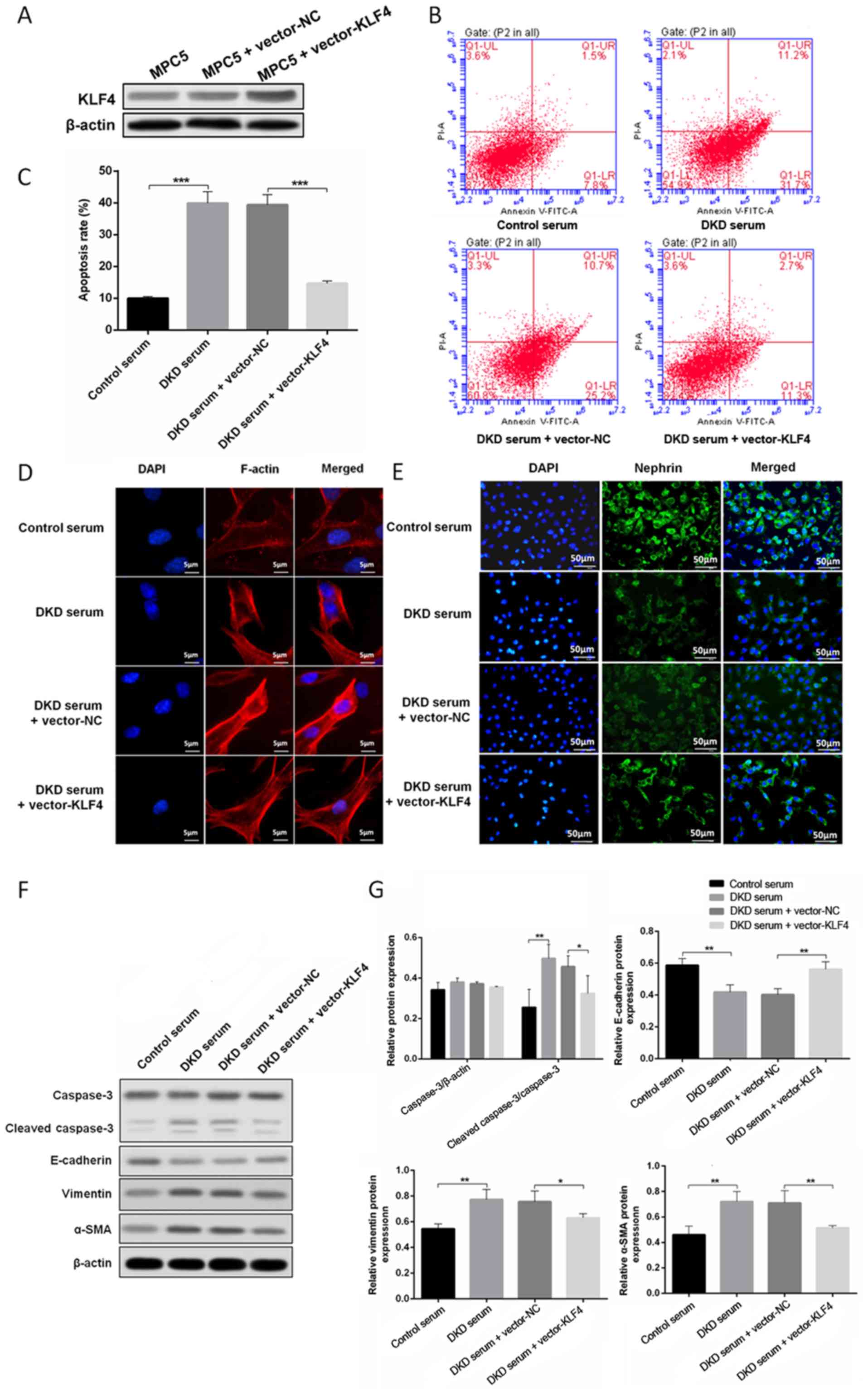 | Figure 1.KLF4 overexpression protects against
DKD mouse serum-induced podocyte injury. (A) Western blotting
showing that transfection with the KLF4 plasmid increased KLF4
expression. (B) The representative images and (C) quantitative
analysis of the apoptotic cells are presented for the individual
groups. (D) Representative photomicrographs of F-actin
immunofluorescence staining. Scale bars, 5 µm. (E) Representative
photomicrographs of nephrin immunofluorescence staining. Scale
bars, 50 µm. (F) Representative bands of caspase-3,
cleaved-caspase-3, vimentin, α-SMA and E-cadherin protein
expression in the individual groups. (G) Bar graphs showing the
relative levels of protein expression. Data are presented as the
mean ± SD (n=5). *P<0.05; **P<0.01; ***P<0.001. α-SMA,
α-smooth muscle actin; KLF4, Krüppel-like factor 4; NC, negative
control; PI, propidium iodide; DKD, diabetic kidney disease. |
KLF4 overexpression protects against
diabetic kidney damage in DKD mice
The western blotting results showed the level of
KLF4 expression was increased in mouse renal tissues after
lentiviral interference (Fig. 2A).
The 24-h proteinuria, Scr and BUN were measured to analyze the
effects on renal function. As shown in Fig. 2B, the transfer of the KLF4 plasmid
resulted in an attenuation of proteinuria in DN mice 30 days after
the injection. Scr and BUN were also decreased by the plasmid
transfer of KLF4 compared with the vector-NC group (Scr in the
vector-NC group, 0.78 0.02 µmol/l; Scr in the vector-KLF4 group,
0.70±0.02 µmol/l; P<0.05). PAS staining of the renal tissues
showed that the glomeruli of DN mice had a greater diameter,
mesangial matrix expansion and mesangial cell proliferation
compared to that of the db/m mice. However, the aforementioned
lesions of the glomeruli were attenuated following KLF4 plasmid
injection compared to the vector-NC group (Fig. 2C).
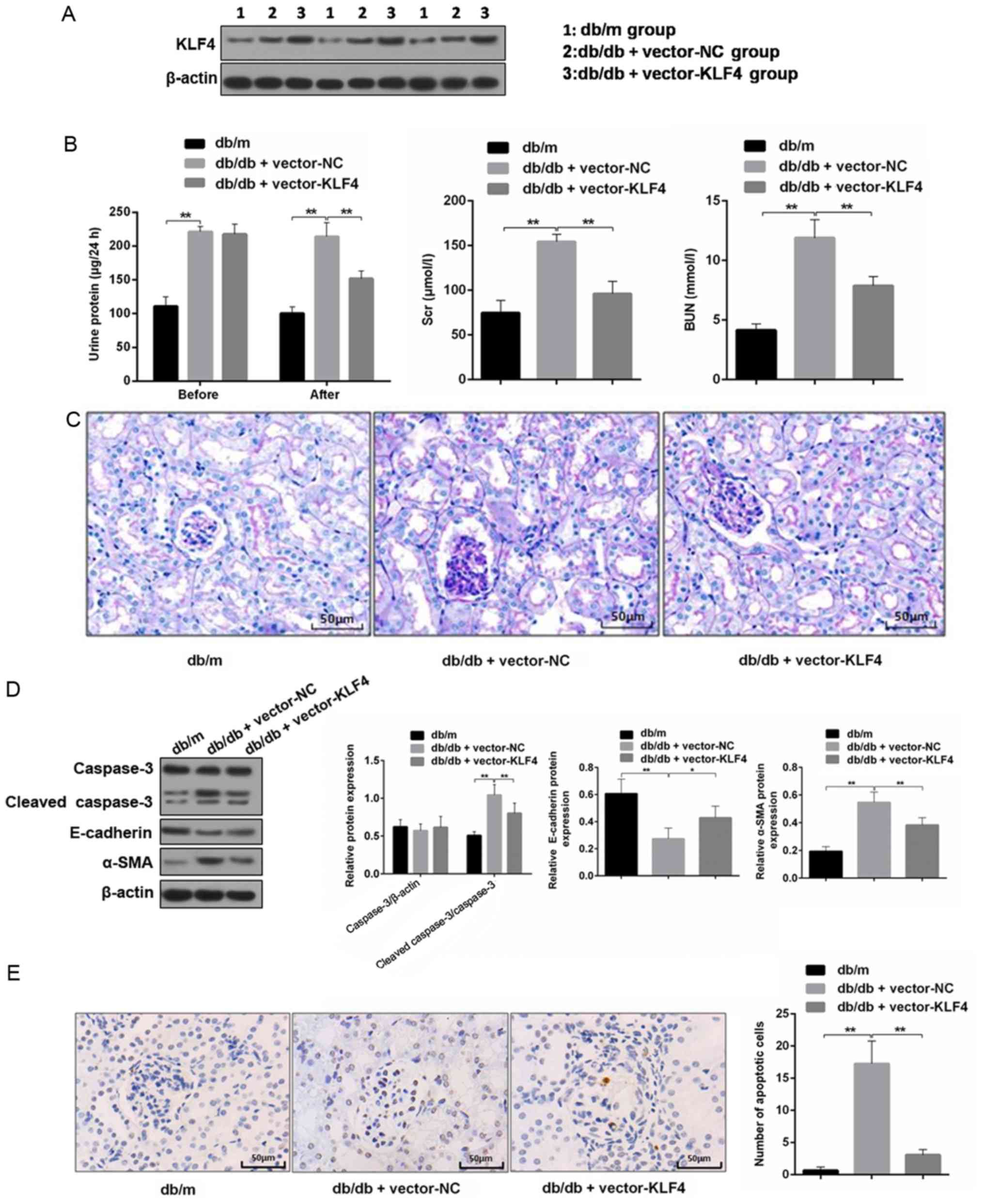 | Figure 2.KLF4 overexpression protects against
kidney damage in diabetic kidney disease model mice. (A)
Representative bands of KLF4 in the individual groups. (B) Bar
graphs showing the quantification of albuminuria, Scr and BUN in
each group. (C) Representative photomicrographs of periodic
acid-Schiff staining of the kidney tissues in each group. Scale
bars, 50 µm. (D) Representative bands and quantitative evaluation
of caspase-3, cleaved caspase-3, α-SMA and E-cadherin in the
individual groups. (E) Representative photomicrographs and
quantitative analysis of apoptotic cells via TUNEL staining of the
kidney tissues in the individual groups. Scale bars, 50 µm. (F)
Representative photomicrographs of immunofluorescence double
staining of nephrin (red) and vimentin (green) in the individual
groups. Scale bars, 50 µm. Data are presented as the mean ± SD
(n=5). *P<0.05; **P<0.01. KLF4, Krüppel-like factor 4; BUN,
blood urea nitrogen; Scr, serum creatinine; NC, negative control;
α-SMA, α-smooth muscle actin. |
Podocyte apoptosis and EMT biomarkers in the renal
tissue were subsequently examined. The TUNEL staining results
showed that the number of apoptotic cells was decreased in the
vector-KLF4 group compared to the vector-NC group (Fig. 2E). The western blotting results
showed that the KLF4 plasmid injection significantly reduced the
expression of cleaved caspase-3 and α-SMA, and increased the
expression of E-cadherin (Fig.
2D). Immunofluorescence double staining showed increased
nephrin expression and attenuated vimentin expression in the
vector-KLF4 group compared to the vector-NC group (Fig. 2F). These findings suggested that
KLF4 has inhibitory effects on podocyte apoptosis and the
progression of EMT in DN.
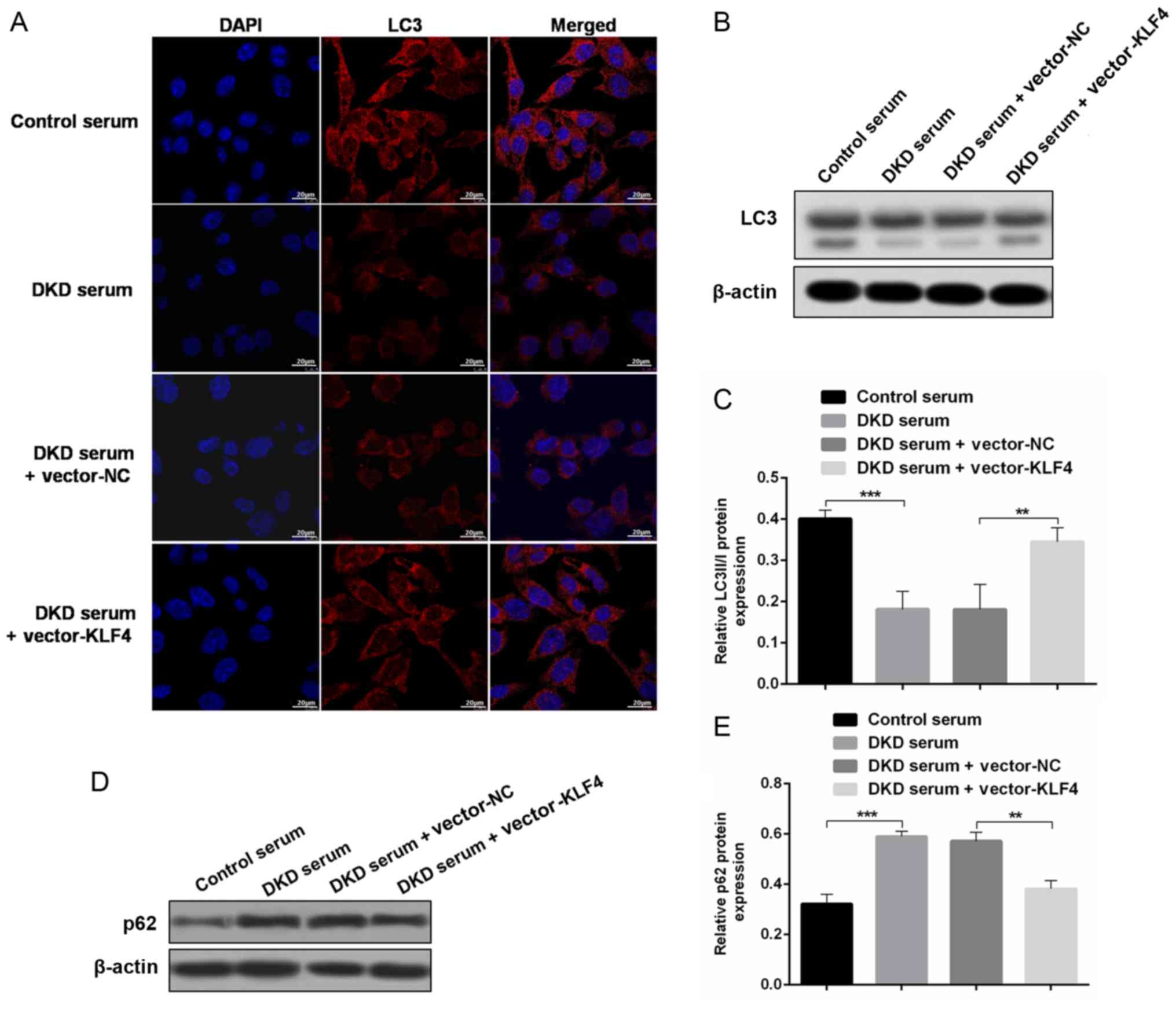
KLF4 overexpression upregulates
podocyte autophagy to protect against diabetic kidney damage
To evaluate the role of autophagy in the protective
effect of KLF4 against diabetic kidney damage, changes in
autophagic markers were detected in vivo and in
vitro. In vitro, the immunofluorescence results showed
that treatment with DKD serum inhibited LC3 expression, but the LC3
fluorescence was substantially increased after the KLF4 plasmid
transfer (Fig. 3A). In addition,
the western blotting results demonstrated that KLF4 overexpression
increased the LC3-II/LC3-I conversion and decreased p62 expression
compared to the DKD serum group (Fig.
3B-E). Similar differences were observed in the in vivo
studies. The LC3 fluorescence area was substantially increased
following KLF4 plasmid injection in DKD mice, as shown by the
immunofluorescence assay (Fig.
3F). Moreover, as shown in Fig. 3G
and H, the LC3-II/LC3-I ratio was significantly increased in
the vector-KLF4 group compared to the vector-NC group.
Additionally, KLF4 overexpression increased the expression of
nephrin compared to the vector-NC group (Fig. 3F). These data further demonstrated
a crucial role for autophagy in modulating the protective effect of
KLF4 against diabetic kidney damage.
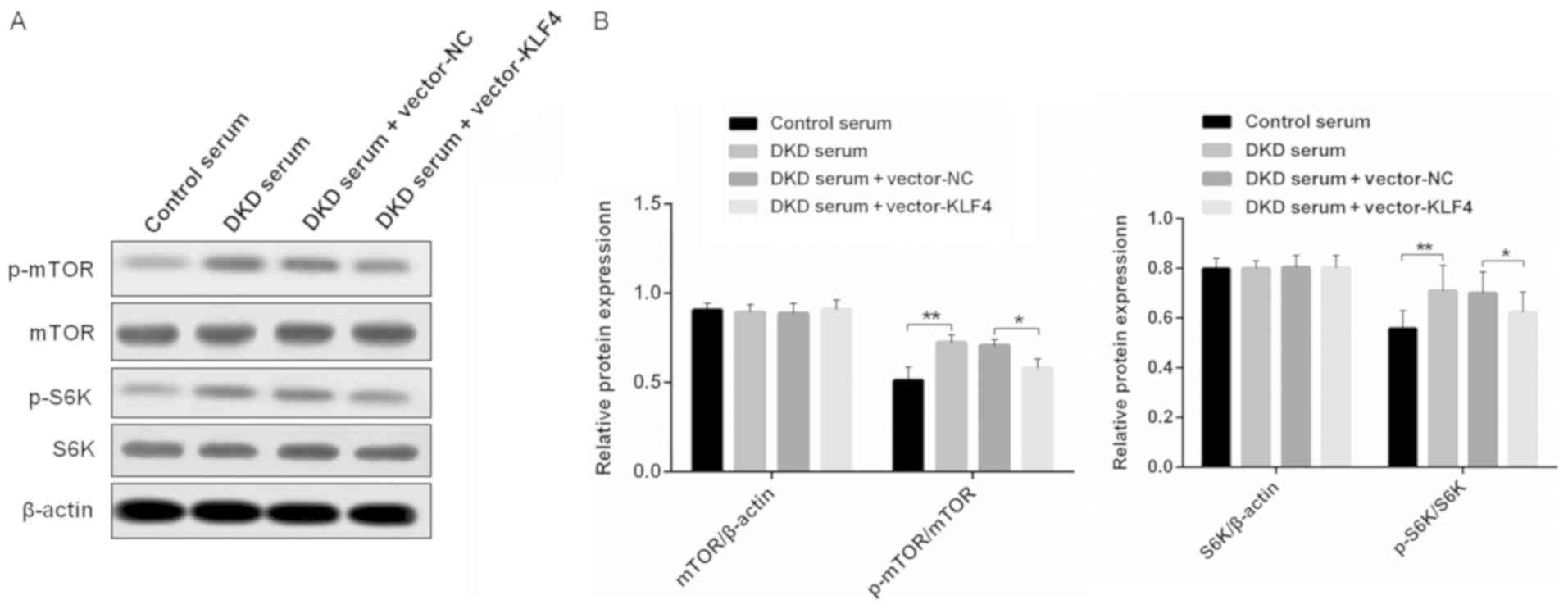
KLF4 activates podocyte autophagy by
negatively regulating the mTOR signaling pathway
To examine the mechanisms of KLF4-induced activation
of podocyte autophagy, western blotting was performed to detect the
level of relative protein expression in the mTOR signaling pathway
in vitro (Fig. 4). It was
found that there were similar levels of mTOR and S6K protein
expression in each group; however, the overexpression of KLF4
resulted in a small but significant decrease in p-mTOR and p-S6K
protein expression, which was increased in the DKD mouse serum
group compared to the group treated with the control mouse
serum.
Discussion
There is emerging evidence that KLFs play a vital
role in the maintenance of normal renal function and are involved
in key physiological processes in the kidneys (e.g., podocyte
differentiation, tubulointerstitial inflammation and the
progression of kidney fibrosis) (11,12).
It was previously confirmed that KLF4 is primarily expressed in the
podocytes of mice and humans, and its expression is decreased in
both animal models and humans with a proteinuric status (8). In the present study, it was found
that the overexpression of KLF4 significantly reduced the level of
urinary albumin, Scr and BUN, whereas mesangial matrix expansion
and mesangial cell proliferation were attenuated in DN mice.
Furthermore, KLF4 overexpression attenuated podocyte apoptosis and
downregulated mesenchymal markers, accompanied by the upregulation
of epithelial cell markers. Moreover, the present findings
suggested that the overexpression of KLF4 may ameliorate DN by
activating podocyte autophagy and inhibiting mTOR signaling.
Previous studies have shown that increased levels of
urinary protein in DN are associated with podocyte injury,
including podocyte apoptosis, detachment and EMT (13). In the present study, it was found
that KLF4 overexpression significantly decreased the apoptosis rate
of podocytes treated with DKD mouse serum and in DN mice,
accompanied by a reduction in cleaved caspase-3, a crucial
proteolytic cleavage enzyme in cellular apoptosis. These data
suggested that KLF4 plays a protective role in podocytes. This role
was confirmed by an increase in nephrin when KLF4 was upregulated
in both the in vivo and in vitro experiments.
Podocyte EMT, which is characterized by the loss of
epithelial cell markers (e.g., E-cadherin) and re-expression of
mesenchymal markers (e.g., vimentin and α-SMA), is widely involved
in the pathological process of DN (14). A previous study demonstrated that
KLF4 overexpression attenuates lung fibrosis and EMT in transgenic
mice (15). The present study
found that KLF4 overexpression reversed the downregulation of
E-cadherin, as well as the upregulation of both vimentin and α-SMA,
in DN mouse podocytes. These results suggested that KLF4 is
involved in the regulation of podocyte EMT in DN. This is in line
with a recent study showing that matrix stiffness-regulated
KLF5/KLF4 is related to the pathogenesis of renal fibrosis
(16). However, the role of KLF4
in EMT remains controversial, especially in certain cancer studies
(17,18). Thus, additional studies are
required to clarify the precise relationship between KLF4 and EMT.
In addition, renal histopathology demonstrated that mesangial
matrix expansion and mesangial cell proliferation were alleviated
in DN mice following the restoration of podocyte KLF4 expression in
the present study. Previously, inflammation was found to play a
central role in the progression of DN (19), and KLF4 has been reported to be a
regulatory factor in lipopolysaccharide-induced inflammation and
tissue damage, both in vivo and in vitro (20). Therefore, it was speculated that
the KLF4-mediated alleviation of the DN mouse pathological lesions
may be associated with the regulation of inflammation, independent
of suppressing podocyte apoptosis.
Autophagy is a major intracellular lysosomal
degradation system and cellular process used to degrade and recycle
proteins and other impaired cell organelles in lysosomes. Moreover,
autophagy plays an important role in the maintenance of
intracellular homeostasis. A study focusing on the potential effect
of autophagy on kidney disease, including DN, was previously
performed, indicating that the level of autophagy decreased in
podocytes cultured in the serum of DN rats (21). In the past few years, numerous
studies have suggested that the activation of autophagy in
podocytes may be a potential therapy used to prevent the
progression of DN (22,23). It has been shown that KLF4
regulates autophagy-associated gene expression by binding to the
promoter regions of the sequestosome-1 gene encoding the p62
protein in multiple myeloma (24).
The present study demonstrated that KLF4 overexpression increased
the expression of the autophagy protein, LC3, and the LC3-II/LC3-I
ratio, but decreased p62 expression in both DN mice and podocytes
stimulated with high glucose serum, suggesting that the activation
of autophagy may play an important role in ameliorating DKD
associated with KLF4.
It has been well-established that the mTOR signaling
pathway is involved in cellular growth, metabolism and the negative
regulation of autophagy; however, the role of mTOR signaling in
KLF4-mediated upregulation of autophagy in DN remains unknown. A
previous study indicated that the transient silencing of KLF4 in
mouse embryonic fibroblasts led to overactive mTOR activity, and
autophagy activity was not restored when rapamycin reduced the
level of mTOR (25). The present
in vitro study demonstrated that the overexpression of KLF4
inhibited mTOR and S6K phosphorylation, an important downstream
protein that leads to the activation of the mTORC1 pathway. The
present findings suggested that KLF4 plays a negative regulatory
role in podocyte mTOR signaling pathways.
In conclusion, the findings of the present study
indicated that KLF4 plays a renoprotective role in diabetic renal
injury, which is associated with the activation of podocyte
autophagy. It was further demonstrated that KLF4 overexpression
inhibited the mTOR/S6K signaling pathway. Therefore, the activation
of podocyte autophagy and inactivation of mTOR/S6K signaling
pathway via KLF4 may be an effective strategy for the treatment of
DN.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the Natural
Science Foundation of Zhejiang Province (grant nos. LZ17H050001,
LY16H050005 and Y18H050024), the Project of the Province and the
Ministry (grant no. WKJ-ZJ-1915), the Project of Scientific
Research Foundation of Chinese Medicine (grant nos. 2017ZA008,
2017ZA010 and 2016ZQ007) and the General Project of the Medical and
Health of Zhejiang Province (grant no. 2016KYA015).
Availability of data and materials
The datasets analyzed during the current study are
avaiable from the corresponding auhtor on reasonable request.
Authors' contributions
JJ and QH designed the study. YL proofread the
article and participated in data analysis. HZ and JG performed the
experiments. WZ performed the statistical analyses. JG wrote the
manuscript. The final version of the manuscript was approved by all
authors.
Ethics approval and consent to
participate
This study was approved by the local ethics
committee of Zhejiang Provincial People's Hospital. The study was
performed in accordance with the UK Animals (Scientific Procedures)
Act, 1986 and associated guidelines, and the EU Directive
2010/63/EU for animal experiments.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cunningham A, Benediktsson H, Muruve DA,
Hildebrand AM and Ravani P: Trends in biopsy-based diagnosis of
kidney disease: A population study. Can J Kidney Health Dis.
5:20543581187996902018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huang YM, Xu D, Long J, Shi Y, Zhang L,
Wang H, Levin A and Zhao MH: The spectrum of chronic kidney disease
in China: A national study based on hospitalized patients from 2010
to 2015. Nephrology (Carlton). 24:725–736. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pontrelli P, Oranger A, Barozzino M,
Divella C, Conserva F, Fiore MG, Rossi R, Papale M, Castellano G,
Simone S, et al: Deregulation of autophagy under hyperglycemic
conditions is dependent on increased lysine 63 ubiquitination: A
candidate mechanism in the progression of diabetic nephropathy. J
Mol Med (Berl). 96:645–659. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dai H, Liu Q and Liu B: Research progress
on mechanism of podocyte depletion in diabetic nephropathy. J
Diabetes Res. 2017:26152862017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yamaguchi Y, Iwano M, Suzuki D, Nakatani
K, Kimura K, Harada K, Kubo A, Akai Y, Toyoda M, Kanauchi M, et al:
Epithelial-mesenchymal transition as a potential explanation for
podocyte depletion in diabetic nephropathy. Am J Kidney Dis.
54:653–664. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ghaleb AM and Yang VW: Krüppel-like factor
4 (KLF4): What we currently know. Gene. 611:27–37. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoshida T, Yamashita M, Iwai M and Hayashi
M: Endothelial krüppel-like factor 4 mediates the protective effect
of statins against ischemic AKI. J Am Soc Nephrol. 27:1379–1388.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hayashi K, Sasamura H, Nakamura M, Azegami
T, Oguchi H, Sakamaki Y and Itoh H: KLF4-dependent epigenetic
remodeling modulates podocyte phenotypes and attenuates
proteinuria. J Clin Invest. 124:2523–2537. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gong J, Jin J, Zhao L, Li Y, Li Y and He
Q: Tripterygium glycoside protects against puromycin amino
nucleoside-induced podocyte injury by upregulating autophagy. Int J
Mol Med. 42:115–122. 2018.PubMed/NCBI
|
|
10
|
Tesch GH and Lim AK: Recent insights into
diabetic renal injury from the db/db mouse model of type 2 diabetic
nephropathy. Am J Physiol Renal Physiol. 300:F301–F3010. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mallipattu SK, Guo Y, Revelo MP, Roa-Peña
L, Miller T, Ling J, Shankland SJ, Bialkowska AB, Ly V, Estrada C,
et al: Krüppel-Like factor 15 mediates glucocorticoid-induced
restoration of podocyte differentiation markers. J Am Soc Nephrol.
28:166–184. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mallipattu SK, Estrada CC and He JC: The
critical role of Krüppel-like factors in kidney disease. Am J
Physiol Renal Physiol. 312:F259–F265. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vogelmann SU, Nelson WJ, Myers BD and
Lemley KV: Urinary excretion of viable podocytes in health and
renal disease. Am J Physiol Renal Physiol. 285:F40–F48. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Loeffler I and Wolf G:
Epithelial-to-mesenchymal transition in diabetic nephropathy: Fact
or fiction. Cells. 4:631–652. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lin L, Han Q, Xiong Y, Li T, Liu Z, Xu H,
Wu Y, Wang N and Liu X: Krüpple-like-factor 4 attenuates lung
fibrosis via inhibiting epithelial-mesenchymal transition. Sci Rep.
7:158472017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen WC, Lin HH and Tang MJ:
Matrix-stiffness-regulated inverse expression of Krüppel-like
factor 5 and Krüppel-like factor 4 in the pathogenesis of renal
fibrosis. Am J Pathol. 185:2468–2481. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pinho AV, Rooman I and Real FX:
p53-dependent regulation of growth, epithelial-mesenchymal
transition and stemness in normal pancreatic epithelial cells. Cell
Cycle. 10:1312–1321. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dong P, Kaneuchi M, Watari H, Hamada J,
Sudo S, Ju J and Sakuragi N: MicroRNA-194 inhibits epithelial to
mesenchymal transition of endometrial cancer cells by targeting
oncogene BMI-1. Mol Cancer. 10:992011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wada J and Makino H: Inflammation and the
pathogenesis of diabetic nephropathy. Clin Sci (Lond). 124:139–152.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Z, Jia Y, Han S, Wang X, Han F, Zhang
J, Zhang W, Guan H and Hu D: Klf4 alleviates
lipopolysaccharide-induced inflammation by inducing expression of
MCP-1 induced protein 1 to deubiquitinate TRAF6. Cell Physiol
Biochem. 47:2278–2290. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jin J, Wu D, Zhao L Zou W, Shen W, Tu Q
and He Q: Effect of autophagy and stromal interaction molecule 1 on
podocyte epithelial-mesenchymal transition in diabetic nephropathy.
Int J Clin Exp Pathol. 5:2450–2459. 2018.
|
|
22
|
Lenoir O, Jasiek M, Hénique C, Guyonnet L,
Hartleben B, Bork T, Chipont A, Flosseau K, Bensaada I, Schmitt A,
et al: Endothelial cell and podocyte autophagy synergistically
protect from diabetes-induced glomerulosclerosis. Autophagy.
11:1130–1145. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tagawa A, Yasuda M, Kume S, Yamahara K,
Nakazawa J, Chin-Kanasaki M, Araki H, Araki S, Koya D, Asanuma K,
et al: Impaired podocyte autophagy exacerbates proteinuria in
diabetic nephropathy. Diabetes. 65:755–767. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Riz I, Hawley TS and Hawley RG:
KLF4-SQSTM1/p62-associated prosurvival autophagy contributes to
carfilzomib resistance in multiple myeloma models. Oncotarget.
6:14814–14831. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu C, DeRoo EP, Stecyk C, Wolsey M,
Szuchnicki M and Hagos EG: Impaired autophagy in mouse embryonic
fibroblasts null for Krüppel-like Factor 4 promotes DNA damage and
increases apoptosis upon serum starvation. Mol Cancer. 14:1012015.
View Article : Google Scholar : PubMed/NCBI
|