Introduction
Patients with diabetes mellitus and chronic
hyperglycemia are at risk of developing complications, including
diabetic retinopathy (DR), nephropathy, neuropathy, cardiomyopathy,
rheumatoid arthritis and osteoporosis (1,2). The
pathogenesis of these complications is strongly associated with the
glycation of plasma proteins, which produces a large number of
advanced glycation end products (AGEs) (3,4).
Protein glycation interferes with their physiological function by
altering molecular conformation, enzymatic activity and receptor
functioning. AGEs interact with their membrane-localized receptors
[receptor for advanced glycation end products (RAGE)] and alter
intracellular signaling to influence various biological processes
within cells (5,6). The mechanisms by which accumulation
of AGEs cause DR has not yet been extensively studied.
Since its initial discovery as a proto-oncogene,
protein kinase B (Akt) has become a major focus of attention, due
to its critical involvement in cell apoptosis regulation,
angiogenesis, autophagy, transcription, protein synthesis and
glucose metabolism (7). The Akt
signaling cascade is activated by multiple cell receptors,
including receptor tyrosine kinases, B and T cell receptors,
cytokine receptors and G-protein coupled receptors (7). Other stimuli that induce the
production of phosphatidylinositol (3,4,5)-trisphosphates via phosphoinositide
3-kinase (PI3K) also induce activation of the Akt signaling cascade
(7). Inhibition of the
PI3K/Akt/mammalian target of rapamycin pathway has potential
therapeutic function in DR pathophysiology (8). However, whether the PI3K/Akt pathway
influences the function of AGEs during the development of DR
remains unclear. In the present study, the mechanisms of AGEs in DR
were investigated. Specifically, the role of Akt signaling in DR
pathophysiology was examined.
Materials and methods
Cell culture
Primary human retinal capillary endothelial cells
(HRCECs) and primary human Müller cells were purchased from the
Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). Müller cells were cultured in high-glucose
Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 100 U/ml penicillin, 100 µg/ml
streptomycin and 10% fetal bovine serum (FBS; Thermo Fisher
Scientific, Inc.). HRCECs were cultured in RMPI-1640 medium (Thermo
Fisher Scientific, Inc.) with the same supplements. Cells were
sub-cultured until 80% confluence was reached. A gradient of AGEs
conjugated to bovine serum albumin (0, 25, 50 and 100 µg/ml; cat.
no. 121800; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was used
to treat the cells for 24 h at 37°C. LY294002 (1 µM; Sigma-Aldrich;
Merck KGaA), a PI3K inhibitor (PI3Ki), was also used to treat cells
for 6 h at 37°C. Akt inhibitor (Akti; 5 µM; Akt 1/2 kinase
inhibitor; cat. no. sc-300173; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) was used to block Akt function in cells (cells
were treated for 6 h at 37°C).
MTT assay
Following treatment, cell media was removed by
careful aspiration. Subsequently, 50 µl serum-free media and 50 µl
MTT solution was added into each well (104 per ml). The
plate was incubated at 37°C for 3 h. Following incubation, 150 µl
MTT solvent (DMSO) was added into each well. The plate was then
warmed in foil and shaken on an orbital shaker for 15 min. The
plate was read at an absorbance of 590 nm after being maintained in
the incubator for 1 h.
Western blotting
In brief, cells were collected and lysed in
radioimmunoprecipitation assay lysis buffer with proteinase
inhibitor (20 nM) and phosphatase inhibitor (20 nM; Thermo Fisher
Scientific, Inc.). The extracted protein was measured and
normalized with a bicinchoninic acid protein assay. Equivalent
amounts of total proteins (30 µg per lane) were subsequently
separated by electrophoresis on 4–20% gradient SDS-PAGE gels and
were transferred to a polyvinylidene fluoride membrane by a
half-dry transferring system. Blocking was performed by incubating
the membrane with 5% BSA in TBST for 15 min at room temperature.
The following primary antibodies were used for target proteins:
RAGE (1:1,000; cat. no. ab37647; Abcam, Cambridge, MA, USA), Akt
(1:1,000; cat. no. ab126811; Abcam), phosphorylated (p)-Akt
(1:1,000; cat. no. ab81283; Abcam), PI3K (1:1,000; cat. no.
ab86714; Abcam), and p-PI3K (1:1,000; cat. no. ab182651; Abcam) and
GAPDH (1:1,000; cat. no. ab181602; Abcam). The primary antibodies
were incubated with the polyvinylidene fluoride membrane at 4°C
overnight. Horseradish peroxidase-conjugated secondary antibody
(1:5,000; cat. nos. ab6721 and ab6728; Abcam) were added to the
membrane to incubated for 1 h at room temperature. Enhanced
chemiluminescence western blotting detection reagents (Thermo
Fisher Scientific, Inc.) were used for imaging. ImageJ software
1.52a (National Institutes of Health, Bethesda, MD, USA) was used
for densitometry.
Invasion assay
Transwells coated with Matrigel (Corning
Incorporated, Corning, NY, USA) with an 8 µm polycarbonate filter
membrane were used for the invasion assay. HRCECs
(3×104) were added into the upper chamber with
serum-free medium (cat. no. 211; Sigma-Aldrich; Merck KGaA). The
lower chamber was filled with 5% serum medium (cat. no. 211;
Sigma-Aldrich; Merck KGaA). HRCECs were subsequently treated with
AGEs (100 µg/ml) and Akti (5 µM) for a 24 h incubation at 37°C.
Cells on the top surface of the filter were removed, and the
remaining cells on the underside of the filter were subsequently
fixed with the fixation buffer (0.1 M sodium cacodylate buffer
supplemented with 4% paraformaldehyde, 2.5% glutaraldehyde and
0.02% picric acid) at room temperature for 1 h, and stained with 5%
crystal violet for 10 min at room temperature. The stained membrane
was washed with PBS three times and the invaded cell number on the
membrane was counted using a light microscope.
BrdU incorporation assay
BrdU incorporation assay was performed to assess the
cell proliferation. A commercial BrdU cell proliferation assay kit
(cat. no. 6813; Cell Signaling Technology, Inc., Danvers, MA, USA)
was used and the manufacture's protocol was carefully followed.
Briefly, 5×104 cells were seeded in 96-well plate 24 h
before the BrdU incorporation assay. Then BrdU solution was added
to the plate well and incubated for 12 h at 37°C. Then, the cells
were collected by centrifuging the plate at 300 × g for 10 min at
4°C. Fixing/denaturing solution was added to each well (100
µl/well) and the plate was kept at room temperature for 30 min.
Following the removal of the fixing/denaturing solution, detection
antibody solution from the kit was added for incubation at room
temperature for 1 h. HRP-conjugated secondary antibody solution was
added to incubate at room temperature for 30 min. Absorbance at 450
nm was read after the stop solution was added.
Flow cytometry
To detect cell apoptosis and necrosis, an Annexin V
and propidium iodide (PI) staining method was utilized. A
commercial kit (cat. no. V1324; Thermo Fisher Scientific, Inc.) was
used in accordance with the manufacturer's protocols. Cells were
washed twice with cold PBS and subsequently resuspended in 1X
binding buffer. Then, 105 cells in 100 µl
annexin-binding buffer were transferred to a 5 ml culture tube and
1 µl annexin V-fluorescein isothiocyanate (FITC) and 2 µl PI was
added to the tube. Samples were gently mixed and incubated for 15
min at room temperature in the dark. Subsequently, 400 µl 1×
binding buffer was added to each tube. Samples were analyzed by
flow cytometry within 30 min following staining. A FACSCanto II
machine (BD Biosciences, Franklin Lakes, NJ, USA) was used.
In additon, the expression of glial fibrillary
acidic protein (GFAP) on Müller cells and cluster of
differentiation (CD)34 protein on HRCECs was measured by flow
cytometry. Cell pellets of Müller cells and HRCECs were harvested
following centrifugation at 500 × g and 5 min at 4°C. Then an equal
number (105) of these cells were collected for
subsequent steps. They were blocked with PBS with 5% bovine serum
albumin (BSA; cat. no. A2058; Sigma-Aldrich; Merck KGaA) for 10 min
at room temperature. Then mouse antibodies of GFAP (1:200; cat. no.
G3893, Sigma-Aldrich; Merck KGaA) and CD34 (1:200; cat. no.
SAB4700160, Sigma-Aldrich; Merck KGaA) were incubated with Müller
cells and HRCECs for 10 min at room temperature, respectively. To
measure the expression of cleaved caspase-3, the 105
harvested HRCECs and Müller cells were washed in cold PBS once, and
2 ml cold 4% paraformaldehyde (Sigma-Aldrich; Merck KGaA).
Following washing with cold PBS for 5 min, permeabilization buffer
(cat. no. 22016, Biotium, Inc.) was added to incubate at 4°C for 30
min. Cells were washed in PBS prior to the addition of cleaved
caspase-3 (1:100; cat. no. ab2302, Abcam) antibodies to the cells.
Following incubation with GFAP, CD34 or cleaved caspase-3
antibodies for 30 min at 4°C, the cells were washed by PBS.
Subsequently, FITC-labeled anti-mouse secondary antibodies
(1:5,000, cat. no. SAB3701081; Sigma-Aldrich; Merck KGaA) and
APC-preadsorbed anti-rabbit secondary antibodies (1:5,000, cat. no.
ab130805, Abcam) were added to the cells to incubate for 20 min at
room temperature. Following three washes in PBS, cells were
analyzed using the FACSCanto II machine (BD Biosciences) and FlowJo
software (version 10.0.7, FlowJo LLC, Ashland, OR, USA) was used to
analyze the data.
Tube formation assay
Prior to the tube formation assay, HRCECs
(1×105 cells/well) were starved in medium containing 1%
FBS for 12 h at 37°C. Cells were incubated with different
concentrations of AGEs for another 12 h at 37°C. Cells were
subsequently seeded into 24 well plates pre-coated with Matrigel
(BD Biosciences) and treated with AGEs (50 µg/ml) or Akti (5 µM)
for 8 h at 37°C. Images were captured under an inverted microscope
at 200× magnification and 5 fields were randomly selected for
assessment.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
PCR was performed to detect expression of vascular
endothelial growth factor (VEGF), VEGF receptor (VEGFR), pigment
epithelium-derived factor (PEDF), angiopoietin (Ang) 1, Ang 2,
fibroblast growth factor (FGF), platelet-derived growth factor
(PDGF) and 18S rRNA. The primers used for these genes are listed in
Table I. RNA was isolated from the
cell lines according to the manufacturer's protocol using TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc.). Total RNA (1 µg) was
reverse transcribed using High Capacity cDNA Reverse Transcription
kit (Thermo Fisher Scientific, Inc.). PCR was performed on a
LightCycler II 480 (Roche Diagnostics, Basel, Switzerland) with
SYBR Green I dye (Roche Diagnostics). The results were normalized
to the housekeeping 18S rRNA gene.
 | Table I.Primers used for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primers used for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| 18S rRNA |
CTACCACATCCAAGGAAGCA |
TTTTTCGTCACTACCTCCCCG |
| VEGF |
GTCCGATTGAGACCCTGGTG |
ACCGGGATTTCTTGCGCTTT |
| VEGFR |
GTGTCTATAGGTGCCGAGCC |
CGGAAGAAGACCGCTTCAGT |
| Ang1 |
TCAGCCTTTGCACTAAAGAAGTTT |
GGCCCTTTGAAGTAGTGCCA |
| Ang2 |
AAGGAAGCCCTTATGGACGA |
CCAGCCATTCTCACAGCCAA |
| FGF |
CACTTTCCCAGGAGGATGGAG |
TCCCCAGCTGAGAAGACACT |
| PDGF |
TACTGAATTTCGCCGCCACA |
GGAGGAGAACAAAGACCGCA |
| PEDF |
TACTCCTCTGGACTGGAGCC |
TGGATCTCAGGCGGTACAGA |
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 5.0 (GraphPad Software, Inc., CA, USA). All experiments were
repeated 3 times. Data were expressed as the mean ± standard error
of the mean. Two-tailed Student's t-test was used to evaluate the
significance of differences between two groups. One-way analysis of
variance was used to compare results with more than three groups.
Tukey's post-hoc test was performed for multiple comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
RAGE expression is upregulated in
HRCECs and Müller cells in response to AGE treatment
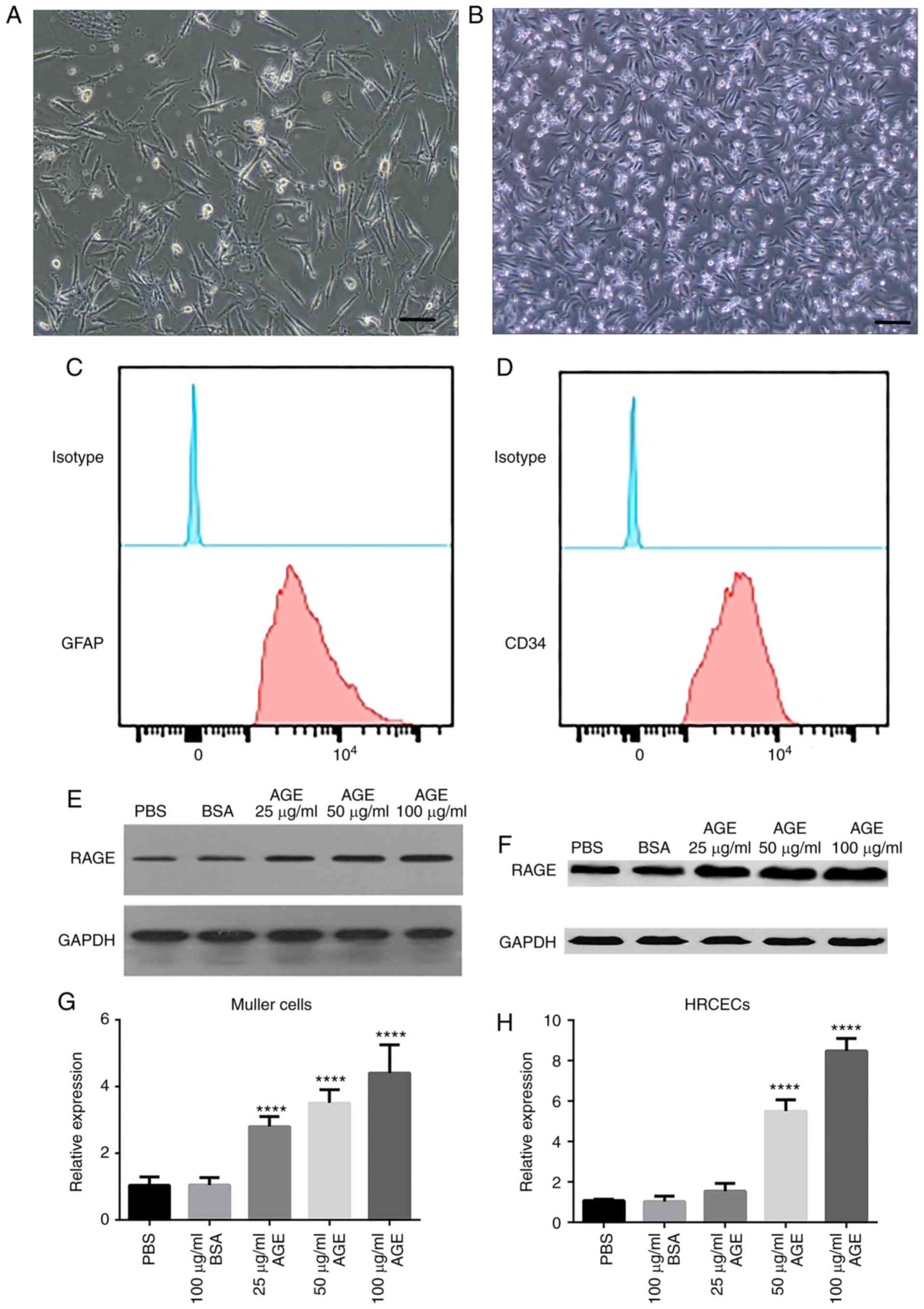
To investigate the effects of AGEs on DR, primary
Müller cells (Fig. 1A) and HRCECs
(Fig. 1B) were cultured in
vitro, and the expression of RAGE was measured. Biomarkers of
human retinal capillary endothelial cells (CD34) and Muller cells
(GFAP) were assessed by flow cytometry. These two biomarkers were
highly expressed in the two kinds of cells, respectively (Fig. 1C and D). The results revealed that
there was a basal level of RAGE expression in Müller cells
(Fig. 1E) and HRCECs (Fig. 1F). Following AGE treatment at 100
µg/ml, RAGE expression was significantly upregulated in the two
cell cultures by more than five-fold, compared with the untreated
cells (P<0.0001; Fig. 1G and
H). The increase in RAGE expression was proportional to the
concentration of AGE treatment. These data provided initial
verification that Müller cells and HRCECs were responsive to AGE
treatment.
Akt inhibition reduces HRCEC
viability
As the expression of RAGE was upregulated by the AGE
treatment in Müller cells and HRCECs, the biological effects of AGE
treatment (100 µg/ml) were examined. The cell viability assay
results demonstrated that the AGE treatment did not exert a
significant effect on Müller cell viability (Fig. 2A), but enhanced HRCEC viability
(Fig. 2B). The PI3K/Akt pathway
has a profound impact on cell proliferation and survival (8). Müller cells and HRCECs were treated
with PI3Ki and Akti, and it was noticed that Akti significantly
reduced the effects induced by AGE treatment (P<0.0001; Fig. 2B), although PI2Ki did not notably
influence the effect of AGE treatment. Western blotting analysis
confirmed that AGE treatment upregulated Akt phosphorylation, but
not PI3K phosphorylation (Fig. 2C and
D).
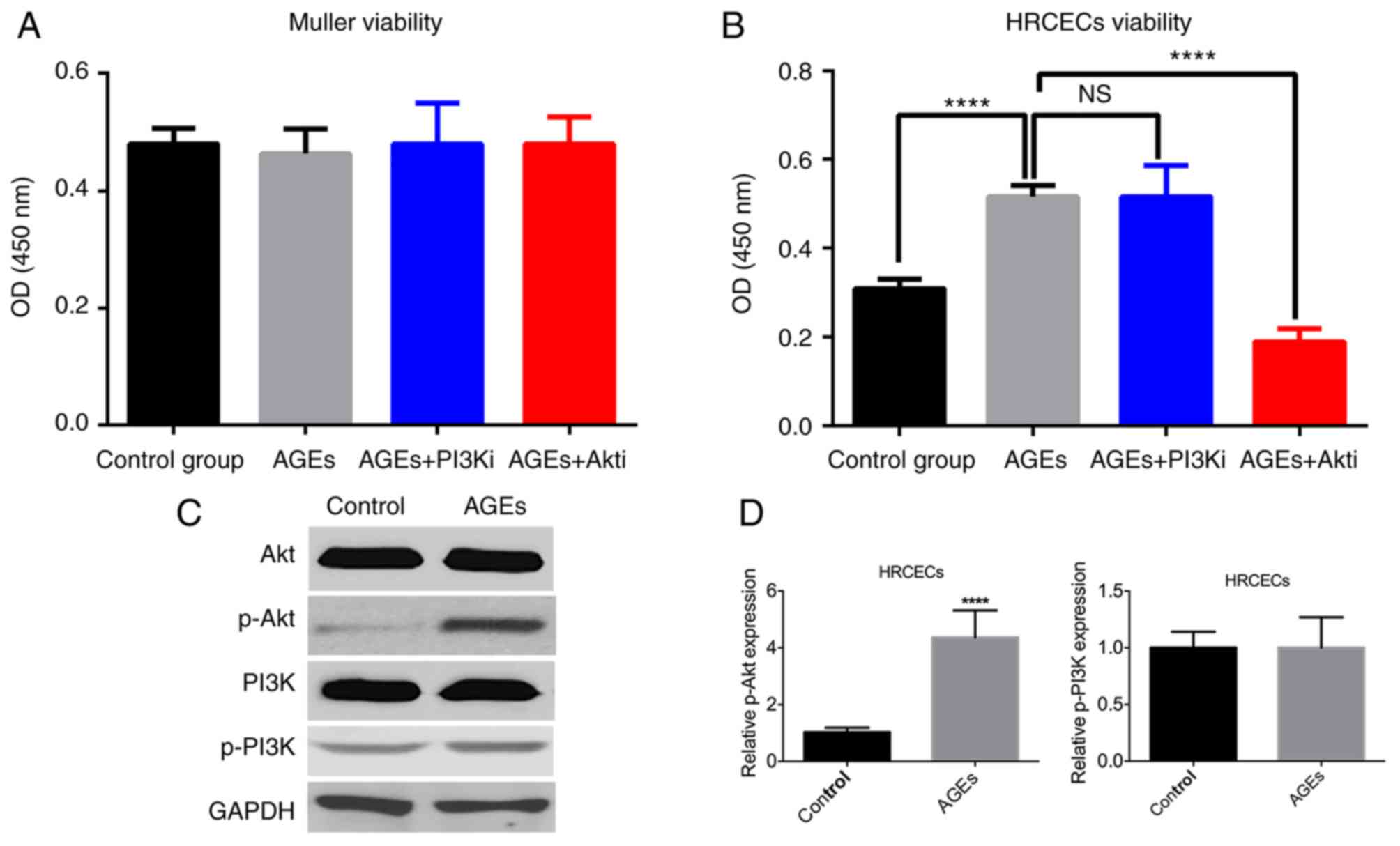 | Figure 2.AGE treatment increases HRCEC
viability. (A) Müller cell viability was not altered by treatment.
(B) AGE treatment enhanced HRCEC viability, whereas Akti treatment
suppressed it. (C) AGE treatment stimulated Akt phosphorylation,
but not PI3K phosphorylation. (D) The differences in Akt
phosphorylation were statistically significant, but that of the
PI3K phosphorylation was not. ****P<0.0001 vs. control group or
the AGEs treated group. AGE, advanced glycation end products;
HRCEC, human retinal capillary endothelial cell; PI3K,
phosphoinositide 3-kinase; Akt, protein kinase B; p,
phosphorylated; i, inhibitor; NS, not significant. |
Akt inhibition suppresses HRCEC
proliferation and induces apoptosis
To understand the mechanisms underlying the
alterations in cell viability as a result of AGE and Akti treatment
in HRCECs, cell proliferation and apoptosis was measured via a BrdU
incorporation assay and flow cytometry, respectively. Flow
cytometry demonstrated that Akti treatment markedly increased the
cell death rate in HRCECs (Fig.
3A). The expression of full length and cleaved caspase-3 was
also quantified in Müller cells and HRCECs following treatment. As
presented in Fig. 3B, neither the
AGE treatment nor the Akti treatment stimulated cell apoptosis in
Müller cells. However, in HRCECs, the Akti treatment induced
cleaved caspase-3 expression (Fig.
3B). The BrdU incorporation assay detected cell proliferation.
In HRCECs, AGE treatment significantly increased cell
proliferation, and Akti treatment suppressed cell proliferation
(Fig. 3C). These results indicated
that AGE treatment enhanced HRCEC viability by increasing cell
proliferation, and that Akti treatment reduced HRCEC viability via
suppressing cell proliferation, as well as via induction of
apoptosis.
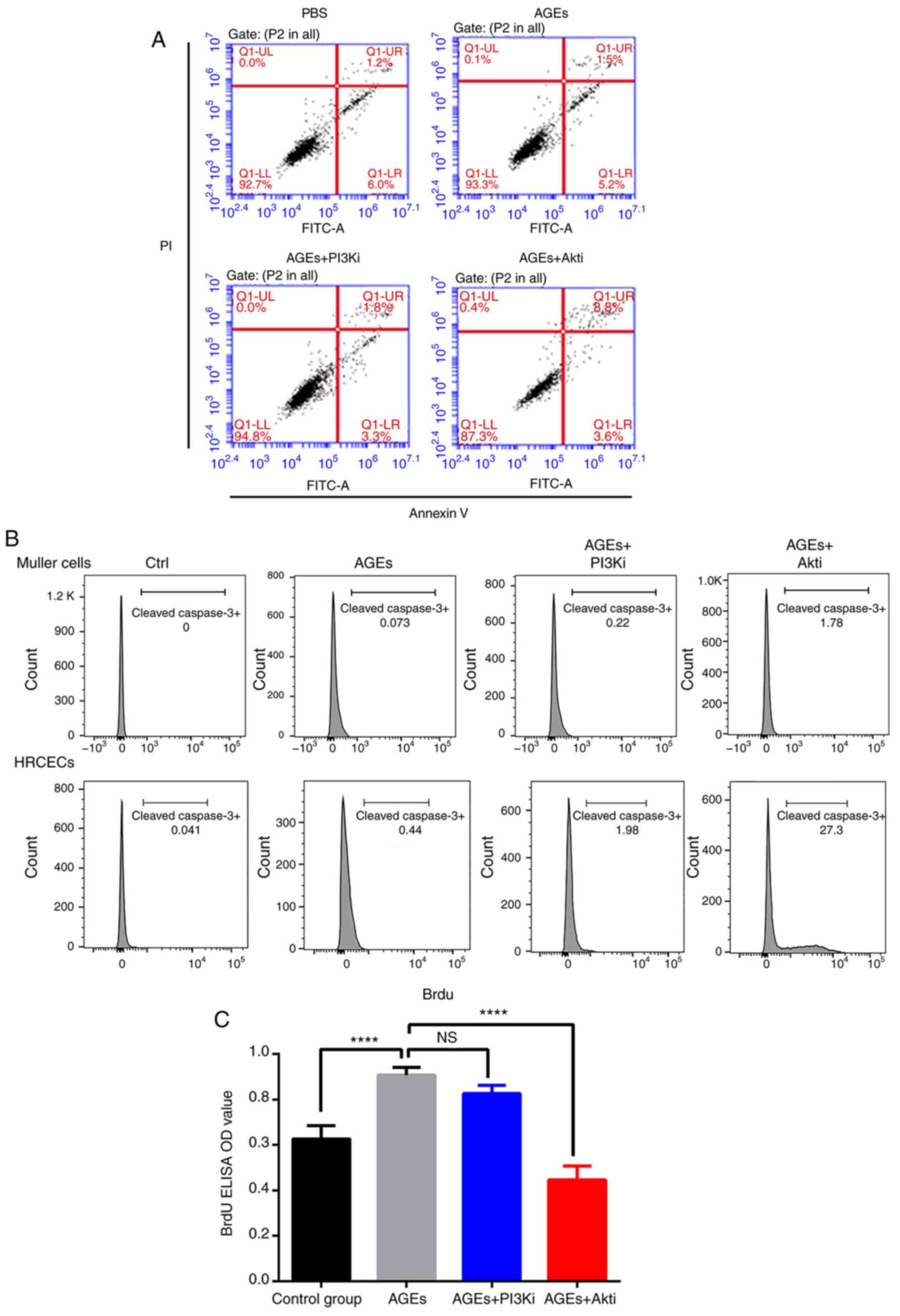 | Figure 3.Akti induces HRCEC apoptosis and
suppresses proliferation. (A) Flow cytometry analysis of HRCEC
apoptosis when treated with AGE, PI3Ki and Akti. Akti treatment
increased the number of apoptotic cells (the upper right quadrant
was designated as apototic cells). (B) Flow cytometry analysis of
cleaved caspase-3 expression in treated HRCECs and Müller cells.
(C) BrdU ELISA indicated that AGE treatment increased HRCEC
proliferation, and Akt inhibitor treatment decreased HRCEC
proliferation. ****P<0.0001 vs. AGEs treated group. Ctrl,
control; BSA, bovine serum albumin; ELISA, enzyme-linked
immunosorbent assay; Q, quadrant; FITC, fluorescein isothiocyanate;
PI, propidium iodide; AGE, advanced glycation end products; HRCECs,
human retinal capillary endothelial cells; NS, not significant; OD,
optical density; PI3K, phosphoinositide 3-kinase; Akt, protein
kinase B; i, inhibitor. |
Akt inhibition reduces
angiogenesis-associated gene expression
The angiogenic function of HRCECs was further
investigated by quantifying the relative expression of
angiogenesis-associated genes using RT-qPCR, including the
pro-angiogenic genes Ang1, Ang2, VEGF, VEGFR, PDGF and FGF, as well
as the anti-angiogenic gene PEDF. The expression level was
normalized by calculating the z scores. Expression levels of these
pro-angiogenic genes were upregulated by AGEs treatment, whereas
PEDF expression was downregulated (Fig. 4). Furthermore, Akti treatment
suppressed pro-angiogenic gene expression induced AGE
treatment.
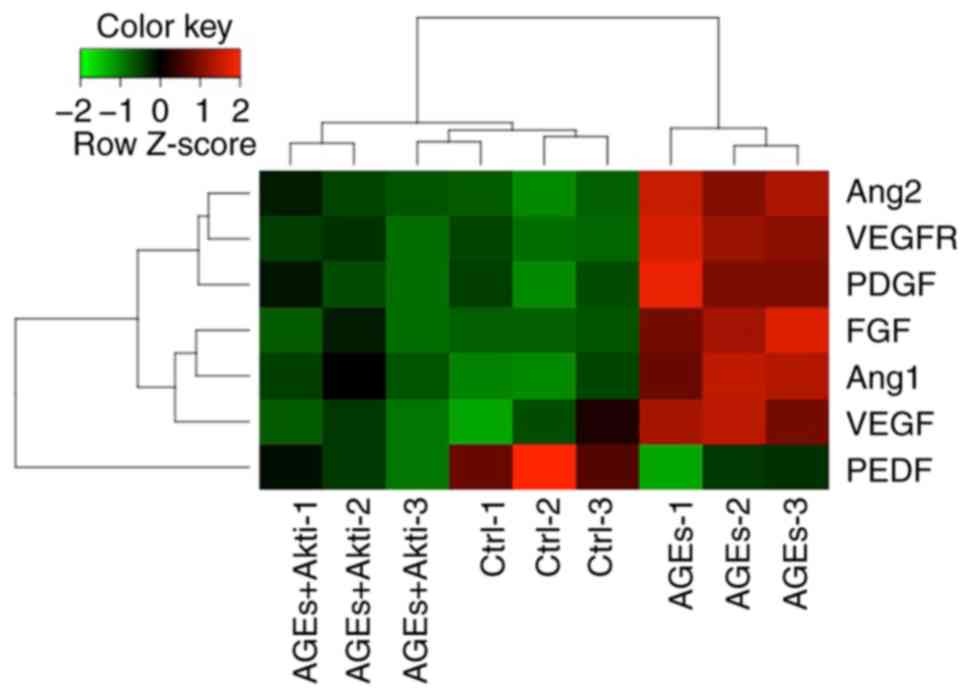 | Figure 4.Expression of angiogenesis-associated
genes in human retinal capillary endothelial cells. mRNA expression
of was plotted as a heatmap. Red indicates relatively high
expression level, and green indicates a relatively low expression
level. AGE, advanced glycation end products; Ctrl, control; VEGF,
vascular endothelial growth factor; VEGFR, vascular endothelial
growth factor receptor; PEDF, pigment epithelium-derived factor;
Ang1, angiopoietin 1; Ang2, angiopoietin 2; FGF, fibroblast growth
factor; PDGF, platelet-derived growth factor; PI3K,
phosphoinositide 3-kinase; Akt, protein kinase B; i, inhibitor;
Ctrl, control. |
Akt inhibition suppresses the tube
formation ability of HRCECs
In addition to measuring the expression of
angiogenesis-associated genes, functional assays were performed to
further assess the HRCEC angiogenesis. In the in vitro
invasion assay, HRCECs treated with AGE (100 µg/ml) had the highest
invasive ability (Fig. 5A),
whereas the AGEs and Akti-treated HRCECs exhibited a comparable
invasive ability to control cells (Fig. 5A). A similar trend was observed in
the tube formation assay (Fig.
5B). Quantitative analysis revealed that these differences were
significant (P<0.01; Fig. 5C and
D). These data, together with the angiogenic gene expression
data, suggested that AGEs had a pro-angiogenic effect in HRCECs,
and Akt inhibition had an anti-angiogenic effect.
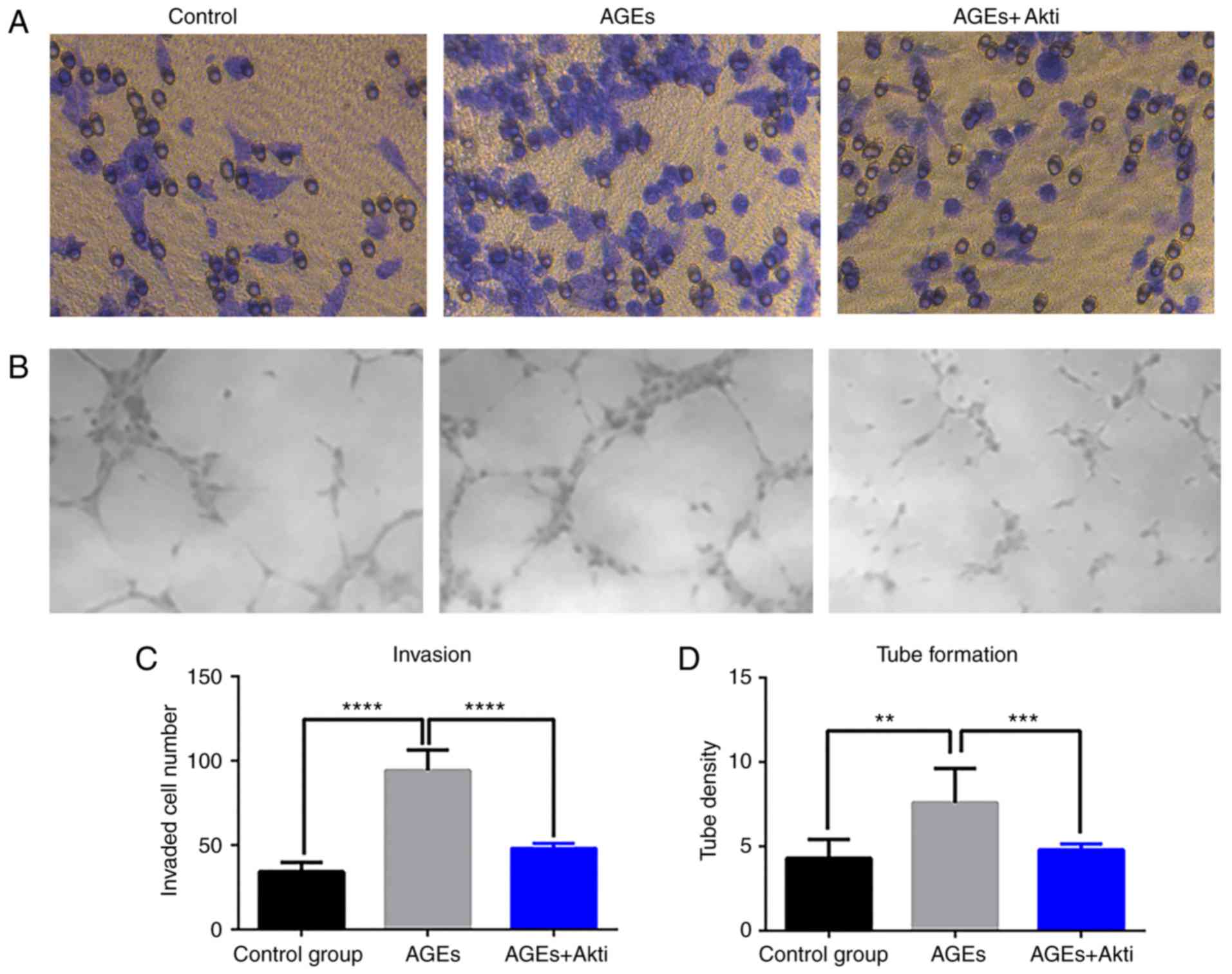 | Figure 5.Invasive and tube forming ability of
HRCECs. (A) Representative results of HRCEC invasion in control
group following AGE and Akti treatment. AGE treatment significantly
increased HRCECs invasion, and Akt inhibitor reduced this effect.
(B) The tube formation ability of HRCECs was measured in control
HRCECs, AGEs treated HRCECs, and Akt inhibitor-treated HRCECs.
Magnification, ×200. (C) Differences in invasive and (D) tube
formation ability were statistically significant. **P<0.01,
***P<0.001, ****P<0.0001 vs. the AGEs treated group. HRCECs,
human retinal capillary endothelial cells; AGE, advanced glycation
end products; PI3K, phosphoinositide 3-kinase; Akt, protein kinase
B; i, inhibitor. |
Discussion
Patients with a long history of diabetes mellitus
frequently have comorbidities, including DR and cardiomyopathy,
which are highly associated with a chronic hyperglycemic state
(1,2,9).
Abnormal, newly formed blood vessels grow from the retina and
result in subsequent tractional retinal detachment and hemorrhage,
which is the major concern of DR (9–11).
Although the underlying mechanisms of aberrant angiogenesis in DR
remains unclear, evidence has demonstrated AGE accumulation is a
major factor that contributes to angiogenesis (12). AGEs interact with RAGEs, therefore
altering intracellular signaling and influencing several essential
biological processes within cells (3–6). The
exact pathological mechanisms of AGE-induced angiogenesis in DR
remain to be elucidated.
Müller cells are a type of retinal glial cell that
have been demonstrated to be critical to retinal development, by
serving as promoters of retinal growth and histogenesis (13). HRCECs are the major cell type that
causes vascularization in DR (14). In the present study; the effects of
AGEs on these two cell types were examined. Initially, the
expression of RAGEs in Müller cells and HRCECs following AGE
treatment was investigated. These two cell types expressed a basal
level of RAGEs without any treatment. When treated with AGEs, they
expressed a much higher level of RAGEs, compared with the control
group. The effects of AGEs on Müller cells and HRCEC viability were
subsequently examined. AGE treatment significantly enhanced HRCEC
viability. The PI3K/Akt pathway has critical roles in regulating
cell proliferation and death, and has been demonstrated to be
involved in a number of AGE-associated biological processes,
including autophagy and cell migration (15,16).
When cells were treated with PI3Ki, however, no obvious alterations
in cell viability were observed in Müller cells or HRCECs. When
cells were treated with Akti, the viability of HRCECs was
dramatically suppressed, whereas Müller cell viability was not
influenced. The following cell proliferation and apoptosis assays
confirmed the findings of the cell viability assay, and suggested
that AGE facilitated the survival of HRCECs in an Akt-dependent
way. These initial results suggested that the AGEs primarily
influenced HRCECs, rather than Müller cells.
Retinal vascularization is a coordinated
collaboration involving several cell types, including endothelial
cells, pericytes and astrocytes, and a dynamic balance of positive
and negative regulatory factors (17–19).
In angiogenesis-associated DR, this delicate balance is disturbed
(10). Since AGEs regulate HRCEC
proliferation via Akt, whether or not AGEs regulate the
vascularization of the retina was investigated. By measuring the
expression of genes associated with angiogenesis, it was
demonstrated that the AGE treatment induced a pro-angiogenic gene
expression, including VEGF, VEGFR, FGF, Ang1, Ang2 and PDGF. In
addition, PEDF expression was decreased. Treatment with Akti
inhibited these effects. Furthermore, the HRCEC invasion and tube
formation assays also indicated the Akt-dependent pro-angiogenic
effects of AGEs.
Studies of HRCECs have shed light on the earliest
stages of DR and other diseases of the retinal microvasculature
(20–22). However, whether or not the PI3K/Akt
pathway influences AGE-mediated DR development remains unclear. The
present study reported that AGEs induced HRCEC proliferation and
angiogenesis via an Akt-dependent mechanism. Inhibiting the Akt
pathway prevented the effects of AGEs on HRCEC proliferation and
vascularization. Therefore, targeted therapies that suppress Akt
function may be a promising treatment for retinal
vascularization-associated diseases, including DR.
Acknowledgements
The authors acknowledge the support from the First
People's Hospital of Yunnan Province during the present study.
Funding
The present study was supported by the Health
Science Program of Yunnan Province (grant no. 2016NS238) and the
Health Science Profession Training Program of Kunming, Yunnan
Province (grant no. 2016-sw-66).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
DT was responsible for study design, major
experiments, data analysis and manuscript preparation. NN was
responsible for experiments, data analysis and manuscript
preparation. TZ conducted some experiments, and performed data
analysis and manuscript preparation. CL was responsible for
literature review, data analysis, and manuscript preparation and
revision. QS was responsible for data interpretation, manuscript
revision, and data collection. LW performed some experiments and
was responsible for manuscript preparation and revision. YM was
responsible for funding collection, study design and manuscript
revision.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tripathi BK and Srivastava AK: Diabetes
mellitus: Complications and therapeutics. Med Sci Monit.
12:RA130–RA147. 2006.PubMed/NCBI
|
|
2
|
Bos M and Agyemang C: Prevalence and
complications of diabetes mellitus in Northern Africa, a systematic
review. BMC Public Health. 13:3872013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vlassara H and Uribarri J: Advanced
glycation end products (AGE) and diabetes: Cause, effect, or both?
Curr Diab Rep. 14:4532014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nowotny K, Jung T, Höhn A, Weber D and
Grune T: Advanced glycation end products and oxidative stress in
type 2 diabetes mellitus. Biomolecules. 5:194–222. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xie J, Méndez JD, Méndez-Valenzuela V and
Aguilar-Hernández MM: Cellular signalling of the receptor for
advanced glycation end products (RAGE). Cell Signal. 25:2185–2197.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ott C, Jacobs K, Haucke E, Navarrete
Santos A, Grune T and Simm A: Role of advanced glycation end
products in cellular signaling. Redox Biol. 2:411–429. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Toker A and Marmiroli S: Signaling
specificity in the Akt pathway in biology and disease. Adv Biol
Regul. 55:28–38. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jacot JL and Sherris D: Potential
therapeutic roles for inhibition of the PI3K/Akt/mTOR pathway in
the pathophysiology of diabetic retinopathy. J Ophthalmol.
2011:5898132011.PubMed/NCBI
|
|
9
|
Yau JW, Rogers SL, Kawasaki R, Lamoureux
EL, Kowalski JW, Bek T, Chen SJ, Dekker JM, Fletcher A, Grauslund
J, et al: Global prevalence and major risk factors of diabetic
retinopathy. Diabetes Care. 35:556–564. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tarr JM, Kaul K, Chopra M, Kohner EM and
Chibber R: Pathophysiology of diabetic retinopathy. ISRN
Ophthalmol. 2013:3435602013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Antonetti DA, Klein R and Gardner TW:
Diabetic retinopathy. N Engl J Med. 366:1227–1239. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zong H, Ward M and Stitt AW: AGEs, RAGE,
and diabetic retinopathy. Curr Diab Rep. 11:244–252. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Goldman D: Müller glial cell reprogramming
and retina regeneration. Nat Rev Neurosci. 15:431–442. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shin ES, Sorenson CM and Sheibani N:
Diabetes and retinal vascular dysfunction. J Ophthalmic Vis Res.
9:362–373. 2014.PubMed/NCBI
|
|
15
|
Qin Q, Niu J, Wang Z, Xu W, Qiao Z and Gu
Y: Heparanase induced by advanced glycation end products (AGEs)
promotes macrophage migration involving RAGE and PI3K/AKT pathway.
Cardiovasc Diabetol. 12:372013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hou X, Hu Z, Xu H, Xu J, Zhang S, Zhong Y,
He X and Wang N: Advanced glycation endproducts trigger autophagy
in cadiomyocyte Via RAGE/PI3K/AKT/mTOR pathway. Cardiovasc
Diabetol. 13:782014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Smith LE, Shen W, Perruzzi C, Soker S,
Kinose F, Xu X, Robinson G, Driver S, Bischoff J, Zhang B, et al:
Regulation of vascular endothelial growth factor-dependent retinal
neovascularization by insulin-like growth factor-1 receptor. Nat
Med. 5:1390–1395. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Majka S, McGuire PG and Das A: Regulation
of matrix metalloproteinase expression by tumor necrosis factor in
a murine model of retinal neovascularization. Invest Ophthalmol Vis
Sci. 43:260–266. 2002.PubMed/NCBI
|
|
19
|
Gupta N, Mansoor S, Sharma A, Sapkal A,
Sheth J, Falatoonzadeh P, Kuppermann B and Kenney M: Diabetic
retinopathy and VEGF. Open Ophthalmol J. 7:4–10. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lois N, McCarter RV, O'Neill C, Medina RJ
and Stitt AW: Endothelial progenitor cells in diabetic retinopathy.
Front Endocrinol (Lausanne). 5:442014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Joussen AM, Murata T, Tsujikawa A,
Kirchhof B, Bursell SE and Adamis AP: Leukocyte-mediated
endothelial cell injury and death in the diabetic retina. Am J
Pathol. 158:147–152. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kady N, Yan Y, Salazar T, Wang Q,
Chakravarthy H, Huang C, Beli E, Navitskaya S, Grant M and Busik J:
Increase in acid sphingomyelinase level in human retinal
endothelial cells and CD34+ circulating angiogenic cells isolated
from diabetic individuals is associated with dysfunctional retinal
vasculature and vascular repair process in diabetes. J Clin
Lipidol. 11:694–703. 2017. View Article : Google Scholar : PubMed/NCBI
|