Introduction
DNA double-strand breaks (DSBs) are the most harmful
type of DNA lesions and can affect various processes including cell
cycle progression, genomic stability and the induction of
tumorigenesis. There are two distinct mechanisms of DNA DSB repair:
Homologous recombination (HR) and non-homologous end-joining (NHEJ)
(1). Three major DNA
damage-activated PI3K-related serine/threonine protein kinases,
DNA-protein kinase (-PK), ataxia-telangiectasia mutated (ATM), and
ATR serine/threonine kinase (ATR) (2), participate in repair pathways. The
DNA-dependent protein kinase catalytic subunit (DNA-PKcs) plays an
important role during the repair of DNA DSBs. The
autophosphorylation of DNA-PKcs represents the activation of DNA-PK
and regulates its own dynamics at DNA DSBs (3).
Posttranslational modifications of proteins such as
ubiquitination, neddylation, acetylation and polyADP-ribosylation
are significant mechanisms that regulate many cellular processes.
PARylation plays crucial roles in DNA repair, replication,
transcription and cell death (4–6). The
poly(ADP-ribosylation) reaction, in which DNA-dependent
poly(ADP-ribose) (PAR) is synthesized from nicotinamide
mononucleotide (NAD) by poly(ADP-ribose) polymerases (PARPs) and
poly(ADP-ribose) glycohydrolase (PARG), regulates the hydrolysis of
PAR and was discovered in 1963 (7,8).
After cells are exposed to ionizing radiation, free radicals and
alkylating agents, PARP1 binds rapidly to DNA DSB sites, resulting
in PAR modification. This process uses NAD+ as a
substrate and leads to the formation of poly(ADP-ribose) polymers
on target proteins, and intracellular NAD+ is depleted
in this process. However, poly(ADP-ribose) has a short half-life
in vivo since it is rapidly degraded by PARG (2–5 min after
polymer formation) (9). The PARP
family has 16 members, but only PARP1 and PARP2 are closely
associated with DSBs (10).
Furthermore, PARP1, a 116 kDa protein, contains a DNA binding
domain, a central auto-modification domain and a C-terminal
catalytic domain (11,12) and has 18 distinct isoforms in
humans (13). PARP1 is more
important than PARP2 in DSB repair as PARP1 affects several key HR
factors, including BRCA1, exonuclease 1 and BRCA2, and acts as a
stress sensor and a stress response mediator in biological systems
(14). PARP1 has been reported to
mediate MRN complex recruitment to DSBs in a γ-histone family
member 2AX (H2AX)- and mediator of DNA damage checkpoint protein
1-independent manner (15). PARP1
and MRN together mediate ATM accumulation and the phosphorylation
of H2AX, and stabilize the DNA damage response factor at the DNA
damage site (16). PARP1
substrates include PARP1 itself, histones, DNA repair proteins,
transcription factors and chromatin modulators (17). PARP1 poly-ADP-ribosylates BRCA1,
targeting its DNA binding domain and reducing its affinity for DNA
(18). DNA-PKcs was previously
reported to be modified by IFNγ-induced PARylation (18). However, it is unclear how PARP1
affects DNA-PKcs in the DNA damage response.
The present study identified the PAR modification of
DNA-PKcs after DNA damage. The inhibition of PARylation increases
the chromatin binding of DNA-PKcs and DNA-PKcs Ser2056
phosphorylation, and the synergistic inhibition of PARylation and
DNA-PK activity suppresses cell survival.
Materials and methods
Cell culture and transfection
Hela cells were purchased from American Type Culture
Collection. These cells were cultivated at 37°C in a humidified
incubator containing 5% CO2. The cells were grown in
DMEM supplemented with 10% FBS (Gibco; Thermo Fisher Scientific,
Inc.), penicillin and streptomycin.
Antibodies and chemicals
The following specific antibodies were used in the
present study: PAR (Abcam; cat. no. ab14459), DNA-PKcs (Invitrogen;
Thermo Fisher Scientific, Inc.; MA5-13238), mouse IgG (Santa Cruz
Biotechnology, Inc.; cat. no. sc-2025), PARP-1 (Santa Cruz
Biotechnology, Inc.; cat. no. sc-7150), ATM (Santa Cruz
Biotechnology, Inc.; cat. no. sc-23921), Ku70 (Abcam; cat. no.
ab3114), Ku80 (Abcam; cat. no. ab119935), DNA-PKcs S2056 (Abcam;
cat. no. ab18192), DNA-PKcs T2609 (Abcam; cat. no. ab4194), γ-H2AX
(Abcam; cat. no. ab11174), phosphorylated (p)-ATM (Cell Signaling
Technology, Inc.; cat. no. 13050S), DAPI (Sigma-Aldrich; Merck
KGaA; cat. no. D9542), β-actin (Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd.; cat. no. TA-09), GAPDH (Beijing Zhongshan
Golden Bridge Biotechnology Co., Ltd.; cat. no. TA-08), Alexa
Flour® 488 goat anti-mouse IgG (H+L; Invitrogen; Thermo
Fisher Scientific, Inc.; cat. no. 1915874), and Alexa
Flour® 568 goat anti-rabbit IgG (H+L; Invitrogen; Thermo
Fisher Scientific, Inc.; cat. no. 1704462), anti-mouse IgG,
AP-linked antibody (Cell Signaling Technology, Inc.; cat. No 7056),
anti-rabbit IgG, AP-linked antibody (Cell Signaling Technology,
Inc.; cat. no. 7054), histone 3.1 (Signalway Antibody LLC.; cat.
no. 21137-1). The chemical inhibitor olaparib (cat. no. AZD2281),
the PARP1 inhibitor UPF1069 (cat. no. S8038), the PARP1 inhibitor
NMS-P118 (cat. no. S8363) and DNA-PK inhibitor NU7441 (cat. no.
S2638) were purchased from Selleck Chemicals. DMSO was purchased
from InnoChem LLC.
Immunoprecipitation and western
blotting
NETN buffer 300 [20 mM Tris-HCL (pH 8.0), 300 mM
NaCl, 1 mM EDTA and 0.5% Nonidet P-40] was used to lyse the cells
at 4°C for 10 min. Then NETN buffer 100 [20 mM Tris-HCL (pH 8.0),
100 mM NaCl, 1 mM EDTA and 0.5% Nonidet P-40] was used to lyse the
cells at 4°C for 5 min. After the removal of the cell debris by
centrifugation (12,000 × g for 10 min at 4°C), the supernatant was
collected and incubated with IgG (1 µg/ml) and protein A/G (Santa
Cruz Biotechnology, Inc.; 20 µl) with rotation for 1 h at 4°C for
preclearing. Then, the precipitate was removed by centrifugation
(12,000 × g for 10 min at 4°C) and the supernatant was collected
and incubated with an antibody against DNA-PKcs (1 µg/ml) and
protein A/G (Santa Cruz Biotechnology, Inc.; 40 µl) with rotation
overnight at 4°C. After that, the protein A/G was washed three
times with NETN 100 buffer and boiled with 5X SDS loading buffer at
100°C for 10 min. The samples were then subjected to SDS-PAGE and
immunoblotting with specific antibodies.
The concentration was measured using a NanoDrop™
2000C (Thermo Fisher Scientific, Inc.) and 40 µg was loaded per
lane on 6% SDS PAG gels. Proteins were transferred to
nitrocellulose membranes and blocked with 5% BSA for 1 h at room
temperature. Membranes were incubated overnight at 4°C with the
following primary antibodies: DNA-PKcs (1:500) and PAR (1:1,000),
GAPDH (1:1,000) and histone 3.1 (1:1,000). After washing, membranes
were incubated with secondary antibodies (1:3,000) at room
temperature for 1 h. The membranes were washed twice and
SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo
Fisher Scientific, Inc.) was uniformly added to the membrane. Bands
were visualized using an ImageQuant LAS 500 and the ImageQuant LAS
500 1.1.0 software (GE Healthcare Life Sciences).
For chromatin fractionation, HeLa cells were lysed
with NETN 100 buffer [20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1 mM
EDTA, and 0.5% Nonidet P-40] for 30 min on ice. The soluble
fractions were then collected after centrifugation at 12,000 × g
for 10 min at 4°C, and the pellets were washed twice with PBS and
once with ddH2O. Then, they were treated with 0.2 M HCl
to release histones and chromatin-bound proteins, which were then
neutralized with 1 M Tris-HCl (pH 8.5). Both fractions were
subjected to electrophoresis and western blotting as
aforementioned, and probed with antibodies as indicated.
Immunofluorescence
HeLa cells (8.8×106) were irradiated with
the indicated doses of irradiation (IR). After incubation for 0, 1,
2, 4 and 8 h, the cells were fixed in 4% paraformaldehyde at room
temperature for 30 min and permeabilized with 0.3% Triton X-100 in
1X PBS for 30 min at room temperature. After blocking nonspecific
antibody binding sites with 3% BSA in 1X PBS, the cells were
incubated with DNA-PKcs S2056 (1:100) and γ-H2AX (1:100) at room
temperature for 60 min. Then, cells were washed with 1X PBS three
times and incubated with a secondary antibodies (1:400; Alexa
Flour® 488 goat anti-mouse IgG and Alexa
Flour® 568 goat anti-rabbit IgG) in the dark at room
temperature for 60 min. Then, the slides were washed three times
with 1X PBS and the cells were stained for 10 min at room
temperature with DAPI to visualize nuclear DNA. Coverslips were
placed on glass slides with anti-fade solution, and the results
were visualized using a ZEISS fluorescence microscope.
Cell colony formation assay
HeLa cells were seeded in 35 mm dishes at different
cell concentrations as indicated and allowed to attach. Then,
different concentrations of olaparib (1 and 10 µM) were added, and
DMSO was used as a control, for 1 h at 37°C. After drug treatment,
the cells were treated with different irradiation doses (0, 0.5, 1,
2, 4 and 8 Gy). The cells were cultured at 37°C in a humidified
incubator in an atmosphere containing 5% CO2, and were
grown in DMEM supplemented with 10% FBS, penicillin, streptomycin
and olaparib. The cells were maintained for 10–14 days. Only
colonies containing ≥50 cells were scored.
Cell proliferation assay
HeLa cells in the logarithmic growth phase
(5×104 cells/ml) were prepared as cell suspensions and
seeded into 6-well cell culture plates (3 ml/dish; n=3). After the
cells had attached, different inhibitors [DMSO, NU7441 (5 µM),
olaparib (10 µM), and olaparib (10 µM) + NU7441 (5 µM)] were added
to the culture for 1 h at 37°C, an equal volume of DMSO was used as
the control. The number of cells on the 1st, 2nd, 3rd, 4th, 5th and
6th days was determined using flow cytometry. Briefly, cells were
collected using 0.25% trypsin and the total number of cells in the
cell suspension was directly measured by flow cytometry (NovoCyte;
ACEA Biosciences, Inc.) and the NovoExpress 1.3.0 software (ACEA
Biosciences, Inc.).
NHEJ assay
Before transfection, a NHEJ-GFP plasmid was digested
with HindIII enzyme overnight at 37°C and recovered using AxyPrep
DNA Gel Extraction kit (Axygen; Corning, Inc.), according to the
manufacturer's instructions. The cells were transfected with 1 µg
of pCherry and 1 µg of the digested NHEJ-GFP plasmid (gifts from Dr
Zhenkun Lou; Division of Oncology Research, Mayo Clinic, USA) and
mixed with 5 µl of Lipofectamine 2000™ (Invitrogen; Thermo Fisher
Scientific, Inc.), as described previously (18). Following 6 h, the culture medium of
the transfected cells was replaced with medium containing olaparib
(10 µM) or NU7441 (5 µM) and further cultured for 20 h at 37°C. The
cells were trypsinized (0.25%) and resuspended in PBS. The cellular
fluorescence was measured by flow cytometry analysis as previously
described (19).
HR assay
HeLa cells (3×105) were pretreated with
NU7441 (5 µM) or olaparib (10 µM) for 1 h at 37°C. Then, they were
transfected with a single copy of a DR-GFP, I-SceI
expression plasmid and with a pCherry plasmid used as a
transfection efficiency control (gifts from Dr Zhenkun Lou;
Division of Oncology Research, Mayo Clinic, USA) (19). The cells were harvested 3 days
after transfection and subjected to flow cytometry analysis
(NovoCyte; ACEA Biosciences, Inc.) and the NovoExpress 1.3.0
software (ACEA Biosciences, Inc.), as previously described
(19); the GFP-positive cell
population was measured. The mean values were obtained from three
independent experiments. Little variation was observed among the
three independent experiments. In addition, cell viability was also
examined before transfection under a microscope using trypan blue
staining for 30 min at room temperature. All of the groups
exhibited >90% viability.
Cell synchronization and cell cycle
analysis
HeLa cells (3×105) were incubated with 2
mM thymidine for 17 h at 37°C, cultured in fresh medium for 10 h,
and then treated with thymidine again for a further 13 h. The cells
were collected at different times (S phase, 4.5 h; G2/M phase 8 h;
G0/G1 phase, 14 h) after release for cell cycle analysis and
western blotting, as aforementioned. The cells were washed with
prechilled PBS, treated with 100 µg/ml RNase in PBS and stained
with 10 µg/ml propidium iodide for 10 min at room temperature. The
cell cycle was analyzed using a flow cytometer (NovoCyte; ACEA
Biosciences, Inc.) and the NovoExpress 1.3.0 software (ACEA
Biosciences, Inc.).
Statistical analysis
Statistical analyses were conducted using SPSS
version 23.0 (IBM Corp.). The statistical significance analysis of
the experimental data was performed by t-test for two group
comparisons or ANOVA followed by Dunnett's post hoc test for
multiple group comparison. P<0.01 was considered to indicate a
statistically significant difference.
Results
DNA-PKcs is modified by PARylation
after DNA damage
Since DNA-PKcs interacts with PARP1, it is possible
that DNA-PKcs is the substrate of PARP1. To test if DNA-PKcs can be
modified by PARylation, the present study examined the DNA-PKcs
PARylation status by immunoprecipitation. First, endogenous
DNA-PKcs was immunoprecipitated from cells after treatment with
different doses of IR. The results revealed that DNA-PKcs
PARylation increased as the IR dose increased (Fig. 1A). Next, DNA-PKcs was
immunoprecipitated by a PAR antibody IP, and IR treatment increased
the amount of DNA-PKcs pulled down (Fig. 1B). These results suggest that
DNA-PKcs PARylation is induced by IR. The present study also
investigated DNA-PKcs PARylation in different phases of the cell
cycle. When the cells were synchronized in the G1, S, and G2
phases, either PAR IP or DNA-PKcs IP was performed. The results
indicated that more DNA-PKcs PARylation was seen in the S phase
(Fig. S1). Furthermore, when the
PARP1/2 inhibitor olaparib was administered to the cells, DNA-PKcs
PARylation was reduced at a concentration of 1 µM and abolished at
a concentration of 10 µM (Fig.
1C). Since olaparib cannot distinguish between PARP1 and PARP2,
the effects of the specific PARP1 inhibitor NMS-P118 and the PARP2
inhibitor UPF1069 on DNA-PKcs PARylation were evaluated. The
results revealed that both PARP1 and PARP2 are required for
DNA-PKcs PARylation (Fig. S2A),
suggesting the redundant roles of PARP1 and PARP2, as previously
reported (20).
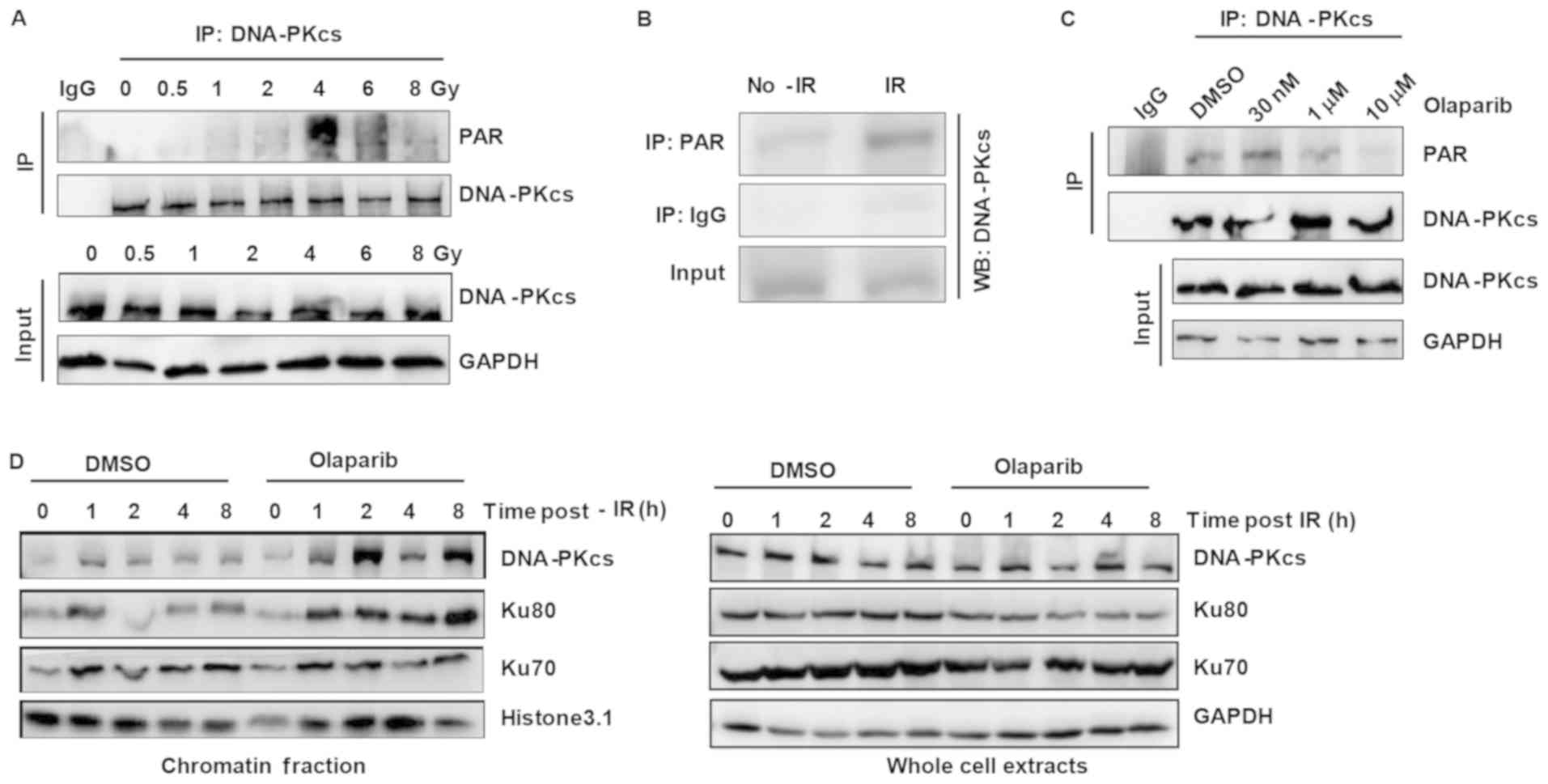 | Figure 1.DNA-PK is modified by PAR in response
to DNA damage. (A) IP with an anti-DNA-PKcs antibody was performed
to detect the PAR modification of DNA-PKcs in HeLa cells after
treatment with different irradiation doses (0, 0.5, 1, 2, 4, 6 and
8 Gy). The cells were harvested 2 h after irradiation and then
lysed. (B) Western blotting was used to detect DNA-PKcs in the IP
product of the anti-PAR antibody from HeLa cells treated with or
without irradiation. (C) The effect of olaparib on the PARylation
of DNA-PKcs. HeLa cells were treated with different concentrations
of olaparib. After 24 h, the cells were harvested and lysed, and IP
was used to detected the PAR modification of DNA-PKcs. (D) The
effect of olaparib on the chromatin binding of DNA-PKcs in
irradiated HeLa cells. DNA-PKcs, DNA-dependent protein kinase
catalytic subunit; PAR, poly(ADP-ribose); IP, immunoprecipitation;
IR, irradiation. |
Next, the present study explored if DNA-PKcs
PARylation can affect the DNA-PKcs/Ku70/Ku80 complex. Since the
DNA-PKcs/Ku70/Ku80 complex binds DNA ends and is activated by
broken DNA ends (21), the
chromatin fraction content of the complex after olaparib treatment
was examined. The results demonstrated that all three proteins were
retained on chromatin (Fig. 1D).
These results indicate that overall PARylation inhibition activates
the DNA-PK complex and that DNA-PKcs PARylation can suppress DNA-PK
activity.
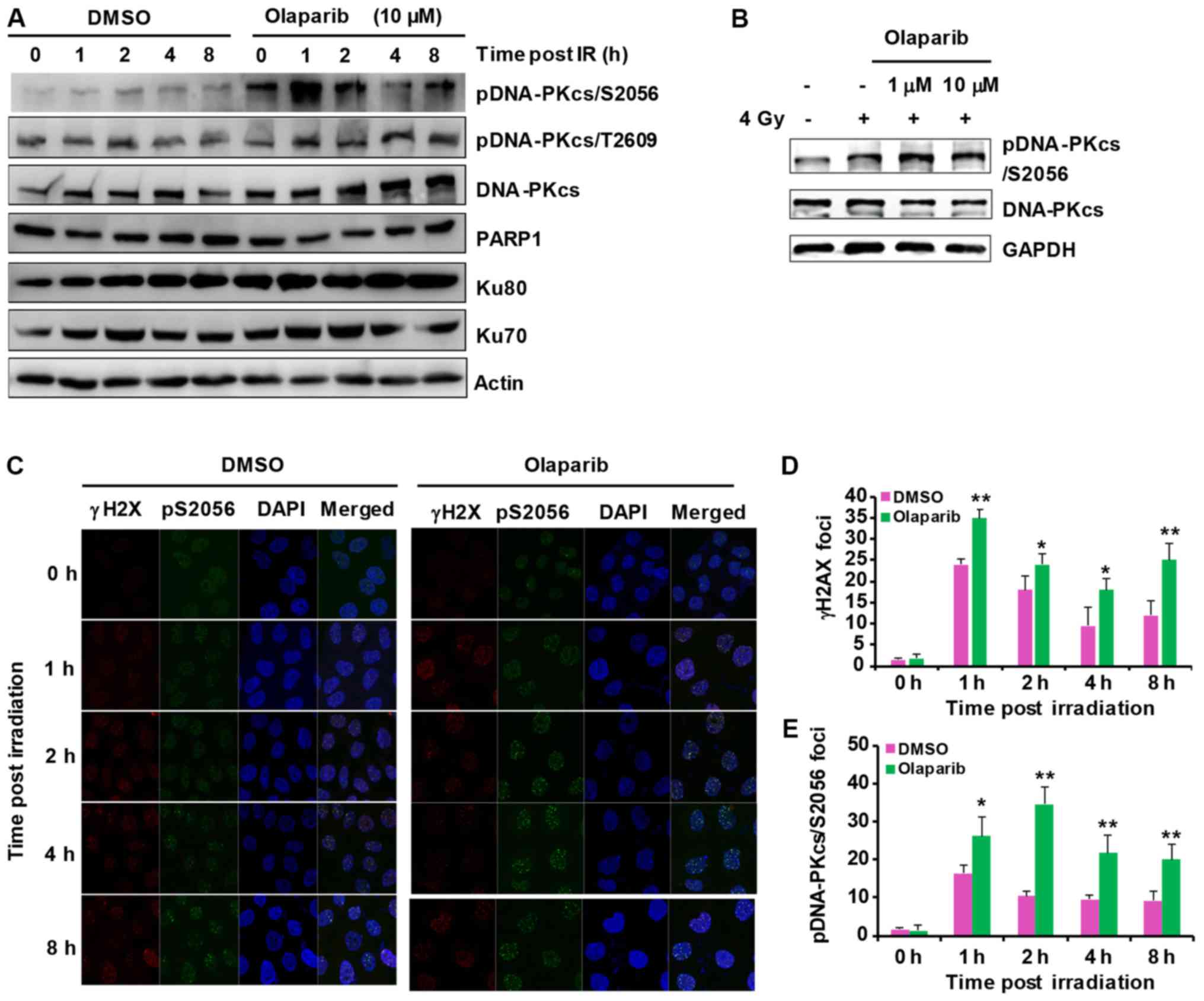
Olaparib treatment increases DNA-PKcs
phosphorylation
DNA-PKcs phosphorylation is critical for DNA-PK
activity and NHEJ repair (22). To
test if the inhibition of DNA-PKcs PARylation by olaparib can
affect DNA-PK activity, the present study compared DNA-PKcs Ser2056
and Thr 2609 phosphorylation with and without olaparib treatment.
The results showed that DNA-PKcs Ser2056 phosphorylation increased
while DNA-PKcs Thr 2609 phosphorylation did not change (Fig. 2A and B). These results indicate
that olaparib treatment promotes DNA-PK activity through both the
inhibition of DNA-PKcs PARylation and the induction of DNA damage.
Likewise, treatment with both the PARP1 inhibitor NMS-P118 and the
PARP2 inhibitor UPF1069 increased DNA-PKcs Ser2056 phosphorylation
(Fig. S2B). Similar results were
observed by immunofluorescence staining. DNA-PKcs Ser2056 increased
more in the inhibitor-treated groups when compared with the DMSO
group and was accompanied by increased γ-H2AX foci (Fig. 2C-E).
Olaparib treatment results in enhanced
NHEJ repair
Based on the above findings, one can deduce that
PARylation regulates DNA-PKcs Ser2056 phosphorylation. DNA-PKcs
Ser2056 phosphorylation is critical for DNA-PK activity and
DNA-PKcs conformation (23). Since
DNA-PK is the initiator of NHEJ repair, the present study explored
NHEJ activity after olaparib treatment. The NHEJ reporter assay
indicated that NHEJ repair was significantly boosted after olaparib
treatment, while NU7441, as a control, inhibited NHEJ. Furthermore,
HR repair was inhibited, suggesting that olaparib treatment can
directly induce DNA-PK activation (Fig. 3).
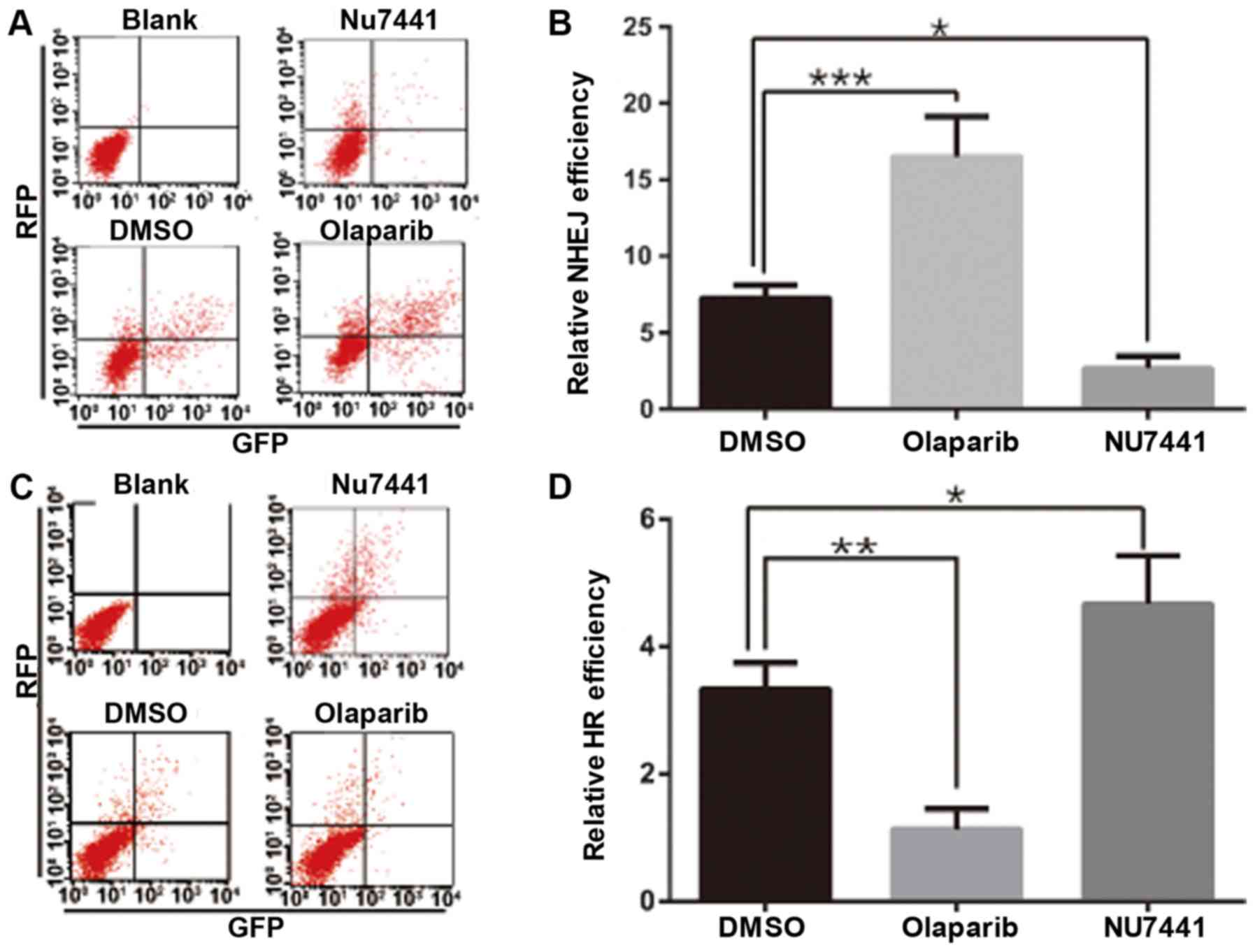 | Figure 3.Olaparib treatment activates NHEJ
repair. (A and B) HeLa cells were transfected with NHEJ-GFP and a
pCherry plasmid for 6 h, the medium was replaced with medium
containing 10 µM olaparib or 10 µM NU7441 for 20 h, and then cells
were collected. Flow cytometry was used to determine the GFP/RFP
double-positive ratio, with the DMSO group as a control. (C and D)
HeLa cells were transfected with Dr-GFP, I-SceI and pCherry
plasmids for 6 h, the medium was replaced with medium containing 10
µM olaparib or 10 µM NU7441 for 20 h, and then cells were
collected. Flow cytometry was used to determine the GFP/RFP
double-positive ratio. *P<0.05, **P<0.01, ***P<0.001.
NHEJ, non-homologous end-joining; GFP, green florescent protein;
RFP, red fluorescent protein; HR, homologous recombination. |
Olaparib increases the
radiosensitivity of cells
Sustained DNA-PK activation can hinder the
completion of NHEJ and threaten cell survival (24). Therefore, the present study sought
to determine if olaparib can increase the radiosensitivity of
cells, which was tested by cell colony formation experiments. The
results showed that 1 µM olaparib sensitized the cells to different
doses of ionizing radiation (Figs.
4A, and S3A and B).
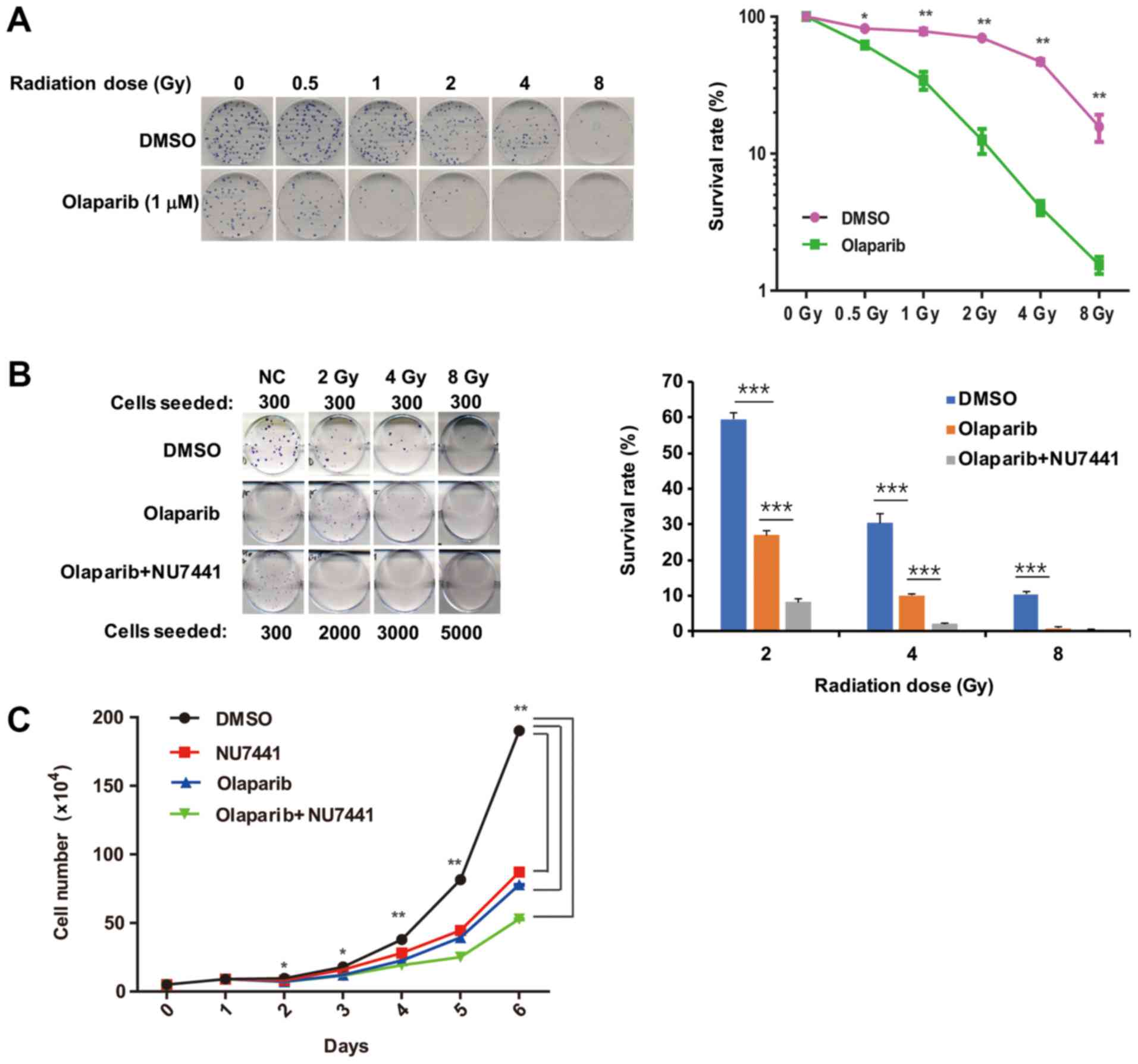 | Figure 4.Olaparib treatment sensitizes cells
to ionizing radiation. (A) Clonogenic assay. After trypsinization,
300 HeLa cells were plated. Once cells had attached, they were
treated with olaparib for 1 h and then irradiated. The DMSO group
was used as a control. The cells were cultured for two weeks, and
the surviving colonies were observed and counted. *P<0.05 and
**P<0.01. (B) The effect of combinational treatment with
olaparib and the DNA-PKcs inhibitor NU7441 on the cell colony
formation of irradiated HeLa cells. After trypsinization, HeLa
cells were seeded at different cell concentrations as indicated (in
DMSO controls 300 cells seeded/plate). After the cells were
attached, the cells were treated with olaparib (10 µM), or olaparib
(10 µM) and NU7441 (5 µM) for 1 h and then irradiated. The DMSO
group was used as a control and cultured continuously for two
weeks. Colonies containing 50 or more cells were scored.
***P<0.001, as indicated. (C) HeLa cells were prepared as cell
suspensions of 5×104 cells/ml and seeded into 6-well
cell culture plates (3 ml/dish, n=3). After the cells were
attached, different inhibitors [DMSO, NU7441 (5 µM), olaparib (10
µM), and olaparib (10 µM) + NU7441 (5 µM)] were added to the
culture. The number of cells was counted by flow cytometry to
calculate the mean concentration of each group on each day.
*P<0.05 and **P<0.01. DNA-PKcs, DNA-dependent protein kinase
catalytic subunit. |
NU7441 is an inhibitor of the kinase activity of
DNA-PKcs; thus, the simultaneous inhibition of DNA-PKcs kinase
activity and PARylation may have an effect on cell clonogenic
formation. The results showed that, when used together, olaparib
and NU7441 more significantly reduced cell survival than treatment
with olaparib alone (Fig. 4B).
In addition, cell proliferation was also examined
after either Olaparib or NU7441 treatment or a combination of both
inhibitors. As indicated in Fig.
4C, both Olaparib and NU7441 decreased the cell proliferation
rate.
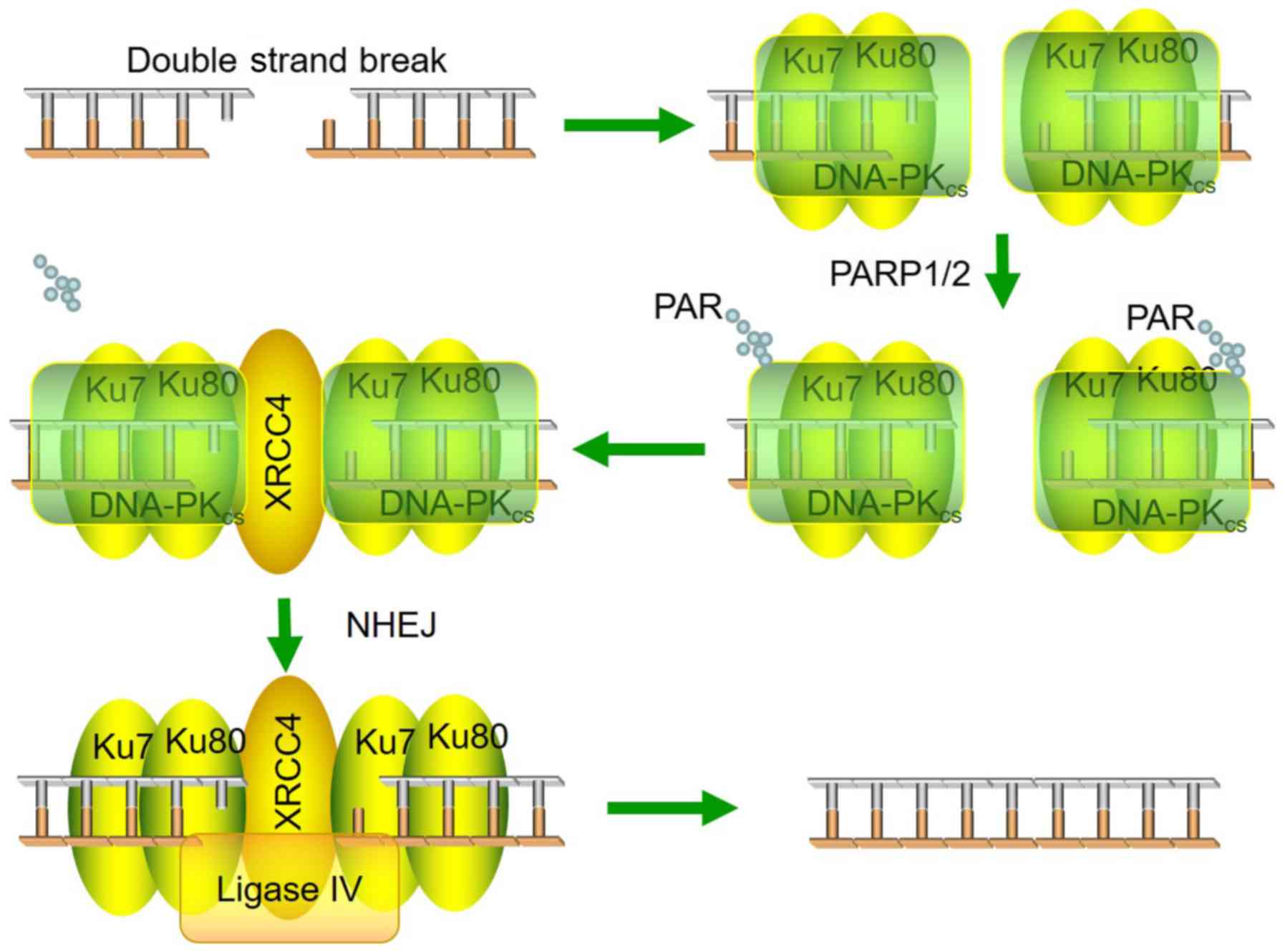
The present study demonstrates a model for DNA-PKcs
PARylation and how PARylation affects DNA-PKcs activity (Fig. 5). Both DNA-PKcs kinase activity and
PARylation are important regulators of radiosensitivity.
Discussion
DNA-PKcs forms a complex with PARP1 (25), and DNA-PKcs is PARylated by PARP-1
in an IFN-γ- and p53-dependent manner (18). The present study reports that
DNA-PKcs is PARylated after DNA damage and that PARylation
inhibition causes enhanced DNA-PKcs autophosphorylation and NHEJ
repair. Our findings therefore link DNA-PKcs PARylation to DNA
damage and substantiates the role of PARP1 in NHEJ repair.
The DNA-PK complex initiates NHEJ by binding broken
DNA ends and phosphorylates downstream NHEJ factors (26). According to a previous study,
DNA-PKcs T2609 is involved in the DNA damage response and
phosphorylated by ATM. However, DNA-PKcs T2609 is not essential for
NHEJ repair (27). DNA-PKcs S2056
is critical for the DNA-PKcs function in NHEJ. DNA-PKcs Ser2056
autophosphorylation is critical for DNA-PKcs detachment from DSB
sites and the completion of NHEJ (28). Based on the present results,
DNA-PKcs PARylation can retain DNA-PKcs on chromatin and cause the
continuous activation of DNA-PK. The aberrant activation of DNA-PK
can block other repair factors from chromatin and hinder the
completion of NHEJ repair. On the other hand, olaparib treatment
can cause DNA damage since HR is repressed. PARylation is possibly
required for DNA-PKcs detachment from chromatin and kinase
deactivation. DNA-PK deactivation leads to deficiencies of the NHEJ
pathway (28) but may be crucial
for successful HR repair.
PARP1 is the major enzyme of the PARP family
responsible for PARylation (11,29).
It is unknown whether other accessory factors also account for
PARylation. The present results also indicated the redundant role
of PARP1 in DNA-PKcs PARylation. TrpRS has been reported as one of
the 10 class I tRNA synthetases that act as bridging proteins
between DNA-PKcs and PARP1 (18).
We previously found that tankyrase 1 binding protein 1 (TNKS1BP1)
functions in DNA DSB repair by facilitating PARP-1-dependent
DNA-PKcs autophosphorylation (30). It would be of interest to determine
whether TNKS1BP1 functions as the bridging protein for DNA-PKcs and
PARP1 in DNA damage-induced DNA-PKcs PARylation.
DNA-PKcs PARylation can alter kinase activity, but
the detailed mechanisms are not understood. Structural analysis is
needed to determine the conformational changes after DNA-PKcs
PARylation. In a study by Sajish et al (18) the DNA-PKcs/Ku70/80/PARP-1 complex,
which forms in the presence of damaged DNA, and the
DNA-PKcs/TrpRS/PARP-1 complex were mutually exclusive. It is
plausible that the DNA-PKcs/Ku70/80/PARP-1 and
DNA-PKcs/TNKS1BP1/PARP-1 complexes are mutually exclusive too
(18). However, it is notable that
TNKS1BP1 promotes DNA-PKcs autophosphorylation while DNA-PKcs
PARylation may suppress DNA-PKcs autophosphorylation.
How DNA-PKcs PARylation suppresses its
autophosphorylation remains unknown. It is known that serine is the
major site for protein PARylation (31). Therefore, it is possible that the
conventional DNA-PKcs autophosphorylation sites are also PARylation
sites. The interaction between autophosphorylation and PARylation
may be an important mechanism of NHEJ repair completion. Olaparib
increases the radiation sensitivity of cells through the activation
of DNA-PK, thereby providing a possible future treatment for
cancer.
In conclusion, DNA-PKcs PARylation is a newly
identified player in regulating NHEJ repair. It may answer the
question of how NHEJ is completed and how the choice between HR and
NHEJ repair is made.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Key Basic Research Program (973 Program) of MOST, China
(grant no. 2015CB910601) and the National Natural Science
Foundation of China (grant nos. 81530085, 31570853, 81602799,
31870847).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YH and FJ carried out the experiments. TM wrote the
manuscript with support from PZ. YX, YL and SH performed cell
culture. HG, YG and XL helped with data analysis, data
interpretation and supervised the project. TM and PZ conceived the
original idea. PZ supervised the project.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhou PK: DNA damage, signaling and repair:
Protecting genomic integrity and reducing the risk of human
disease. Chin Sci Bull. 56:3119–3121. 2011. View Article : Google Scholar
|
|
2
|
Durocher D and Jackson SP: DNA-PK ATM and
ATR as sensors of DNA damage variations on a theme? Curr Opin Cell
Biol. 13:225–231. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dobbs TA, Tainer JA and Lees-Miller SP: A
structural model for regulation of NHEJ by DNA-PKcs
autophosphorylation. DNA Repair (Amst). 9:1307–1314. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Witze ES, Old WM, Resing KA and Ahn NG:
Mapping protein post-translational modifications with mass
spectrometry. Nat Methods. 4:798–806. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brown JS and Jackson SP: Ubiquitylation,
neddylation and the DNA damage response. Open Biol. 5:1500182015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oliver FJ, Menissier-de-Murcia J and de
Murcia G: Poly(ADP-ribose) polymerase in the cellular response to
DNA damage, apoptosis, and disease. Am J Hum Genet. 64:1282–1288.
1999. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chambon P, Weill JD and Mandel P:
Nicotinamide mononucleotide activation of new DNA-dependent
polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res
Commun. 11:39–43. 1963. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cohen MS and Chang P: Insights into the
biogenesis, function, and regulation of ADP-ribosylation. Nat Chem
Biol. 14:236–243. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Slade D, Dunstan MS, Barkauskaite E,
Weston R, Lafite P, Dixon N, Ahel M, Leys D and Ahel I: The
structure and catalytic mechanism of a poly(ADP-ribose)
glycohydrolase. Nature. 477:616–620. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lüscher B, Bütepage M, Eckei L, Krieg S,
Verheugd P and Shilton BH: ADP-Ribosylation, a multifaceted
posttranslational modification involved in the control of cell
physiology in health and disease. Chem Rev. 118:1092–1136. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barkauskaite E, Jankevicius G and Ahel I:
Structures and mechanisms of enzymes employed in the synthesis and
degradation of PARP-dependent protein ADP-ribosylation. Mol Cell.
58:935–946. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Krishnakumar R and Kraus WL: The PARP side
of the nucleus: Molecular actions, physiological outcomes, and
clinical targets. Mol Cell. 39:8–24. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gibson BA and Kraus WL: New insights into
the molecular and cellular functions of poly(ADP-ribose) and PARPs.
Nat Rev Mol Cell Biol. 13:411–424. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Luo X and Kraus WL: On PAR with PARP:
Cellular stress signaling through poly(ADP-ribose) and PARP-1.
Genes Dev. 26:417–432. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Haince JF, McDonald D, Rodrigue A, Dery U,
Masson JY, Hendzel MJ and Poirier GG: PARP1-dependent kinetics of
recruitment of MRE11 and NBS1 proteins to multiple DNA damage
sites. J Biol Chem. 283:1197–1208. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Haince JF, Kozlov S, Dawson VL, Dawson TM,
Hendzel MJ, Lavin MF and Poirier GG: Ataxia telangiectasia mutated
(ATM) signaling network is modulated by a novel
poly(ADP-ribose)-dependent pathway in the early response to
DNA-damaging agents. J Biol Chem. 282:16441–16453. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hu Y, Petit SA, Ficarro SB, Toomire KJ,
Xie A, Lim E, Cao SA, Park E, Eck MJ, Scully R, et al: PARP1-driven
poly-ADP-ribosylation regulates BRCA1 function in homologous
recombination-mediated DNA repair. Cancer Discov. 4:1430–1447.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sajish M, Zhou Q, Kishi S, Valdez DM Jr,
Kapoor M, Guo M, Lee S, Kim S, Yang XL and Schimmel P: Trp-tRNA
synthetase bridges DNA-PKcs to PARP-1 to link IFN-γ and p53
signaling. Nat Chem Biol. 8:547–554. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tan W, Guan H, Zou LH, Wang Y, Liu XD,
Rang WQ, Zhou PK, Pei HD and Zhong CG: Overexpression of TNKS1BP1
in lung cancers and its involvement in homologous recombination
pathway of DNA double-strand breaks. Cancer Med. 6:483–493. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ali SO, Khan FA, Galindo-Campos MA and
Yélamos J: Understanding specific functions of PARP-2: New lessons
for cancer therapy. Am J Cancer Res. 6:1842–1863. 2016.PubMed/NCBI
|
|
21
|
Spagnolo L, Rivera-Calzada A, Pearl H and
Llorca O: Three-dimensional structure of the human
DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications
for DNA DSB repair. Mol Cell. 22:511–519. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Davis AJ, Chen BP and Chen DJ: DNA-PK: A
dynamic enzyme in a versatile DSB repair pathway. DNA Repair
(Amst). 17:21–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Uematsu N, Weterings E, Yano K,
Morotomi-Yano K, Jakob B, Taucher-Scholz G, Mari PO, van Gent DC,
Chen BP and Chen DJ: Autophosphorylation of DNA-PKCS regulates its
dynamics at DNA double-strand breaks. J Cell Biol. 177:219–229.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dong J, Zhang T, Ren Y, Wang Z, Ling CC,
He F, Li GC, Wang C and Wen B: Inhibiting DNA-PKcs in a
non-homologous end-joining pathway in response to DNA double-strand
breaks. Oncotarget. 8:22662–22673. 2017.PubMed/NCBI
|
|
25
|
Spagnolo L, Barbeau J, Curtin NJ, Morris
EP and Pearl LH: Visualization of a DNA-PK/PARP1 complex. Nucleic
Acids Res. 40:4168–4177. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Meek K, Dang V and Lees-Miller SP: DNA-PK:
The means to justify the ends? Adv Immunol. 99:33–58. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Povirk LF, Zhou RZ, Ramsden DA,
Lees-Miller SP and Valerie K: Phosphorylation in the
serine/threonine 2609–2647 cluster promotes but is not essential
for DNA-dependent protein kinase-mediated nonhomologous end joining
in human whole-cell extracts. Nucleic Acids Res. 35:3869–3878.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiang W, Crowe JL, Liu X, Nakajima S, Wang
Y, Li C, Lee BJ, Dubois RL, Liu C and Yu X: Differential
phosphorylation of DNA-PKcs regulates the interplay between
end-processing and end-ligation during nonhomologous end-joining.
Mol Cell. 58:172–185. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wei H and Yu X: Functions of PARylation in
DNA damage repair pathways. Genomics Proteomics Bioinformatics.
14:131–139. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zou LH, Shang ZF, Tan W, Liu XD, Xu QZ,
Song M, Wang Y, Guan H, Zhang SM, Yu L, et al: TNKS1BP1 functions
in DNA double-strand break repair though facilitating DNA-PKcs
autophosphorylation dependent on PARP-1. Oncotarget. 6:7011–7022.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Palazzo L, Leidecker O, Prokhorova E,
Dauben H, Matic I and Ahel I: Serine is the major residue for
ADP-ribosylation upon DNA damage. Elife. 7:343342018. View Article : Google Scholar
|