Introduction
In intensive care units (ICUs), sepsis and its
complications are the most common causes of mortality (1). Sepsis-associated encephalopathy (SAE)
is clinically characterized by mental alterations that ranges from
a transient brain damage to severe and irreversible encephalopathy
(2). The mechanism of SAE is
elusive but is considered to be multifactorial. A recent study has
demonstrated that the mitochondrial respiratory chain and creatine
kinase activities are involved in cecal ligation and perforation
(CLP) rats (3). Additionally, the
uncoupling of mitochondrial oxidative phosphorylation also takes
place in the septic brain (4).
These previous results suggested that mitochondrial dysfunction and
neuronal apoptosis may be involved in the pathogenesis of SAE.
Furthermore, SAE has also been reported to be associated with
long-term learning and memory deficits (5).
Dexmedetomidine is a potent and highly selective
α2-adrenergic receptor agonist with sedative, amnestic,
sympatholytic and analgesic properties (6). Dexmedetomidine is reported to reduce
intraoperative anesthetic requirements and offers good
perioperative hemodynamic stability (7). In addition, dexmedetomidine decreases
psychosis, shortens hospital stays and may improve cognitive
dysfunctions (8). Therefore,
dexmedetomidine is widely used for sedation, especially in ICUs
(9). Dexmedetomidine has been
shown to protect organs against stimuli in various models. For
example, dexmedetomidine combined with ketamine reduces ventilator
induced lung injury in endotoxemia rats (10). In addition, dexmedetomidine possess
a protective effect on ischemia-reperfusion injury in heart,
kidney, testis and intestine in various animal models (11–14).
However, whether dexmedetomidine can improve neuronal apoptosis and
protect brain against sepsis in SAE model remains to be
elucidated.
The serine/threonine kinase AKT, also known as
protein kinase B (PKB), is a pro-survival protein and involved in
the regulation of cell apoptosis and caspase-3 activation (15). The heat-shock protein 90 (Hsp90) is
a constitutively expressed stress protein located in the cytosol of
eukaryotic cells, including neuronal cells (16). Hsp90 serves an essential role in
controlling AKT functions. The formation of chaperone-substrate
protein complex leads to a reduction in the binding between Hsp90
and AKT, resulting in AKT kinase inactivation (17). In myocardial cells,
lipopolysaccharide (LPS) treatment contributes to the degradation
of Hsp90 and AKT (18,19).
The Hsp90/AKT pathway is an important survival and
antiapoptotic pathway in cells. For example, the cleavage of Hsp90
in the AKT/Hsp90 complex appears to be vital in the AKT/Hsp90
complex destabilization and in triggering the apoptotic cascade
(20,21). Dexmedetomidine has been shown to
upregulate the AKT signaling pathway in a traumatic brain injury
model (22). The present study
explored the neuronal protective role of dexmedetomidine in an
experimental septic model using CLP rats and LPS-treated primary
neuronal cultures. In addition, the present study investigated the
possible involvement of the Hsp90/AKT signaling pathway in this
process.
Materials and methods
Animals and regents
A total of 146 adult male Sprague-Dawley rats (mean
weight, 250 g, weight range: 233–272 g; age, 8 weeks) were acquired
from Tianjin Medical University. All studies performed on animals
were approved by the local Institutional Animal Care and Use
Committee (Tianjin Baodi Hospital, Baodi Clinical College Of
Tianjin Medical University, Tianjin, China). All animal experiments
complied with the ARRIVE guidelines (23) and the National Institutes of Health
guide for the care and use of Laboratory animals (NIH Publications
No. 8023, revised 1978) (24). The
animals were housed at 22±1°C with 50–70% humidity, and maintained
on a 12-h light/dark cycle throughout the experiments. Every effort
was made to minimize suffering and the number of animals used in
the present study. All animals were given food and water ad
libitum. DMEM, 10% FBS, poly-L-lysine, Neurobasal media,
L-glutamine and B27 were purchased from Gibco (Thermo Fisher
Scientific, Inc.). Trypsin-EDTA, Cell Counting Kit-8 (CCK-8),
lactate dehydrogenase (LDH), pentobarbital, prazosin hydrochloride,
yohimbine hydrochloride and wortmannin were purchased from
Sigma-Aldrich (Merck KGaA). TUNEL staining reaction solution was
obtained from Roche Applied Science, 17-AAG was purchased from
Tocris BioScience, and Rhodamine (TRITC)-Streptavidin from Jackson
ImmunoResearch Laboratories, Inc. Dexmedetomidine hydrochloride
injection was acquired from Jiangsu Hengrui Medicine Co., Ltd.
Hippocampal neuronal cultures and cell
transfection
Primary hippocampal neuronal cells were isolated
from embryonic day 18 Sprague-Dawley rats as previously described
(25). In brief, the fetuses were
removed from pregnant rats (a total of 18 rats provided by Nanjing
University, weight 455–529 g, aged 10 weeks) euthanized by
CO2 anoxia at concentrations of 100%. Hippocampal
tissues were dissociated with 0.25% Trypsin-EDTA. The digestion was
terminated using DMEM combined with 10% FBS. Neuronal cells were
cultured in96-well plates (for LDH and CCK-8 detection), coverslips
(for TUNEL-staining) or dishes (for western blotting) that were
precoated with poly-L-lysine. Neurons were fed with Neurobasal
media supplemented with 500 µM of L-glutamine and 2% of B27, and
maintained in a humidified atmosphere of 5% CO2 and 95%
O2 at 37°C. After 3 days of incubation, half of the
medium was replaced with fresh medium. A short hairpin RNA (shRNA)
was used to downregulate AKT, and the shRNA sequence (starting
point 221–239) was cloned into a lentiviral expression vector. The
concentration of shRNA transfected was 1 µg/ml according to the
instruction of the manufacturer. The AKT shRNA sequence was the
follow: Sense,
5′-GATCCGTCAGCTGATGAAGACAGATTCAAGACGTCTGTCTTCATCAGCTGACTTTTTTGTCGACA-3′;
anti-sense,
5′-AGCTTGTCGACAAAAAAGTCAGCTGATGAAGACAGACGTCTTGAATCTGTCTTCATCAGCTGACG-3′.
Recombinant AKT shRNA lentiviral vector and negative control
lentiviral vector were diluted into 108 TU/ml with the
high-efficiency lentivirus transfection enhancement solution
(Invitrogen, Thermo Fisher Scientific, Inc.). At 14 days in
vitro, neuronal cells were infected with the recombinant AKT
shRNA lentiviruses using the Lipofectamine 2000® reagent
(Invitrogen, Thermo Fisher Scientific, Inc.). The shRNA
oligonucleotides were synthesized by Shanghai Sangon Biological
Engineering Technology & Services Co., Ltd. (Shanghai, China).
Different dose of dexmedetomidine (0.1, 0.5 or 1.0 µM) or LPS (1
µg/ml) were used to treat cells 48 h after cell transfection.
Neuronal survival evaluation
The neuronal survival was evaluated by measuring the
level of LDH released into the culture media and this evaluation
was confirmed by CCK-8 assay. These experiments were conducted 24 h
after the drug treatments. Briefly, for the LDH assay, 50 µl
neurobasal medium was mixed with equal volume of substrate
maintaining at 37°C. After incubation for 30 min, 50 µl of stop
solution was then added. The absorbance at 490 nm was recorded
using a microtiter plate reader (Quant; Bio-Tek Instruments Inc.).
For the CCK-8 assay, 10 µl of CCK-8 solution was incubated with
cells at 37°C for 2.5 h. Absorbance at 450 nm was then
measured.
Cecal ligation and perforation
surgery
An animal model of SAE was established by cecal
ligation and perforation (CLP) surgery as previously described
(26). Briefly, after
intraperitoneally injecting with 50 mg/kg of pentobarbital for
anesthesia, a midline incision (~3 cm) was performed to allow the
exposure of the cecum. The cecum was isolated carefully, and
ligated tightly below the ileocecal junction. The cecum was
punctured twice with a 16-gauge needle, and gently compressed to
remove a small amount of cecal contents. The cecum was then put
back into the abdominal cavity. Finally, the abdominal wall was
closed. For sham-operated animals, which were used as control, the
rats underwent the same procedure but without ligation and
perforation. After surgery, the CLP rats were subcutaneously (s.c.)
injected with a solution containing saline (50 ml/kg) immediately
and 12 h after CLP performance. In addition, the rats were injected
with 30 mg/kg of ceftriaxone and 25 mg/kg of clindamycin every 6 h
for consecutive 3 days. The sham-operated group received only
saline (50 ml/kg immediately and 12 h after CLP surgery). During
their recovery from sepsis, all animals were observed after CLP to
detect signs of infection (including piloerection, lethargy,
tachypnea and weight loss). If a rat demonstrated constant
lethargic sleep or/and did not take food or water for 48
consecutive h, the rat was evaluated by two researchers. If both
researchers confirmed that the rat would succumb to sepsis, the rat
was euthanized by being deeply anesthetized with sodium
pentobarbital (120 mg/kg, i.p.) followed by cervical dislocation.
The researchers in the present study used utmost efforts to
minimize the quantity and the suffering of all experimental
animals. Survival rateswere100% in the sham group and approximately
36% in the sepsis group, which is in accordance with a previous
study (27).
Drug administration
Following surgery, all animals were administrated
with isotonic saline solution (50 ml/kg s.c.) and antibiotics
(ceftriaxone, 30 mg/kg; clindamycin, 25 mg/kg) immediately after
and 12 h after CLP surgery. The sham-surgery rats received the same
volume of isotonic saline solution without antibiotics. At 6 h
after surgery, the CLP rats were intraperitoneally injected with
different doses of dexmedetomidine (1, 5 and 10 µg/kg) every 12 h
consecutively for a week. The control rats were administrated with
the same volume of vehicle. 17-AAG (dose, 10 mg/kg), wortmannin
(dose, 50 mg/kg) or same volume of vehicle was also injected
intraperitoneally once a day for 1 week. The whole hippocampal
tissues were subjected to western blot analysis 1 week after
surgery.
Western blot analysis
Whole hippocampal tissues and primary neuronal
cultures were collected and subjected to lysis buffer (Tris-HCl: 50
mM, pH 7.4; NP-40: 1%; Sodium deoxycholate: 0.25%; NaCl: 150 mM;
EDTA: 1 mM; PMSF: 1 mM; Aprotinin, leupeptin, pepstatin: 1 µg/ml
each; Na3VO4: 1 mM; NaF: 1 mM). Bradford Protein Assay (Bio-Rad
Laboratories, Inc.) was used to quantify the protein concentration
of samples. Lysates (30 µg) were loaded and separated by 10–15%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) (Beyotime Institute of Biotechnology) and transferred
them onto PVDF membranes (Beyotime Institute of Biotechnology). The
membranes were blocked by 5% skimmed milk for 1 h at room
temperature, and then incubated overnight at 4°C with the
appropriate primary antibodies. After three washes with PBS-T (0.1%
Tween-20), membranes were incubated with horseradish peroxidase
(HRP)-conjugated appropriate secondary antibodies for 2 h at room
temperature. Polyclonal rabbit anti-cleaved caspase-3 (C9598;
1:2,000), polyclonal rabbit anti-Bcl-2 (SAB4500005; 1:1,500) and
monoclonal mouse anti-Hsp90 (SAB1305541; 1:2,000) were from
Sigma-Aldrich (Merck KGaA). Polyclonal rabbit anti-AKT (sc-55523;
1:1,000) and anti-phosphorylated(p)-AKT-Ser 473 (sc-24500; 1:1,000)
were obtained from Santa Cruz Biotechnology, Inc. Monoclonal
antibody anti-Actin (58169; 1:1,000) was purchased from Cell
Signaling Technology, Inc. Goat anti-rabbit HRP-conjugated IgG
(A0208; 1:1,000) and goat anti-mouse HRP-conjugated IgG (A0216;
1:1,000) were from Beyotime Institute of Biotechnology. The
membranes were washed and processed with ECL kit (Gibco; Thermo
Fisher Scientific, Inc.) and then read digitally with Image
ReaderLAS-4000 (FujiFilm Life Science). The bands were quantified
using Multi Gauge version 3.0 software (FujiFilm Life Science).
TUNEL assay
TUNEL assay was used to determine neuronal DNA
damage. Briefly, the neuronal cells on coverslips were
permeabilized with 0.01% Triton X-100 (Sigma-Aldrich; Merck KGaA)
in PBS containing 1% sodium citrate. The TUNEL reaction mixture was
added into and co-incubated with the neuronal cultures at 37°C for
30 min. Following washing with PBS, the coverslips were then
incubated with Rhodamine (TRITC)-Streptavidin at 37°C for 1 h.
Hoechst 33342 (Beyotime Institute of Biotechnology) was used for
nuclei staining. In brief, 1% of Hoechst 33342 was added on
coverslips and incubated for 5 min at room temperature according to
the instructions of the manufacturer. Three separate cultures and
20 fields (magnification, ×40) per culture were examined in each
group. Immunofluorescence of neuronal cells was captured and
analyzed by a laser scanning confocal microscope (Zeiss AG). The
TUNEL-positive cells were considered to be apoptotic.
Behavioral studies
Open-field test
The behavior study was performed 2 weeks after
surgery. The open-field test was used to detect the possible
locomotor activity deficits. In brief, each animal was released in
the center of the arena, and allowed to walk freely for 10 min. At
the end of each test, the arena was carefully cleaned with alcohol
to avoid the presence of olfactory cues. The total distance
traveled in 10 min was measured, and considered as locomotor
activity of the experimental animal.
Morris water maze
A Morris water maze (Shanghai Bio-will Co.) was used
to evaluate spatial cognitive functions as previously described
(28). Place trials were performed
to determine the spatial learning capability. All rats received two
trails each day for consecutive three days. The rats were
individually placed into the pool facing the sidewall. They were
allowed 1 min to find the platform that was 2.5 cm below the water
surface. The time to reach the platform (latency) and the swimming
speed were recorded. Probe test was carried out to determine
capability of memory retention. After the last place trial, the
platform was removed. All rats were allowed to swim for 1 min
starting from the same quadrant. The number of crossings over the
platform and the swimming time spent in each quadrant were
recorded. All swimming paths were tracked by a video monitoring
system (Coulbourn Instruments, LLC).
Fear conditioning
The contextual and cued fear conditioning experiment
was performed to examine the capability of emotional learning and
memory as previously described (29). The fear conditioning consisted of a
5-min free exploration period followed by three conditioned
stimulus and unconditioned stimulus pairings separated by 1 min
each. At the end of the conditioned stimulus presentation (80 db
noise for 20 sec), 1 mA of foot shock was delivered for 1 sec as
unconditioned stimulus. The contextual test was carried out in the
conditioning chamber for 5 min in the absence of noise at 24 h
after conditioning. The cued test was conducted 48 h after
conditioning by presentation of a cue (80 db noise for 3 min) in an
alternative context with distinct visual and tactile cues. The
context was changed in several ways to test conditioned fear of the
tone CS in the absence of contextual cues associated with shock.
First, the walls and floor of chamber were covered by black and
white plastic inserts. In addition, the chamber was scented with
limonene, and the experimenter wore a different style of gloves.
Furthermore, all rats were kept in a different holding room before
testing. The rate of freezing time was recorded.
Statistical analysis
All data are expressed as mean and standard error of
the mean. Normal distribution of the residuals was assessed by the
Shapiro-Wilk test. Comparisons among multiple groups were analyzed
using one-way ANOVA followed by Tukey's multiple comparison test.
For Morris water maze analysis, two-way ANOVA (treatment as
between-groups and time as repeated measures factors) followed by
Bonferroni multiple comparison testing was used. Data analysis was
generated using GraphPad Prism 5.0 (GraphPad Software, Inc.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
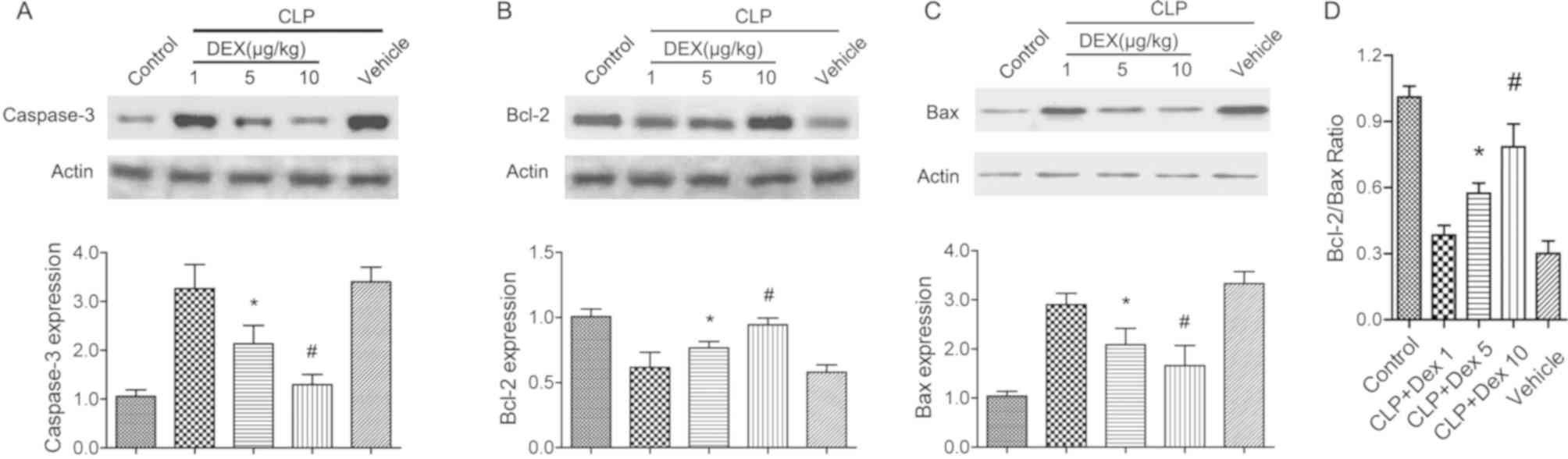
Administration of dexmedetomidine
attenuates neuronal apoptosis in CLP rats
Mitochondria-mediated neuronal apoptosis has been
reported to be involved in experimental sepsis (30). The protein level of cleaved
caspase-3 and Bcl-2 were detected to evaluate the neuronal
protective role of dexmedetomidine in CLP rats. Following CLP
surgery and dexmedetomidine treatment (1, 5 and 10 µg/kg), the
hippocampal tissues were harvested for western blot analysis. It
was found that CLP treatment significantly increased the expression
of cleaved caspase-3 and Bax (P<0.05; Fig. 1A and C), but decreased Bcl-2
(P<0.05; Fig. 1B). Application
of 5 or 10 µg/kg dexmedetomidine significantly restored the
alterations of cleaved caspase-3, Bcl-2 and Bax (P<0.05;
Fig. 1A-C). In addition,
application of dexmedetomidine (5 or 10 µg/kg) also significantly
increased the Bcl-2/Bax ratio (P<0.05; Fig. 1D). The present results suggested
that dexmedetomidine may protect brain against sepsis by
suppressing the intrinsic cell apoptosis signaling pathway.
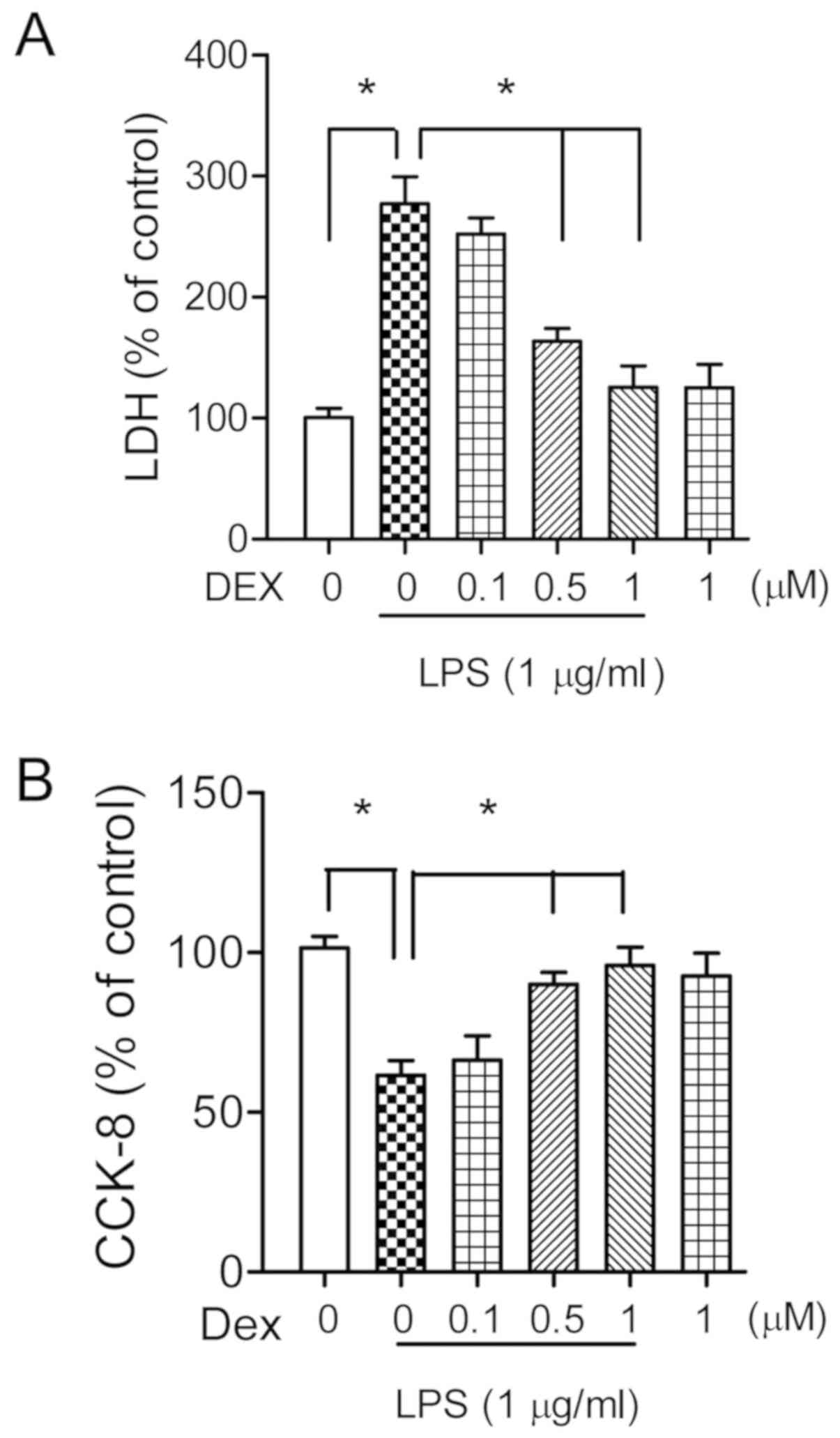
Dexmedetomidine increases neuronal
survival in LPS treated hippocampal cultures
To investigate the possible neuronal protective
effects of dexmedetomidine against endotoxemia, the primary
cultured hippocampal neurons (14 DIV) were co-incubated with LPS (1
µg/ml) and different doses of dexmedetomidine (0.1, 0.5 or 1 µM)
for 24 h. The neuronal injury was quantitatively evaluated through
the measurement of LDH activity in the medium. Analysis of LDH
assay suggested that LPS treatment significantly increased LDH
levels (273±29%, P<0.05; Fig.
2A), which were statistically decreased following treatment
with 0.5 or 1 µM of dexmedetomidine (163±11 and 127±18%,
respectively, P<0.05; Fig. 2A).
Administration of dexmedetomidine (1 µM) alone had no effects on
neuronal survival (P>0.05; Fig.
2A). These results were confirmed by CCK-8 assay, which showed
similar results (Fig. 2B).
Therefore, 1 µM of dexmedetomidine was selected for the following
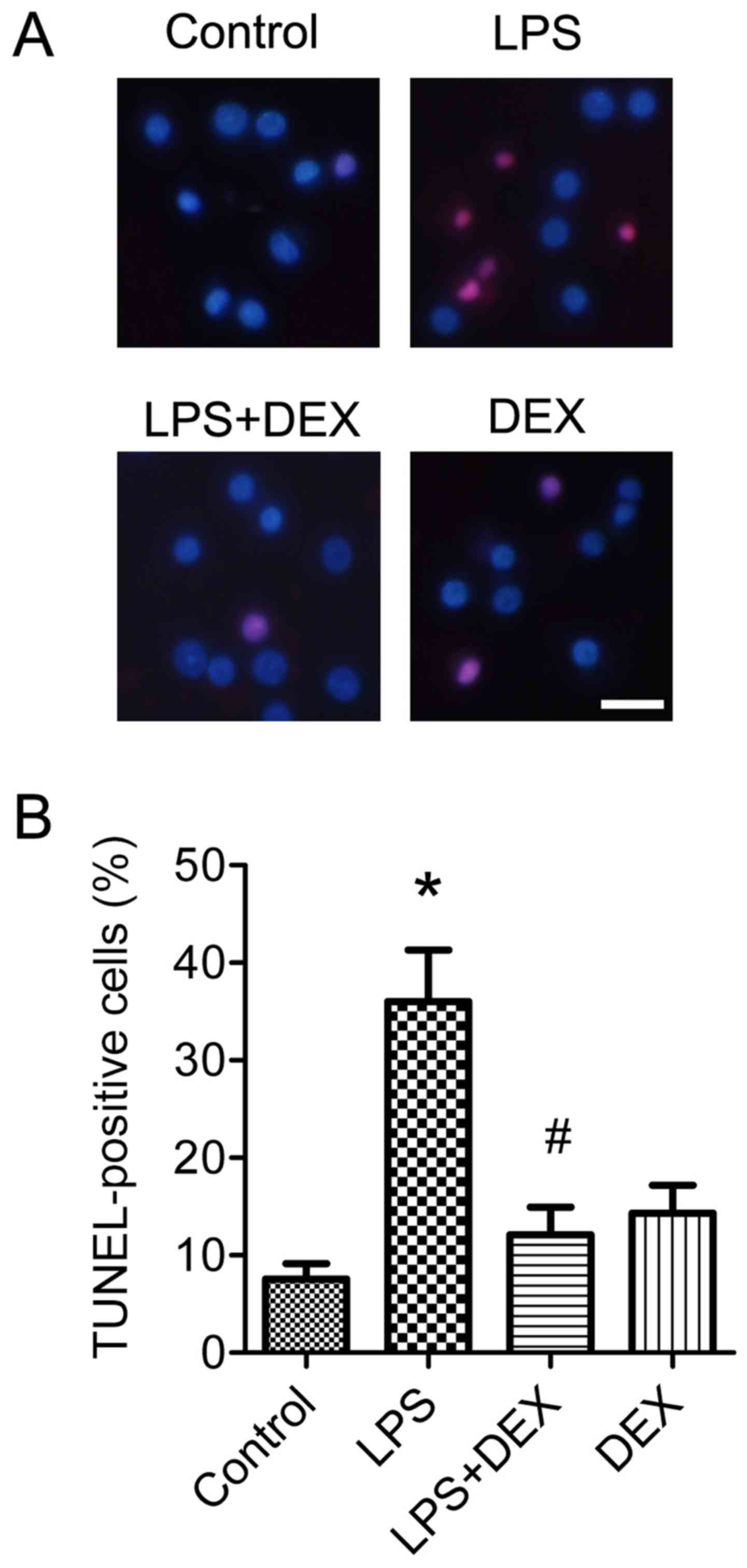
in vitro experiments. The neuronal protective role of
dexmedetomidine was also morphologically assessed by
TUNEL-staining. Analysis of data indicated that 1 µM of
dexmedetomidine significantly improved the neuronal apoptosis
induced by LPS treatment (P<0.05; Fig. 3A and B).
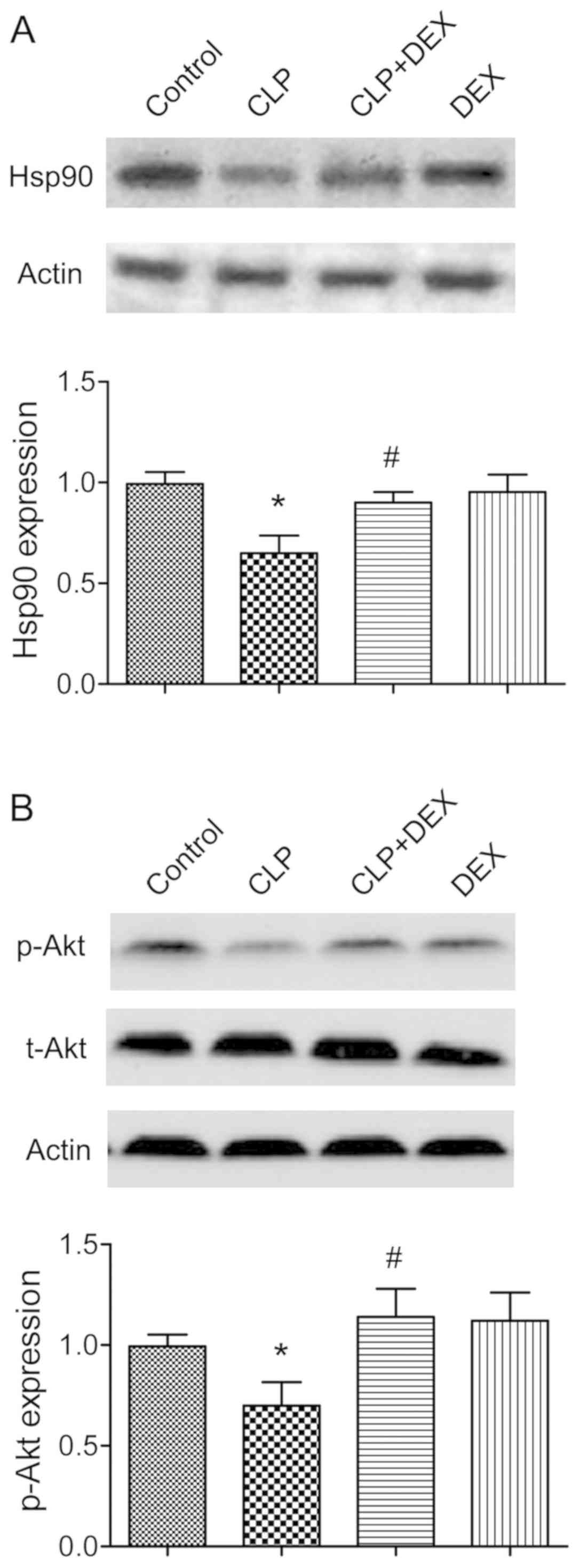
Hsp90/AKT signaling pathway is
involved in the neuronal protective effects of dexmedetomidine
Several previous studies have demonstrated that AKT
can be phosphorylated by Hsp90 at the Thr 308 site, and serves an
essential role in regulating neuronal fate (15,16).
The involvement of the Hsp90/AKT signaling pathway in the neuronal
protective effects of dexmedetomidine in rat models of CLP was
therefore examined. In the present study, the expressions of Hsp90
and p-AKT Thr 308 were significantly downregulated in CLP rats
(P<0.05; Fig. 4).
Administration of dexmedetomidine significantly ameliorated the
inhibition of Hsp90 and p-AKT Thr 308 (P<0.05; Fig. 4A and B).
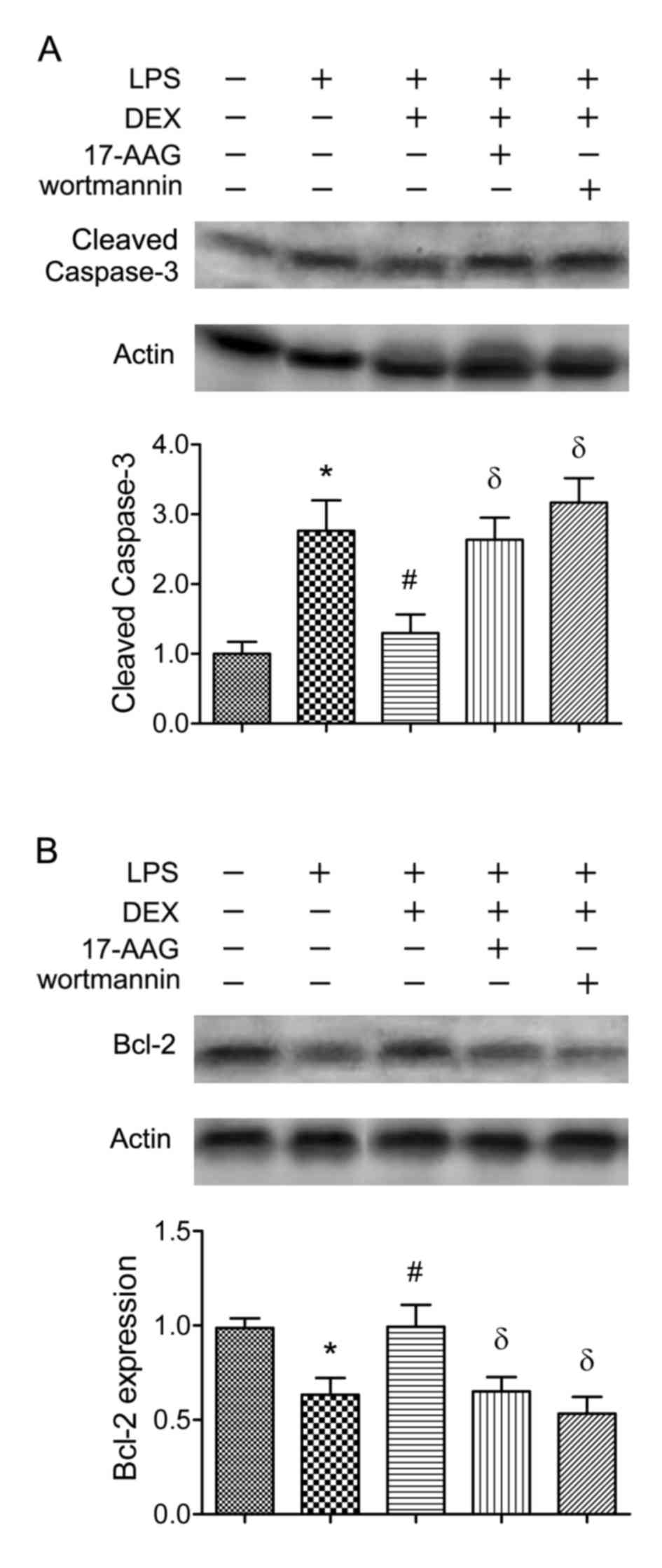
To further determine the involvement of the
Hsp90/AKT signaling pathway, 17-AAG (a Hsp90 inhibitor) or
wortmannin (a PI3K inhibitor) was injected in CLP rats. It was
found that the neuronal protective effects of dexmedetomidine were
abolished by 17-AAG or wortmannin (P<0.05; Fig. 5). To confirm these results, AKT
shRNA was transfected into hippocampal neurons for 48 h. The
transfection efficiency was shown in Fig. 6B. It was found that the
anti-apoptotic role of dexmedetomidine was also reversed by 17-AGG,
PI3K inhibitor wortmannin, or AKT shRNA transfection, as assessed
by TUNEL-assay (P<0.05; Fig.
6A).
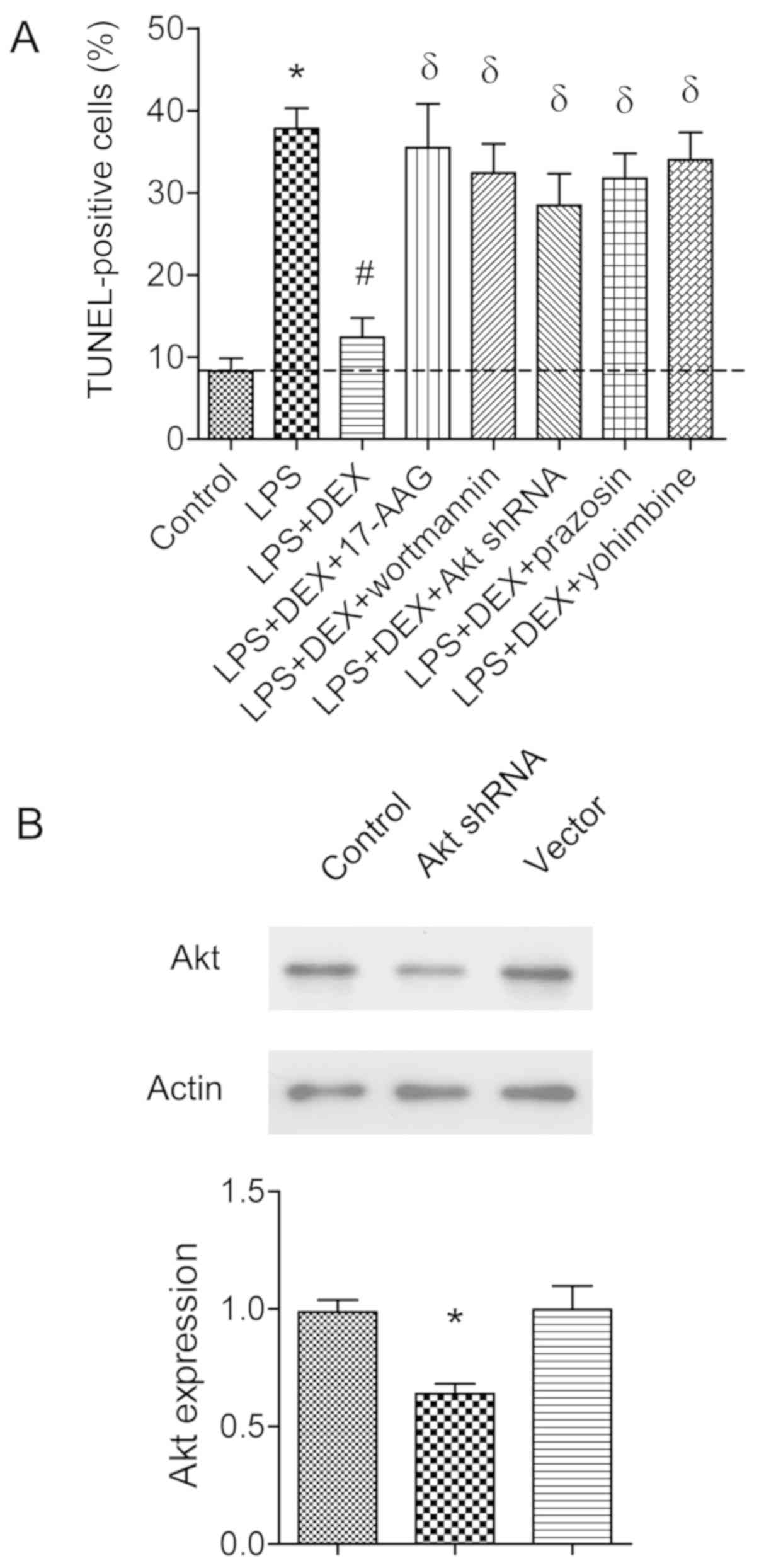 | Figure 6.Assessment of apoptotic cells by
TUNEL assay in primary hippocampal neuronal cultures. (A) Neuronal
cultures were co-incubated with LPS (1 µg/ml), or combined with
dexmedetomidine (1 µM), 17-AAG (1 µM), wortmannin (5 µM), prazosin
(0.5 µM) or yohimbine (0.3 µM) as indicated. Apoptotic neurons were
evaluated by TUNEL assay. The histogram shows the percentage of
TUNEL-positive cells. *P<0.05 vs. control group,
#P<0.05 vs. LPS group, δP<0.05 vs. LPS
+ DEX group, (n=8). (B) Neuronal cultures were transfected with AKT
shRNA or vector as control. The AKT protein expression was detected
by western blot analysis. *P<0.05 vs. control group, (n=5). LPS,
lipopolysaccharide; DEX, dexmedetomidine; shRNA, short hairpin
RNA. |
Dexmedetomidine is an α2-adrenergic
receptor agonist (31), so the
hypothesis that the neuronal protective role of dexmedetomidine
could be abolished by α2-adrenergic receptor antagonist
was then tested. Prazosin (0.5 µM) or yohimbine (0.3 µM) was
coincubated with LPS-treated neuronal cultures. It was found that
either prazosin or yohimbine had the capability to revert the
anti-apoptotic effects of dexmedetomidine (P<0.05; Fig. 6A).
Dexmedetomidine improves the spatial
and emotional disorders in CLP rats
The behavior study was performed 2 weeks after CLP
surgery, according to the protocol. In the open-field test, it was
found that there was no difference among groups as for the traveled
distance (P>0.05; Fig. 7A). For
the place trials, CLP rats were found to spend significant more
time to find the submerged platform at trial 3 day compared with
control rats (P<0.05; Fig. 7B).
The rats in the CLP + DEX group were compared with the control
group in terms of latency to find the submerged platform
(P>0.05; Fig. 7B). There were
no statistical differences in terms of average swimming speeds
(P>0.05; Fig. 7C), suggesting
that dexmedetomidine did not alter the locomotor activity. As for
the probe test in Morris water maze, the CLP rats had fewer
crossings compared with those of control rats (P<0.05; Fig. 7D). Rats of CLP + DEX group had
significantly more crossings compared with rats of CLP group
(P<0.05; Fig. 7D). In addition,
CLP rats spent significantly less time searching the target
quadrant than those of control group (P<0.05; Fig. 7E). Rats administered
dexmedetomidine spent significantly more time in the target
quadrant than CLP animals (P<0.05; Fig. 7E). In the fear-conditioning test,
it was found that the freezing time of CLP group was significantly
reduced compared with control rats both in the 24-h context and
48-h cue tests (P<0.05; Fig. 7F and
G). However, the freezing time in rats treated with
dexmedetomidine was significantly increased compared with the rats
of the CLP group in the 24-h context and 48-h cue tests (P<0.05;
Fig. 7F and G). Collectively, the
behavioral results of the present study suggested that
dexmedetomidine ameliorated both the spatial and emotional deficits
induced by systemic sepsis.
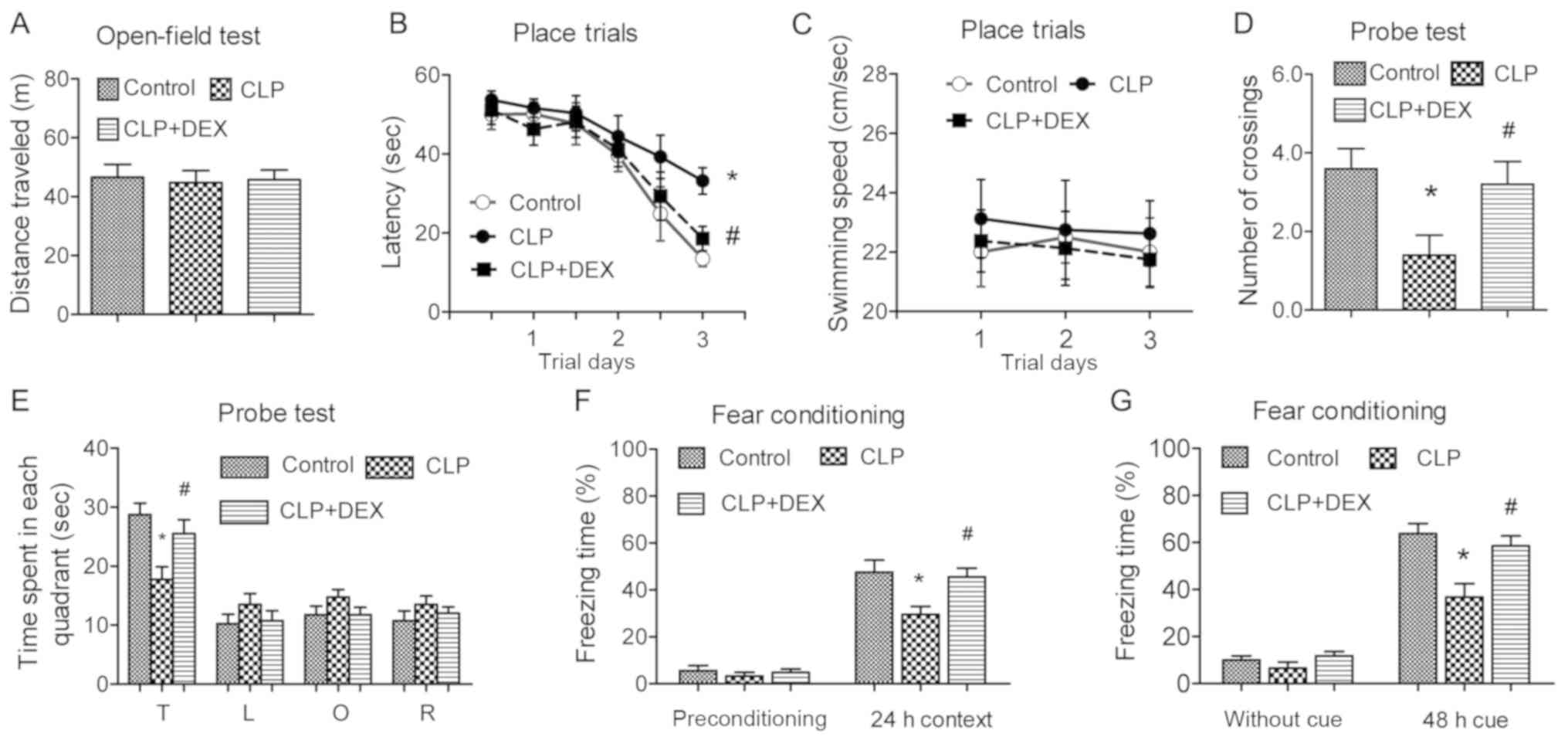 | Figure 7.Dexmedetomidine ameliorates the
spatial and emotional cognitive disorders in CLP rats. The behavior
study was performed 2 weeks after CLP surgery. The Open-field test,
Morris water maze and fear-conditioning test were performed to
examine the locomotor activity, spatial and emotional cognitive
disorders respectively. (A) The histogram shows the total distance
of each group rats traveled in 10 min. (B) In place trials of
Morris water maze, all rats received two blocks of trails (two
trials per block and 2 h rest between blocks) each day for
consecutive 3 days. Latency was defined as the time to reach the
submerged platform. (C) Each swimming speed of rat was recorded,
and the average in place trials of Morris water maze was
calculated. (D) The histogram represents the number of crossings of
each group in probe test of Morris water maze. (E) In the probe
test, the platform was removed and all rats commenced to swim (1
min) in the same quadrant. The histogram represents the percentage
of swimming time spent in each quadrant. The fear conditioning
consisted of a 5-min free exploration period followed by three
conditioned stimulus (80 db noise for 20 sec) and unconditioned
stimulus (1 mA for 1 sec) pairings. (F) The contextual test was
carried out in the conditioning chamber for 5 min in the absence of
noise at 24 h after conditioning. (G) The cued test was conducted
48 h after conditioning by presentation of a cue (80 db noise for 3
min) in an alternative context with distinct visual and tactile
cues. The rate of freezing time was recorded. All data are
expressed as mean ± SEM. The experimental rats were 17, 21 and 19
in control, CLP and CLP+DEX group respectively. Statistical
significance was determined by *P<0.05 vs. control group, and
#P<0.05 vs. CLP group. CLP, cecal ligation and
perforation; DEX, dexmedetomidine; T, target; L, left; O, opposite;
R, right. |
Discussion
SAE is the neurological manifestation of sepsis and
has been reported to occur in 9–71% of patients with sepsis
(32). Patients with SAE have a
higher mortality rate compared with those without brain
complications (33). Although SAE
could be caused by multiple factors including inflammation, reduced
cerebral blood flow, disruption of the blood-brain barrier, cell
injury of endothelium and cerebral edema (34,35),
the molecular mechanism underlying this process remains to be
elucidated. The present study demonstrated that administration of
dexmedetomidine decreased neuronal apoptosis and enhanced cell
viability in vitro and in vivo. In addition, it was
also found that dexmedetomidine markedly improved both the spatial
and emotional dysfunctions in CLP rats. Furthermore, the present
study provided evidence that Hsp90/AKT pathway may be involved in
the neuronal protective effects of dexmedetomidine in septic model.
To the best of the authors' knowledge, this is the first study
demonstrating the anti-apoptotic role of consecutive
dexmedetomidine exposure (1 week) in SAE model.
Dexmedetomidine exposure is an off-label therapy in
clinical contexts. Accumulating literature has shown the neuronal
protective role of dexmedetomidine in brain impairment (36,37).
The current study demonstrated that dexmedetomidine improved
neuronal apoptosis induced by systemic sepsis. Of note, the
sedative property of dexmedetomidine is dose-dependent. A recent
study demonstrated that 100% of rats given 10 µg/kg dexmedetomidine
were still able to right themselves with in 2 sec. However, only
33% of rats were still able to right themselves at 30 min when the
concentration increased to 25 µg/kg (38). In the present study, consecutive
dexmedetomidine exposure (10 µg/kg, i.p.) up to 1 week was chosen
for the rats of the CLP model. In recent years, dexmedetomidine has
been increasingly used for consecutive sedation of ICU patients
(39). This prolonged
dexmedetomidine exposure during recovering from systemic sepsis was
adopted in order to simulate an actual clinical practice in ICUs.
Prolonged dexmedetomidine exposure (average 2–3 weeks) has been
investigated to be safe in human study (38).
AKT is a central node in cell signaling downstream
of cytokines, growth factors, and other cellular stimuli (40). Aberrant loss or gain of AKT
activation underlies the pathophysiological properties of a variety
of complex diseases, including neuronal apoptosis and survival
(41). The present study
identified that AKT activity inhibition or AKT shRNA transfection
reversed the neuronal protective effects of dexmedetomidine,
indicating that AKT signaling serves an essential role in the
neuroprotection of dexmedetomidine against sepsis. In addition, AKT
pathway has also been reported to be involved in the
dexmedetomidine's biological effects encountering transient focal
ischemia/reperfusion injury (24),
isoflurane induced neuronal apoptosis (42), neuropathic pain (43) and LPS-induced acute lung injury
(10). Activation of α2-adrenergic
receptor is associated with increased AKT signaling in lung
apoptosis (44), heart
ischemia/reperfusion injury (45)
and arachidonic acid metabolism by cytochrome P450-dependent
epoxygenase (46). Taken together,
these previous studies indicate that AKT signaling pathway may
serve a critical function in molecular mechanisms of the
α2-adrenergic receptor.
Caspase-3 and Bcl-2 are important factors in
intrinsic neuronal apoptosis. The present study demonstrated that
dexmedetomidine treatment enhanced Bcl-2, but decreased caspase-3
expression, suggesting that dexmedetomidine protects the brain
against sepsis mainly through inhibiting mitochondria-mediated
neuronal apoptosis. Bad, as a proapoptotic Bcl-2 family member, can
also be phosphorylated by AKT, and allow Bcl-xl to bind to the
pro-apoptotic protein Bax (47,48).
Hypophosphorylated Bad interacts with the pro-survival Bcl-2 family
proteins, which frees Bak and Bax to induce apoptosis at the
mitochondria (49). In addition to
Bcl-2, AKT activation controls cell survival through
phosphorylation of downstream effector proteins, such as glycogen
synthase kinase 3β, Bad, Bid, Bax, caspase-9, and possibly
apoptosis inducing factor (50).
Therefore, further studies are required to identify the involvement
of other Bcl-2 family members and AKT downstream signaling in a
dexmedetomidine-exposed SAE model.
In the present study, the improvements of spatial
and emotional dysfunctions are mainly attributed to the decrease of
apoptotic hippocampal neurons. However, to fully appreciate these
results, the following points should be considered. First, AKT
signaling itself is important for learning and memory (51,52).
In addition to reducing neuronal apoptosis, the AKT activity
restored by dexmedetomidine exposure may participate in the
improvements of cognitive deficits. Second, 5 or 10 µg/kg of
dexmedetomidine was reported to improve spatial working memory in
α2C-adrenoceptor knockout mice (53). In addition, repeated administration
of dexmedetomidine demonstrated decreased anxiety-like behaviors,
impaired fear conditioning memory and improved spatial cognitive
impairments in post-traumatic stress disorder model (54). Therefore, the amelioration of
spatial and emotional disorders improved by dexmedetomidine
administration may be multifactorial, and further studies are
necessary to identify the relationship between consecutive
dexmedetomidine exposure and long-term cognitive effects in the
model of SAE.
Finally, one limitation of the present study was
that the concentration of dexmedetomidine was not measured in the
brain. A recent study determined the plasma and brain
pharmacokinetics of dexmedetomidine in neonatal rats, and
demonstrated the corresponding correlations between brain and
plasma dexmedetomidine concentrations in the inset. Uptake of
dexmedetomidine from the brain was rapid, and equilibration between
plasma and brain was apparent by 15 min after dexmedetomidine
injection (38). In summary, the
present study suggested that application of dexmedetomidine could
decrease neuronal apoptosis and increase neuronal survival in
vitro and in vivo. It was also demonstrated that
dexmedetomidine treatment markedly ameliorated the spatial and
emotional dysfunctions induced by systemic sepsis. Taken together,
the results suggested that application of dexmedetomidine could
improve neuronal apoptosis and cognitive disorders in septic
model.
Acknowledgements
The authors thank Professor Shoulin Zhang (Tianjin
Medical University) for kind assistance in investigating behavior
performance.
Funding
This project was supported by Tianjin Medical
University (grant no. 2017-FM-00016).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LY designed the present study and prepared the draft
manuscript. XC prepared the primary neuronal cultures and conducted
cell transfection. HJ performed all the molecular biological
experiments. SG performed the behavioral study, and data collection
and analysis.
Ethics approval and consent to
participate
All studies performed on animals were approved by
the local Institutional Animal Care and Use Committee (Tianjin
Baodi Hospital, Baodi Clinical College of Tianjin Medical
University, Tianjin, China). All animal experiments complied with
the ARRIVE guidelines and the National Institutes of Health guide
for the care and use of Laboratory animals (NIH Publications no.
8023, revised 1978) (24).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vincent JL, Sakr Y, Sprung CL, Ranieri VM,
Reinhart K, Gerlach H, Moreno R, Carlet J, Le Gall JR, Payen D, et
al: Sepsis in European intensive care units: Results of the SOAP
study. Crit Care Med. 34:344–353. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Iacobone E, Bailly-Salin J, Polito A,
Friedman D, Stevens RD and Sharshar T: Sepsis-associated
encephalopathy and its differential diagnosis. Crit Care Med. 37
(10 Suppl):S331–S336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Comim CM, Rezin GT, Scaini G, Di-Pietro
PB, Cardoso MR, Petronilho FC, Ritter C, Streck EL, Quevedo J and
Dal-Pizzol F: Mitochondrial respiratory chain and creatine kinase
activities in rat brain after sepsis induced by cecal ligation and
perforation. Mitochondrion. 8:313–318. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
d'Avila JC, Santiago AP, Amâncio RT,
Galina A, Oliveira MF and Bozza FA: Sepsis induces brain
mitochondrial dysfunction. Crit Care Med. 36:1925–1932. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weberpals M, Hermes M, Hermann S, Kummer
MP, Terwel D, Semmler A, Berger M, Schäfers M and Heneka MT: NOS2
gene deficiency protects from sepsis-induced long-term cognitive
deficits. J Neurosci. 29:14177–14184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hall JE, Uhrich TD, Barney JA, Arain SR
and Ebert TJ: Sedative, amnestic, and analgesic properties of
small-dose dexmedetomidine infusions. Anesth Analg. 90:699–705.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Arcangeli A, D'alo C and Gaspari R:
Dexmedetomidine use in general anaesthesia. Current Drug Targets.
10:687–695. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pandharipande PP, Pun BT, Herr DL, Maze M,
Girard TD, Miller RR, Shintani AK, Thompson JL, Jackson JC, Deppen
SA, et al: Effect of sedation with dexmedetomidine vs lorazepam on
acute brain dysfunction in mechanically ventilated patients: The
MENDS randomized controlled trial. JAMA. 298:2644–2653. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Martin E, Ramsay GJ, Mantz J and Sum-Ping
ST: The role of the alpha2-adrenoceptor agonist dexmedetomidine in
postsurgical sedation in the intensive care unit. J Intensive Care
Med. 18:29–41. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang CL, Chen CH, Tsai PS, Wang TY and
Huang CJ: Protective effects of dexmedetomidine-ketamine
combination against ventilator-induced lung injury in endotoxemia
rats. J Surg Res. 167:e273–e281. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Okada H, Kurita T, Mochizuki T, Morita K
and Sato S: The cardioprotective effect of dexmedetomidine on
global ischaemia in isolated rat hearts. Resuscitation. 74:538–545.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kocoglu H, Ozturk H, Ozturk H, Yilmaz F
and Gulcu N: Effect of dexmedetomidine on ischemia-reperfusion
injury in rat kidney: A histopathologic study. Ren Fail. 31:70–74.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Engelhard K, Werner C, Eberspacher E,
Bachl M, Blobner M, Hildt E, Hutzler P and Kochs E: The effect of
the alpha2-agonist dexmedetomidine and the N-methyl-D-aspartate
antagonist S(+)-ketamine on the expression of apoptosis-regulating
proteins after incomplete cerebral ischemia and reperfusion in
rats. Anesth Analg. 96:524–531. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hanci V, Erol B, Bektas S, Mungan G,
Yurtlu S, Tokgöz H, Can M and Ozkoçak Turan I: Effect of
dexmedetomidine on testicular torsion/detorsion damage in rats.
Urol Int. 84:105–111. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Downward J: PI 3-kinase, AKT and cell
survival. Semin Cell Dev Biol. 15:177–182. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gass P, Schröder H, Prior P and Kiessling
M: Constitutive expression of heat shock protein 90 (HSP90) in
neurons of the rat brain. Neurosci Lett. 182:188–192. 1194.
View Article : Google Scholar
|
|
17
|
Sato S, Fujita N and Tsuruo T: Modulation
of AKT kinase activity by binding to Hsp90. Proc Natl Acad Sci USA.
97:10832–10837. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li X, Luo R, Jiang R, Meng X, Wu X, Zhang
S and Hua W: The role of the Hsp90/AKT pathway in myocardial
calpain-induced caspase-3 activation and apoptosis during sepsis.
BMC Cardiovas Dis. 13:82013. View Article : Google Scholar
|
|
19
|
Shen E, Fan J and Peng T: Glycogen
synthase kinase-3beta suppresses tumor necrosis factor-alpha
expression in cardiomyocytes during lipopolysaccharide stimulation.
J Cell Biochem. 104:329–338. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Basso AD, Solit DB, Chiosis G, Giri B,
Tsichlis P and Rosen N: AKT forms an intracellular complex with
heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by
inhibitors of Hsp90 function. J Biol Chem. 277:39858–39866. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang R, Luo D, Miao R, Bai L, Ge Q, Sessa
WC and Min W: Hsp90-AKT phosphorylates ASK1 and inhibits
ASK1-mediated apoptosis. Oncogene. 24:3954–3963. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu YM, Wang CC, Chen L, Qian LB, Ma LL,
Yu J, Zhu MH, Wen CY, Yu LN and Yan M: Both PI3K/AKT and ERK1/2
pathways participate in the protection by dexmedetomidine against
transient focal cerebral ischemia/reperfusion injury in rats. Brain
Res. 1494:1–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McGrath JC, Drummond GB, McLachlan EM,
Kilkenny C and Wainwright CL: Guidelines for reporting experiments
involving animals: The ARRIVE guidelines. Br J Pharmacol.
160:1573–1576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bayne K: Revised guide for the care and
use of laboratory animals available. American Physiological
Society. Physiologist. 39:199, 208–211. 1996.
|
|
25
|
Kaech S and Banker G: Culturing
hippocampal neurons. Nat Protoc. 1:2406–2415. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang X, Venet F, Wang YL, Lepape A, Yuan
Z, Chen Y, Swan R, Kherouf H, Monneret G, Chung CS and Ayala A:
PD-1 expression by macrophages serves a pathologic role in altering
microbial clearance and the innate inflammatory response to sepsis.
Proc Natl Acad Sci USA. 106:6303–6308. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ritter C, Andrades ME, Reinke A,
Menna-Barreto S, Moreira JC and Dal-Pizzol F: Treatment with
Nacetylcysteine plus deferoxamine protects rats against oxidative
stress and improves survival in sepsis. Crit Care Med. 32:342–249.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vorhees CV and Williams MT: Morris water
maze: Procedures for assessing spatial and related forms of
learning and memory. Nat Protoc. 1:848–858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Satomoto M, Satoh Y, Terui K, Miyao H,
Takishima K, Ito M and Imaki J: Neonatal exposure to sevoflurane
induces abnormal social behaviors and deficits in fear conditioning
in mice. Anesthesiology. 110:628–637. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Brealey D, Karyampudi S, Jacques TS,
Novelli M, Stidwill R, Taylor V, Smolenski RT and Singer M:
Mitochondrial dysfunction in a long-term rodent model of sepsis and
organ failure. Am J Physiol Regul Integr Comp Physiol.
286:R491–R497. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kamibayashi T and Maze M: Clinical uses of
alpha2-adrenergic agonists. Anesthesiology. 93:1345–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Siami S, Annane D and Sharshar T: The
encephalopathy in sepsis. Crit Care Clin. 24:67–82. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pytel P and Alexander JJ: Pathogenesis of
septic encephalopathy. Cur Opin Neurol. 22:283–287. 2009.
View Article : Google Scholar
|
|
34
|
Semmler A, Hermann S, Mormann F, Weberpals
M, Paxian SA, Okulla T, Schäfers M, Kummer MP, Klockgether T and
Heneka MT: Sepsis causes neuroinflammation and concomitant decrease
of cerebral metabolism. J Neuroinflam. 5:382008. View Article : Google Scholar
|
|
35
|
Taccone FS, Su F, Pierrakos C, He X, James
S, Dewitte O, Vincent JL and De Backer D: Cerebral microcirculation
is impaired during sepsis: An experimental study. Crit Care.
14:R1402010. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schoeler M, Loetscher PD, Rossaint R,
Fahlenkamp AV, Eberhardt G, Rex S, Weis J and Coburn M:
Dexmedetomidine is neuroprotective in an in vitro model for
traumatic brain injury. BMC Neurol. 12:202012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Degos V, Charpentier TL, Chhor V, Brissaud
O, Lebon S, Schwendimann L, Bednareck N, Passemard S, Mantz J and
Gressens P: Neuroprotective effects of dexmedetomidine against
glutamate agonist-induced neuronal cell death are related to
increased astrocyte brain-derived neurotrophic factor expression.
Anesthesiology. 118:1123–1132. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
McAdams RM, McPherson RJ, Kapur R,
Phillips B, Shen DD and Juul SE: Dexmedetomidine reduces cranial
temperature in hypothermic neonatal rats. Pediatr Res. 77:772–778.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Park JH, Derry K and Owens R: 896:
Dexmedetomidine as adjunctive sedation in mechanically ventilated
patients. Crit Care Med. 47:4272019. View Article : Google Scholar
|
|
40
|
Martini M, De Santis MC, Braccini L,
Gulluni F and Hirsch E: PI3K/AKT signaling pathway and cancer: An
updated review. Ann Med. 46:372–383. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dudek H, Datta SR, Franke TF, Birnbaum MJ,
Yao R, Cooper GM, Segal RA, Kaplan DR and Greenberg ME: Regulation
of neuronal survival by the serine-threonine protein kinase AKT.
Science. 275:661–665. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li Y, Zeng M, Chen W, Liu C, Wang F, Han
X, Zuo Z and Peng S: Dexmedetomidine reduces isofluraneinduced
neuroapoptosis partly by preserving PI3K/AKT pathway in the
hippocampus of neonatal rats. PLoS One. 9:e936392014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Farghaly HS, Mahmoud AM and Abdel-Sater
KA: Effect of dexmedetomidine and cold stress in a rat model of
neuropathic pain: Role of interleukin-6 and tumor necrosis
factor-α. Eur J Pharmacol. 776:139–145. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li J, Chen Q, He X, Alam A, Ning J, Yi B,
Lu K and Gu J: Dexmedetomidine attenuates lung apoptosis induced by
renal ischemia-reperfusion injury through α2AR/PI3K/AKT
pathway. J Transl Med. 16:782018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ibacache M, Sanchez G, Pedrozo Z, Galvez
F, Humeres C, Echevarria G, Duaso J, Hassi M, Garcia L, Díaz-Araya
G and Lavandero S: Dexmedetomidine preconditioning activates
pro-survival kinases and attenuates regional ischemia/reperfusion
injury in rat heart. Biochim Biophys Acta. 1822:537–545. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Karkoulias G, Mastrogianni O,
Lymperopoulos A, Paris H and Flordellis C: alpha(2)-Adrenergic
receptors activate MAPK and AKT through a pathway involving
arachidonic acid metabolism by cytochrome P450-dependent
epoxygenase, matrix metalloproteinase activation and
subtype-specific transactivation of EGFR. Cell Signal. 18:729–739.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cai W, Rudolph JL, Harrison SM, Jin L,
Frantz AL, Harrison DA and Andres DA: An evolutionarily conserved
Rit GTPase-p38 MAPK signaling pathway mediates oxidative stress
resistance. Mol Biol Cell. 22:3231–3241. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Koh PO: Nicotinamide attenuates the
ischemic brain injury-induced decrease of AKT activation and Bad
phosphorylation. Neurosci Lett. 498:105–109. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mendoza MC, Er EE and Blenis J: The
Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends
Biochem Sci. 36:320–328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Parcellier A, Tintignac LA, Zhuravleva E
and Hemmings BA: PKB and the mitochondria: AKTing on apoptosis.
Cell Signal. 20:21–30. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Franke TF: PI3K/Akt: Getting it right
matters. Oncogene. 27:6473–6488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Horwood JM, Dufour F, Laroche S and Davis
S: Signalling mechanisms mediated by the phosphoinositide
3-kinase/AKT cascade in synaptic plasticity and memory in the rat.
Eur J Neurosci. 23:3375–3384. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Björklund M, Siverina I, Heikkinen T,
Tanila H, Sallinen J, Scheinin M and Riekkinen P Jr: Spatial
working memory improvement by an alpha2-adrenoceptor agonist
dexmedetomidine is not mediated through alpha2C-adrenoceptor. Prog
Neuropsychopharmacol Biol Psychiatry. 25:1539–1554. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ji MH, Jia M, Zhang MQ, Liu WX, Xie ZC,
Wang ZY and Yang JJ: Dexmedetomidine alleviates anxiety-like
behaviors and cognitive impairments in a rat model of
post-traumatic stress disorder. Prog Neuropsychopharmacol Biol
Psychiatry. 54:284–288. 2014. View Article : Google Scholar : PubMed/NCBI
|