Introduction
Distal arthrogryposis (DA), a group of autosomal
dominant disorders with clinical and genetic heterogeneity, is
characterized by congenital non-progressive contractures of the
distal upper and/or lower limbs (1,2).
Typically, the distinctive clinical features of DA include severe
camptodactyly, overriding fingers, thumb adduction, ulnar
deviation, hypoplastic or absent interphalangeal creases of hands,
calcaneovalgus deformities, clubfoot and short stature (2). It should be noted that patients with
DA usually exhibit normal levels of intelligence, without primary
neurological or muscular diseases (2,3).
To date, the most common classification of DA was
introduced by Bamshad et al (2) in 1996, who generalized DA into nine
types (types 1 to 9) according to clinical features. In this
classification system, only DA1 was not associated with extra
abnormalities, whereas the other forms of DA had various additional
phenotypic features (2). In 1997,
Krakowiak et al (4)
reported a large family containing 21 patients over 5 generations
that were affected with an intermediate disorder between DA1 and
DA2. They termed this novel type as DA type 2B (DA2B), which is
also known as Sheldon-Hall syndrome, and altered the previous type
DA2 to DA2A (4). Following this,
Stevenson et al (5)
reported on a family affected with plantar flexion contractures,
which was defined as DA10 in 2006. On the basis of different
causative genes, DA1 was further divided into DA1A and DA1B
(6). The newest DA classification
system was established by various physical and genetic features
(Table I) (7). It should be noted that there are some
overlapping features among the different types of DA, which has
added difficulties to diagnosis.
 | Table I.Classification of DA. |
Table I.
Classification of DA.
| Type | Subtype | Characteristic
phenotype | Relative genes | OMIM number |
|---|
| DA1 | DA1A | Camptodactyly,
tight clenched fists neonatally | TPM2, TNNI2, TNNT3,
MYH3 | 108120 |
|
| DA1B | Camptodactyly,
ulnar deviation of the fingers | MYBPC1 | 614335 |
| DA2 | DA2A | H-shaped chin
dimple, ‘Whistling’ appearance | MYH3 | 193700 |
|
| DA2B | Intermediate
between DA1 and DA2A | TNNI2, TNNT3, MYH3,
TPM2 | 601680 |
| DA3 | / | Short stature,
cleft palate | PIEZO2 | 114300 |
| DA4 | / | Severe
scoliosis | / | 609128 |
| DA5 | / | Ocular
abnormalities, ptosis, ophthalmoplegia | PIEZO2 | 108145 |
| DA6 | / | Sensorineural
deafness | / | 108200 |
| DA7 | / | Trismus | MYH8 | 158300 |
| DA8 | / | Multiple
pterygia | MYH3 | 178110 |
| DA9 | / | Congenital
contractural arachnodactyly | FBN2 | 121050 |
| DA10 | / | Congenital plantar
contracture | / | 187370 |
DA2B, one of the most common types of DA, was first
described by Krakowiak et al (4) in 1997. In addition to the common
manifestations of DA, the typical phenotypes of DA2B also include
several facial features such as a triangular face, micrognathia,
down-slanting palpebral fissures, prominent nasolabial folds and a
small mouth (2,8–10).
In recent years, mutations in four genes have been successively
reported to be responsible for DA2B. In 2003, Sung et al
(11,12) determined that DA2B may be caused by
mutations in the troponin I, fast-twitch skeletal muscle isoform
(TNNI2; MIM 191043) and troponin T3, fast skeletal
(TNNT3; MIM 600692) genes; this work established the
foundation for studying the genetic mechanism underlying DA2B.
Missense mutations in myosin heavy chain 3 (MYH3; MIM
160720), which encodes embryonic MYH, were then identified by
Toydemir et al (9) in 2006.
It has been hypothesized that mutations in the MYH3, TNNI2
and TNNT3 genes collectively account for ~50% of all cases
of DA2B. In addition, Tajsharghi et al (3) identified a heterozygous mutation in
the tropomyosin 2 (TPM2; MIM 190990) gene, further enriching
the genetic background of DA2B. In general, these mutations cause
DA2B by altering the adenosine triphosphatase (ATPase) activity of
myosin or by influencing the interactions between myosin and other
proteins (9,13,14).
To date, all of the previous cases of DA2B
associated with MYH3 have been caused by missense mutations,
except for a 3-bp deletion reported in a small number of cases
(9,15–18).
Furthermore, mutations in MYH3 have also been observed in
DA1, DA2A and DA8 (9,16,19,20).
In contrast to the other types of DA, all affected residues in DA2B
are located within the head and neck domains of embryonic MYH
(9,15–17),
and it interacts with other proteins of the contractile apparatus
including light chain, actin and troponin (14,21).
The present study identified a novel causative
missense mutation of the MYH3 gene in a Chinese family with
DA2B. Consistent with previous studies (9,20),
the mutation site identified in the present results was located in
the neck domain of embryonic MYH.
Materials and methods
Patients
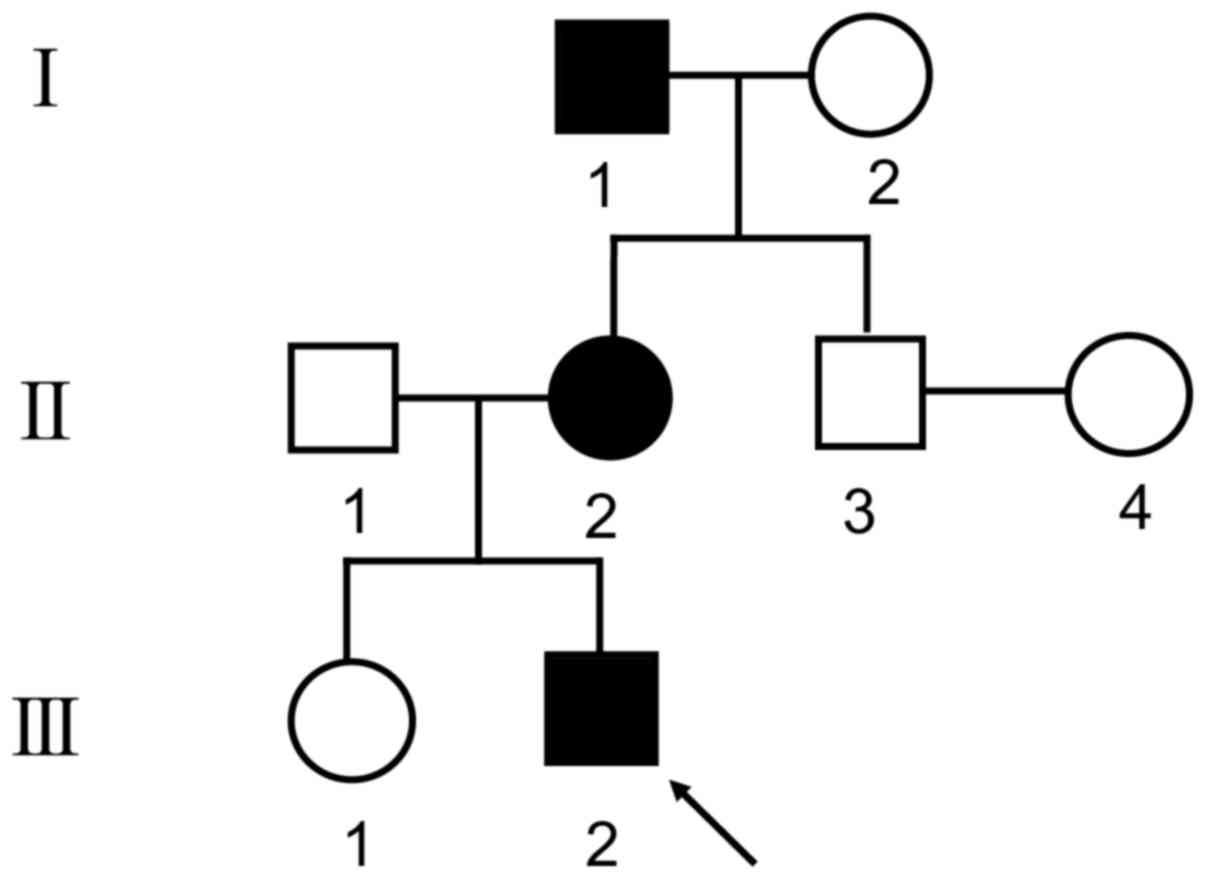
A three-generation non-consanguineous family
(Fig. 1) containing three DA2B
patients, identified by two independent orthopedic surgeons, was
investigated in the present study. The three patients were a
4-year-old boy (III2), a 31-year-old female
(II2) and a 64-year-old male (I1). This
family was recruited from the outpatient department of Shanghai
Jiao Tong University Affiliated Sixth People's Hospital (Shanghai,
China) when they sought treatment for the proband
(III2). All of the 8 family members underwent systematic
physical assessment and the proband was additionally subjected to
X-ray examination. Written informed consent was obtained from all
of the participants. The present study was approved by the Ethics
Committees of Shanghai Jiao Tong University Affiliated Sixth
People's Hospital (Shanghai, China).
Whole-exome sequencing and mutation
prediction
Peripheral blood samples from all of the available
family members (except II4) and 100 unrelated healthy
donors aged 18–50 (50 males and 50 females) were collected and
stored at −80°C until use in subsequent experiments. Genomic DNA
was then extracted using the QuickGene DNA whole blood kit (Kurabo
Industries Ltd., Osaka, Japan). The present study sequenced the
whole-exome of the three affected individuals in order to determine
the mutated gene that induces this disorder. The SureSelect Human
All Exon 57Mb Kit (Agilent Technologies, Inc., Santa Clare, CA,
USA) and the HiSeq 2000 platform (Illumina, Inc., San Diego, CA,
USA) were used to capture the whole-exome sequence, following the
manufacturer's protocol. Illumina base-calling software v1.7 was
then applied to convert the raw image files into 90-base-paired-end
reads. BioAnalyzer 2100 (Agilent Technologies, CA, USA) was
subsequently employed to analyze the bioinformatics and all of the
mutations were validated.
All detected variants were filtered against six
single-nucleotide polymorphism databases, including dbSNP144
(hgdownload.cse.ucsc.edu/goldenPath/hg19/database/snp144)
(22), the HapMap Project
(ftp.ncbi.nlm.nih.gov/hapmap)
(23), the 1,000 Genomes Project
(www.internationalgenome.org/), the
YanHuang database (yh.genomics.org.cn/), the Exome Variant Server
(evs.gs.washington.edu/EVS/) and the
Exome Aggregation Consortium database (exac.broadinstitute.org/). Noncoding variants were
initially excluded and the most likely pathogenic candidate
mutations were presented in accordance with previous studies
(3,9,11,12).
The present study further checked the conservation of altered amino
acid residues and predicted the risk of missense mutations by
utilizing Polymorphism Phenotyping version 2 (Polyphen-2;
genetics.bwh.harvard.edu/pph2/) and
Protein Variation Effect Analyzer (PROVEAN; provean.jcvi.org/index.php/).
Sanger sequencing
The DNA sequence for the MYH3 gene was
obtained from NCBI gene database (Gene ID: 4621). The two mutation
regions of the MYH3 gene that were identified following
whole-exome sequencing were amplified using a standard polymerase
chain reaction protocol (24). The
primers were designed using Primer-3 software
(bioinfo.ut.ee/primer.3-0.4.0/). Direct sequencing was then
performed using the BigDye Terminator Cycle Sequencing Ready
Reaction Kit, version 3.1 (Applied Biosystems; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), and the sequences were
analyzed with an ABI Prism 3130 automated sequencer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Mutations were checked
using Polyphred program (droog.mbt.washington.edu/poly_get.html).
Protein spatial model
construction
The wild type ribbon structure of embryonic MYH was
initially constructed using DeepView and SWISS-MODEL (swissmodel.expasy.org/repository/uniprot/P11055). In
addition, to illustrate the position of the pathogenic missense
mutation p.K836E, the amino acid residue substitution was further
incorporated into the sequence and the mutant protein structure was
constructed. Then, the interactive protein associated with the
altered position was added to the structure.
Results
Clinical features
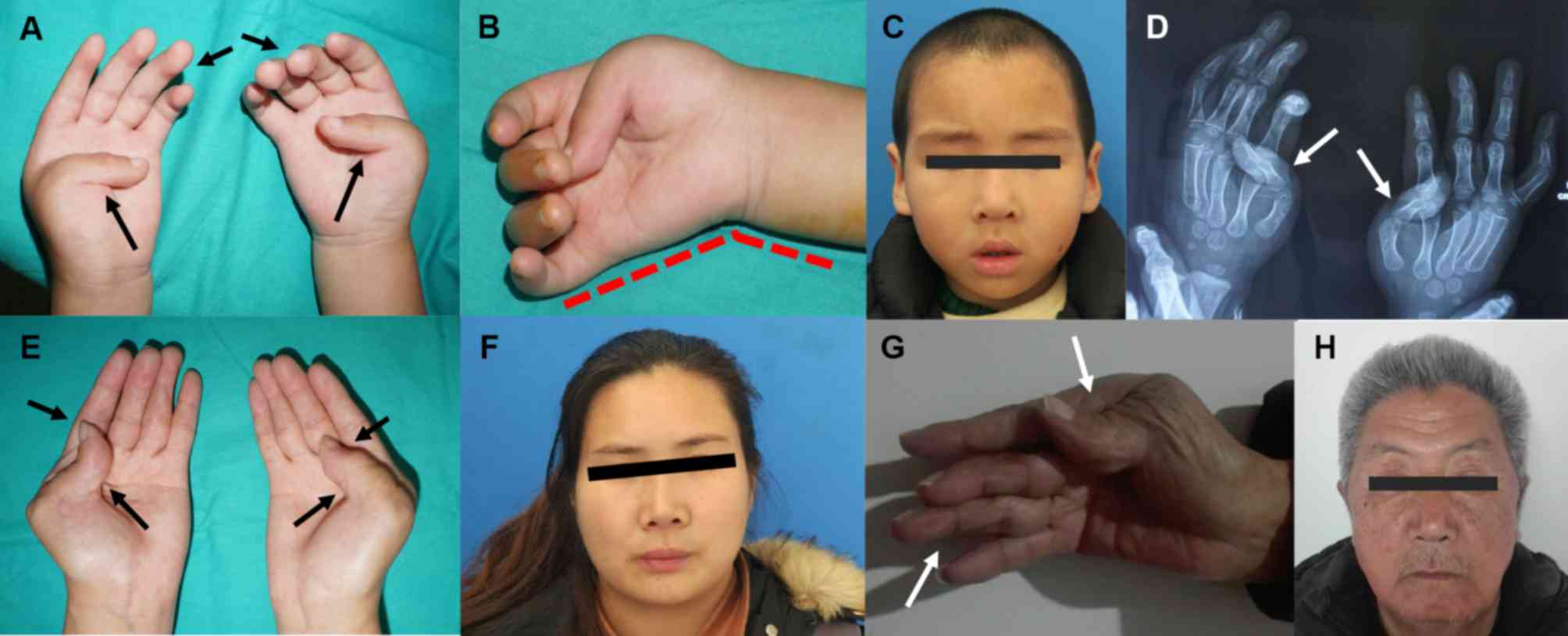
The primary phenotypes of the 4-year-old
(III2) proband and his mother (II2) were
highly consistent (Fig. 2),
including bilaterally medium camptodactyly, thumb adduction,
evident ulnar deviation and hypoplastic interphalangeal creases of
the hands (Fig. 2A, B, D and E).
As for facial features, only slight nasolabial folds and small
mouths without down-slanting palpebral fissures were observed
(Fig. 2C and F).
Notably, the proband's grandfather (I1)
had a normal left hand, while his right hand exhibited similar
manifestations to those of the other aforementioned patients
(Fig. 2G). Furthermore, the facial
features of I1 were also asymmetric, including the
prominent right nasolabial fold, the down-slanting palpebral
fissure and the relatively small mouth (Fig. 2H). In addition, short statures were
observed in all three of the affected family members when compared
with their peers. All unaffected individuals in this family had
normal hands and medium statures without any characteristic facial
appearances.
Mutation analysis
Following systematic screening, >100 potentially
disease-causing candidate single nucleotide variants among the
three patients remained. Considering the function of the genes and
the results of previous studies (9,20),
the present study focused on the novel heterozygous missense
mutation c.2506A>G (p.K836E) in exon 22 of the MYH3 gene.
The Polyphen-2 and PROVEAN scores of the mutation c.2506A>G
(p.K836E) were 0.785 and −3.277, respectively, which indicated a
‘deleterious’ function. Notably, another missense mutation
c.5263A>G (p.K1755E) of MYH3 was identified in the
proband's genomic DNA; however, it was not observed in the other
two patients. According to the single-nucleotide polymorphism (SNP)
databases, c.5263A>G (p.K1755E) was recorded as an SNP
(rs546653497) with a rather low minor allele frequency (MAF;
0.0002). However, c.2506A>G (p.K836E) was not observed in the
SNP database and its MAF was far <1% (0.00%) in the control
population; thus, the present study identified it as a
mutation.
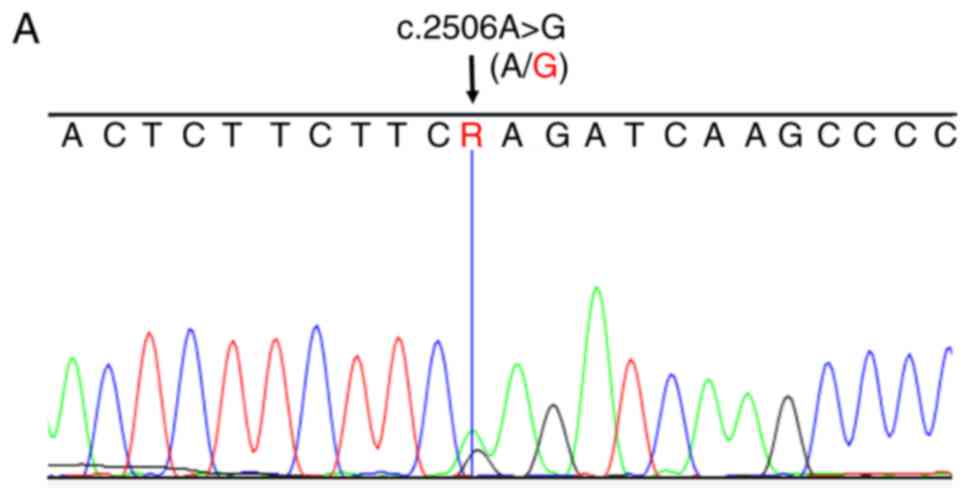
To confirm the two mutations of the MYH3 gene
revealed through whole-exome sequencing, the present study
performed Sanger sequencing for all of the available family members
and the 100 healthy donors. In accordance with the results of
whole-exome sequencing, the mutation c.2506A>G (p.K836E) was
identified in the three affected individuals (Fig. 3A); however, not in the available
unaffected family members and the 100 healthy donors (Fig. 3B). The p.K1755E SNP was only
identified in the proband and his father, and not in the other
available family members (Fig. 3C and
D). In addition, the mutation c.2506A>G (p.K836E) also
affected highly conserved amino acid residues in other species
(Fig. 3E).
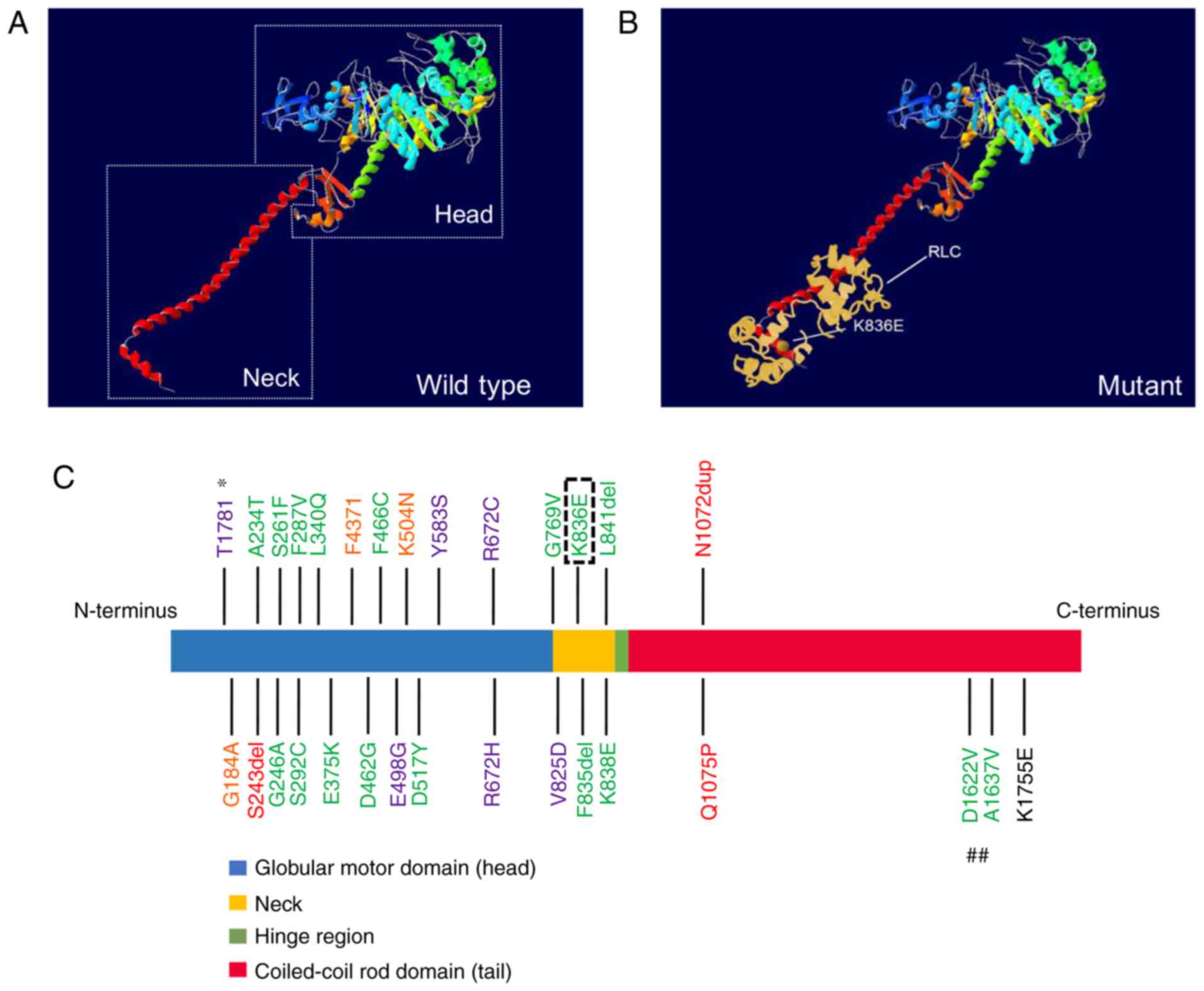
Protein structural model
According to the spatial ribbon structure of the
protein, the mutant p.K836E is located in the neck, and spatially,
on the surface of embryonic MYH, adjacent to several previous
mutation sites in DA2B (Fig. 4A-C)
(9,16). Although there is no apparent
difference between the wild-type protein and the mutant one, the
changed region would still tightly interact with the regulatory
light chain (RLC) (Fig. 4B). In
addition, the SNP p.K1755E is located at the tail far away from the
other mutation sites associated with DA (Fig. 4C).
Discussion
DA2B is an autosomal dominant inherited congenital
disorder, resulting from mutations in genes associated with
embryonic limb morphogenesis (10). On account of the similar clinical
features, DA2B, DA1 and DA2A are occasionally considered to be a
phenotypic continuum of one syndrome (25). Due to this, DA2B is difficult to
diagnose, as its clinical symptoms, including camptodactyly,
overriding fingers, thumb adduction, ulnar deviation, facial
features, and hypoplastic interphalangeal creases of hands, are
also present in DA1 and DA2A (2,10).
The affected individuals in the present study exhibited typical
limb manifestations including medium camptodactyly, thumb
adduction, evident ulnar deviation and slight facial features
without scoliosis. Therefore, the diagnosis of DA2B was clear.
Notably, only the unilateral limb and face of
I1 were affected, while the other two patients exhibited
a symmetric appearance. In addition, all of the siblings of
I1 and their offspring were not affected (data not
shown). The intra-familial difference may be a result of various
factors such as incomplete penetrance and variable expressivity,
which are common in congenital developmental diseases. These
phenomena are usually based on differing susceptibilities due to
discrepancies in gene background and the uterine environment during
the embryonic period. Furthermore, the asymmetric phenotype of
I1 may also be explained by p.K836E as a de novo
mutation, occurring during the embryonic development period,
affecting the right half of his body with a certain number of
reproductive cells, which may have enabled him to transmit the
pathogenic mutation to his descendants (18). However, this hypothesis of
chimerism was hard to confirm in the present study as tissues from
different parts of the patient's body were not available due to
individual reluctance and ethical concerns.
In recent years, it has been reported that a total
of four genes are associated with DA2B, including TNNI2, TNNT3,
MYH3 and TPM2, all of which encode the fast-twitch
skeletal muscle myofibers complex and participate in skeletal
muscle contraction (3,9,11,12).
The MYH3 gene, which is mapped to chromosome 17p13.1, is
composed of 41 exons, 39 of which are responsible for protein
coding (26). Embryonic MYH is the
product of the MYH3 gene, which consists of 1,940 amino
acids, and serves a vital role in the embryonic morphogenesis of
the limbs and face (9). The
embryonic MYH is divided into a globular motor domain (amino acid
residues 1- ~767), a short neck (~767-~847) and a long coiled-coil
rod domain (~847-1,940) attached to the neck by a hinge (~847)
(9). When variants occur in the
protein coding regions of the MYH3 gene, the spatial
structure of embryonic MYH may be altered, leading to interaction
impairment with the light chain or actin, as well as dysfunction of
ATPase activity (9,15,21).
The present study detected the mutation c.2506A>G
(p.K836E) in exon 22 of MYH3, where the short neck of
embryonic MYH was affected, according to the protein structure
modeling. This mutation may impair the interaction between the
embryonic MYH and RLC, damaging the regulatory mechanism and
skeletal muscle activity (14,21).
In addition, the SNP c.5263A>G (p.K1755E) was located in the
myosin tail domain, where the change was rarely relevant for
binding ATP or other proteins (20). Therefore, the SNP c.5263A>G
(p.K1755E) appeared not to be critical for MYH3 functioning.
Additionally, the proband's father (II1) exhibited a
normal appearance, which also provided further evidence to confirm
that the disease-causing mutation was c.2506A>G (p.K836E) in the
present study.
Thus far, 28 mutations in the MYH3 gene have
been reported to be associated with DA1, DA2A, DA2B or DA8
(9,15–17,19–20,27).
It is of note that there may be an association between the position
of the mutations in MYH3 and the type of disease. For
instance, all mutations resulting in DA1 are located in the
globular motor domain (16,19),
and the majority of pathogenic mutations for DA2A and DA2B are
situated within the head and neck of embryonic MYH (9,15–17).
In DA8, one small deletion has been reported in the globular motor
domain of MYH3; however, the remaining two mutations
associated with DA8 are located in the tail of the embryonic MYH
near the hinge (20). In the
spatial structure, the mutations causing DA2A are mainly located in
the groove lying between two large domains in the head, which are
involved in the binding and hydrolyzing of ATP (9,28).
When mutations occur in this area, the ATP metabolic process is
disrupted. Conversely, almost all DA2B mutations are on the surface
of the embryonic MYH directly exposed to the outside, leading to
dysfunction in the interactions made with other proteins such as
myosin light chain or actin (9,14–16,28).
Consistent with this, the novel pathogenic mutation p.K836E
identified in the present study is located in the neck of embryonic
MYH corresponding to the surface of the spatial model, which is
also adjacent to a mutation site (p.K838E) reported by Toydemir
et al (9) in 2006. However,
the underlying mechanism contributing to this phenomenon is still
unclear.
In conclusion, the present study identified a novel
pathogenic missense mutation of the MYH3 gene in a Chinese
family with DA2B. To the best of our knowledge, this was the first
report of an MYH3 gene mutation leading to DA2B in the Asian
population. Furthermore, based on the construction and analysis of
the protein structure model, the mechanism underlying the DA
formation induced by MYH3 mutation was also assessed. The
present study expanded the mutation spectrum of MYH3 and
supported the results of previous studies regarding the association
between mutation locations and DA types; thereby, contributing to
developments in the clinical and genetic diagnosis of DA.
Acknowledgements
The authors would like to thank Professor Qinglin
Kang (Department of Orthopedic Surgery, Shanghai Jiao Tong
University Affiliated Sixth People's Hospital, Shanghai, P.R.
China) for his support and advice.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81572121).
Availability of data and materials
The primary data generated or analyzed in this study
are included in this published article. The additional data used
and analyzed in the current study are available from the
corresponding author on reasonable request.
Authors' contributions
WW collected the clinical information and peripheral
blood samples of all of the participants and contributed to the
analysis of sequencing outcomes. LK primarily analyzed the outcomes
of whole-exome sequencing and Sanger sequencing and was a major
contributor in writing the manuscript. RZ contributed to the
conducting of the whole-exome sequencing and Sanger sequencing for
the participants. QK conceived and designed the study and
experimental methods. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all of
the participants. The present study was approved by the Ethics
Committees of Shanghai Jiao Tong University Affiliated Sixth
People's Hospital (Shanghai, China).
Patient consent for publication
Written informed consent was obtained from all of
the participants approving this non-commercial research and the
publication of any associated data/images.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DA
|
distal arthrogryposis
|
|
DA2B
|
distal arthrogryposis type 2B
|
|
MYH3
|
myosin heavy chain 3
|
|
TNNT3
|
troponin T3, fast skeletal
|
|
RLC
|
regulatory light chain
|
References
|
1
|
Hall JG, Reed SD and Greene G: The distal
arthrogryposes: Delineation of new entities-review and nosologic
discussion. Am J Med Genet. 11:185–239. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bamshad M, Jorde LB and Carey JC: A
revised and extended classification of the distal arthrogryposes.
Am J Med Genet. 65:277–281. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tajsharghi H, Kimber E, Holmgren D,
Tulinius M and Oldfors A: Distal arthrogryposis and muscle weakness
associated with a beta-tropomyosin mutation. Neurology. 68:772–775.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Krakowiak PA, O'Quinn JR, Bohnsack JF,
Watkins WS, Carey JC, Jorde LB and Bamshad M: A variant of
Freeman-Sheldon syndrome maps to 11p15.5-pter. Am J Hum Genet.
60:426–432. 1997.PubMed/NCBI
|
|
5
|
Stevenson DA, Swoboda KJ, Sanders RK and
Bamshad M: A new distal arthrogryposis syndrome characterized by
plantar flexion contractures. Am J Med Genet A. 140:2797–2801.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gurnett CA, Desruisseau DM, McCall K, Choi
R, Meyer ZI, Talerico M, Miller SE, Ju JS, Pestronk A, Connolly AM,
et al: Myosin binding protein C1: A novel gene for autosomal
dominant distal arthrogryposis type 1. Hum Mol Genet. 19:1165–1173.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ma L and Yu X: Arthrogryposis multiplex
congenita: Classification, diagnosis, perioperative care, and
anesthesia. Front Med. 11:48–52. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bamshad M, Van Heest AE and Pleasure D:
Arthrogryposis: A review and update. J Bone Joint Surg Am. 91
(Suppl 4):S40–S46. 2009. View Article : Google Scholar
|
|
9
|
Toydemir RM, Rutherford A, Whitby FG,
Jorde LB, Carey JC and Bamshad MJ: Mutations in embryonic myosin
heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall
syndrome. Nat Genet. 38:561–565. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Toydemir RM and Bamshad MJ: Sheldon-Hall
syndrome. Orphanet J Rare Dis. 4:112009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sung SS, Brassington AM, Grannatt K,
Rutherford A, Whitby FG, Krakowiak PA, Jorde LB, Carey JC and
Bamshad M: Mutations in genes encoding fast-twitch contractile
proteins cause distal arthrogryposis syndromes. Am J Hum Genet.
72:681–690. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sung SS, Brassington AM, Krakowiak PA,
Carey JC, Jorde LB and Bamshad M: Mutations in TNNT3 cause multiple
congenital contractures: A second locus for distal arthrogryposis
type 2B. Am J Hum Genet. 73:212–214. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Robinson P, Lipscomb S, Preston LC, Altin
E, Watkins H, Ashley CC and Redwood CS: Mutations in fast skeletal
troponin I, troponin T, and beta-tropomyosin that cause distal
arthrogryposis all increase contractile function. FASEB J.
21:896–905. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Alamo L, Ware JS, Pinto A, Gillilan RE,
Seidman JG, Seidman CE and Padrón R: Effects of myosin variants on
interacting-heads motif explain distinct hypertrophic and dilated
cardiomyopathy phenotypes. Elife. 6:e246342017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tajsharghi H, Kimber E, Kroksmark AK,
Jerre R, Tulinius M and Oldfors A: Embryonic myosin heavy-chain
mutations cause distal arthrogryposis and developmental myosin
myopathy that persists postnatally. Arch Neurol. 65:1083–1090.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Beck AE, McMillin MJ, Gildersleeve HI,
Kezele PR, Shively KM, Carey JC, Regnier M and Bamshad MJ: Spectrum
of mutations that cause distal arthrogryposis types 1 and 2B. Am J
Med Genet A 161A. 550–555. 2013. View Article : Google Scholar
|
|
17
|
Scala M, Accogli A, De Grandis E, Allegri
A, Bagowski CP, Shoukier M, Maghnie M and Capra V: A novel
pathogenic MYH3 mutation in a child with Sheldon-Hall syndrome and
vertebral fusions. Am J Med Genet A. 176:663–667. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hague J, Delon I, Brugger K, Martin H,
Abbs S and Park SM: Molecularly proven mosaicism in phenotypically
normal parent of a girl with Freeman-Sheldon syndrome caused by a
pathogenic MYH3 mutation. Am J Med Genet A. 170:1608–1612. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Alvarado DM, Buchan JG, Gurnett CA and
Dobbs MB: Exome sequencing identifies an MYH3 mutation in a family
with distal arthrogryposis type 1. J Bone Joint Surg Am.
93:1045–1050. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chong JX, Burrage LC, Beck AE, Marvin CT,
McMillin MJ, Shively KM, Harrell TM, Buckingham KJ, Bacino CA, Jain
M, et al: Autosomal-dominant multiple pterygium syndrome is caused
by mutations in MYH3. Am J Hum Genet. 96:841–849. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Al-Numair NS, Lopes L, Syrris P, Syrris P,
Monserrat L, Elliott P and Martin AC: The structural effects of
mutations can aid in differential phenotype prediction of
beta-myosin heavy chain (Myosin-7) missense variants.
Bioinformatics. 32:2947–2955. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: dbSNP: The NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Thorisson GA, Smith AV, Krishnan L and
Stein LD: The international hapmap project web site. Genome Res.
15:1592–1593. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lorenz TC: Polymerase chain reaction:
Basic protocol plus troubleshooting and optimization strategies. J
Vis Exp. 22:e39982012.
|
|
25
|
Baumann M, Steichen-Gersdorf E, Krabichler
B, Petersen BS, Weber U, Schmidt WM, Zschocke J, Müller T, Bittner
RE and Janecke AR: Homozygous SYNE1 mutation causes congenital
onset of muscular weakness with distal arthrogryposis: A
genotype-phenotype correlation. Eur J Hum Genet. 25:262–266. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eller M, Stedman HH, Sylvester JE, Fertels
SH, Wu QL, Raychowdhury MK, Rubinstein NA, Kelly AM and Sarkar S:
Human embryonic myosin heavy chain cDNA. Interspecies sequence
conservation of the myosin rod, chromosomal locus and isoform
specific transcription of the gene. FEBS Lett. 256:21–28. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Al-Haggar M, Yahia S, Damjanovich K, Ahmad
N, Hamada I and Bayrak-Toydemir P: p.R672C mutation of MYH3 gene in
an Egyptian infant presented with Freeman-Sheldon syndrome. Indian
J Pediatr. 78:103–105. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Allingham JS, Smith R and Rayment I: The
structural basis of blebbistatin inhibition and specificity for
myosin II. Nat Struct Mol Biol. 12:378–379. 2005. View Article : Google Scholar : PubMed/NCBI
|