Introduction
Aspirin which was synthesized and patented in 1897
for its analgesic and anti-inflammatory properties has been used
now for more than a century, and is one of the most widely used
drugs in medicine (1). Its
mechanism of action was established in 1971 when Sir John Vane in
England demonstrated its ability to decrease the synthesis of
prostaglandins responsible for pain, inflammation and fever
(2). In 1975, it was discovered
that aspirin decreases prostaglandin synthesis by acetylating and
inactivating cyclooxygenases (COX) (3). In the years that followed thereafter,
aspirin was recommended for the prevention of secondary
cardiovascular diseases such as myocardial infarction and stroke,
due to its ability to decrease thromboxane A2 in platelets
(4). Studies conducted in the past
three decades have established yet another therapeutic use for
aspirin in the prevention of epithelial cancers, particularly
colorectal cancer (CRC), and this was observed even with the low
dose aspirin (75 mg) (5–7). Owing to the growing global prevalence
of cancer, the chemo-preventive effects of aspirin has gained much
attention among physicians, scientists and the public alike. In
fact, in 2016 the United States preventive services task force
(USPSTF) recommended ‘initiating low dose aspirin use for the
prevention of cardiovascular disease and CRC in adults aged 50–59
years’ (8). This recommendation
was based on increasing evidences on low-dose aspirin's ability to
decrease CRC and other cancers (9,10).
Though numerous reports have described potential
mechanisms of cancer-prevention by aspirin, its primary mode of
action still remains elusive. One of the most discussed mechanism
involves the inhibition of COX in platelets (platelet hypothesis)
(9–14). Independent of this, other
mechanisms have been proposed that includes inhibition of NF-kB
(15), induction of polyamine
catabolism (16), activation of
AMP-kinase (17,18), inhibition of Wnt signaling and
β-catenin phosphorylation (19),
downregulation of c-myc (20),
induction of DNA mismatch repair proteins (21), induction of autophagy (22), and acetylation of p53 and
glucose-6-phosphate dehydrogenase (23,24)
to name a few. Many of these effects were detected mainly at high
concentrations of aspirin (often at millimolar concentrations)
which are not reached in vivo in the systemic circulation
(maximum reported concentration is 142 µM with 1.2 g tablets/4-6h)
(16). The inability to accurately
pinpoint the mechanism involved also stems from the failure to
differentiate the primary proximal effects from its associated
downstream signaling events and the subsequently observed
biological responses (25).
Additionally, studies show that aspirin is more effective in
decreasing the incidence of CRC as compared to other cancers such
as breast, prostate, lung and skin (26–28);
however, it is still not clear why aspirin is more effective
against CRC as compared to other cancers.
Our laboratory has been focusing on determining a
role for aspirin and salicylic acid metabolites
2,3-dihydroxybenzoic acid (2,3-DHBA) and 2,5-dihydroxybenzoic acid
(2,5-DHBA), known to be generated in the body by the cytochrome
P-450 (CYP450) catalyzed reactions (29), in the prevention of CRC.
Interestingly, similar dihydroxybenzoic acids have also been
reported to be generated from aspirin by the gut microflora
(30). In a previous study we
demonstrated the ability of 2,3-DHBA and 2,5-DHBA to inhibit cyclin
dependent kinase 1 (CDK1) activity in vitro (31). However, an extended study on the
effect of these metabolites on other CDKs involved in cell-cycle
regulation (CDK2, CDK4 and CDK6) as well as the demonstration of
their ability to inhibit cancer cell growth were not reported. As a
dysregulated cell cycle is one of the hallmarks of cancer, it was
important to determine if 2,3-DHBA and 2,5-DHBA affected cancer
cell growth by inhibiting these CDKs to gain a comprehensive
understanding of their mechanism of action. In the present study,
we investigated the effect of 2,3-DHBA and 2,5-DHBA on CDKs 1, 2, 4
and 6 enzyme activities and determined their potential sites of
interaction within these enzymes. In addition, we also performed
studies to determine the effect of these metabolites on cancer cell
proliferation. Our results show that although aspirin and salicylic
acid showed potential interactions with the CDK enzymes, only their
metabolites were effective in inhibiting CDK enzyme activity and
cancer cell proliferation. More specifically, these metabolites
inhibited CDK1 and CDK6 enzyme activity. Our results show that
among the two aspirin metabolites, 2,5-DHBA is highly effective in
inhibiting cell proliferation in three different cell lines
(HCT-116, HT-29 and MDA-MB-231) whereas 2,3-DHBA was capable of
inhibiting cell growth only in MDA-MB-231 cells. Our findings
suggests a role for these metabolites in aspirin's chemo-preventive
actions.
Materials and methods
Cell lines, recombinant proteins and
reagents
HCT-116 (colorectal carcinoma), HT-29 (colorectal
adenocarcinoma) and MDA-MB-231 (breast adenocarcinoma) cell lines
were purchased from American Type Culture Collection (ATCC).
MDA-MB-231 cells were grown in RPMI media while HCT-116 and HT-29
cells were cultured in McCoy's 5A media, both supplemented with 10%
FBS and antibiotics for 24 h before treatment with specified
compounds for indicated times. Authentication of cell lines was
done by ATCC through their DNA-STR profile. CDK1/Cyclin B1,
CDK2/Cyclin A2, CDK4/Cyclin D1, Retinoblastoma (C-term) and kinase
buffer were purchased from SignalChem. Aspirin, salicylic acid and
trypsin-EDTA solution were purchased from Sigma-Aldrich; H1
Histones were obtained from EMD Millipore; [32P] γ-ATP
from PerkinElmer; 2,3-DHBA, 2,5-DHBA and all other reagents
mentioned in this study were obtained from Thermo Fischer
Scientific, Inc.
In vitro CDK assay
In vitro CDK assays, to measure enzyme
activity, were performed as described by the manufacturer (32–35)
and previously published protocols (31,36,37).
Briefly, purified enzyme was aliquoted into the reaction buffer and
incubated with indicated compounds (aspirin, salicylic acid,
2,3-DHBA and 2,5-DHBA) at various concentrations for 10 min. at
room temperature. The reaction mixture was incubated with kinase
buffer containing 15 µM ATP, 2 µCi of [32P] γ-ATP, 5 µg
of H1 Histone (7.5 µg/reaction added as substrate for CDKs 1 and 2)
or retinoblastoma (1.5 µg/reaction added as substrate for CDKs 4
and 6), at 30°C for 20 min in a final volume of 50 µl. The
reactions were halted by adding EDTA to a final concentration of 20
mM and addition of 4X loading buffer. The samples were boiled for
10 min, analyzed by 8 or 10% SDS-PAGE, stained using coomassie
brilliant blue (R250) to confirm equal loading of the H1 histones
and Retinoblastoma protein. The molecular weight of the proteins
were confirmed by molecular weight markers. The gel was dried, and
exposed to X-ray film. NIH ImageJ software was used to quantify the
intensities of the bands. The normalization of the band intensities
of the phosphorylated H1 histones/ Retinoblastoma proteins were
determined by comparing to the control, which is the
phosphorylation in the absence of these compounds.
Molecular docking studies
The crystallographic three dimensional structures of
CDK1 (4Y72 A chain), CDK2 (1FIN A chain), CDK4 (3G33 A chain) and
CDK6 (4AUA) were retrieved from the Protein Data Bank (PDB). Energy
minimization for these proteins was performed using Gromacs 3.3.1
package utilizing GROMOS96 force field (38). The energy-minimized molecules were
used as the receptors for virtual small molecule docking with
aspirin, salicylic acid, 2,3-DHBA, 2,5-DHBA using AutoDock Vina.
The results were visualized by PYMOL molecular graphics system
version 1.3.
Cell proliferation
Approximately 250,000 cells were seeded per 100 mm
plates containing 10% FBS and grown overnight. Cells were left
untreated (control) or were treated with drugs at various
concentrations and incubated for 48 h. The floating cells in the
spent media (if any) were collected, pooled with the trypsinized
cells, and counted using the Nexcelom Cellometer Auto T4 cell
counter.
Clonogenic assay
Clonogenic assays were performed as previously
described (36). Cells were seeded
at a density of 500 cells/100 mm plate and grown for 48 h following
which cells were left untreated (control) or treated with compounds
(aspirin, salicylic acid, 2,3-DHBA and 2,5-DHBA) at the
concentrations indicated. The spent media was replaced with fresh
media containing the respective compounds every 5–6 days. Cells
were incubated for 21 days, fixed with 100% methanol for 20 min,
and stained with 0.5% crystal violet prepared in 25% methanol. The
colonies were then photographed and quantified using NIH ImageJ
software.
Statistical analysis
All experiments were repeated 3–6 times
independently of each other. One-way ANOVA followed by Tukey's
post-hoc tests were adopted to compare group differences with the
control. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effect of aspirin, salicylic acid,
2,3-DHBA and 2,5-DHBA on CDK enzyme activity
We initiated this study by determining the
dose-dependent effect of aspirin, salicylic acid, 2,3-DHBA and
2,5-DHBA on recombinant CDKs 1, 2, 4 and 6 by measuring the
phosphorylation pattern through in vitro kinase assays.
Fig. 1 shows that CDK1 activity
remained unaffected following treatment with aspirin and salicylic
acid at all concentrations tested (125 µM to 1 mM); however,
2,3-DHBA and 2,5-DHBA inhibited CDK1 activity, confirming our
previously published results (31). In both cases, inhibition was around
20% at 500 µM, 40% at 750 µM and 60% at 1,000 µM. CDK2 activity
remained unchanged upon exposure to aspirin and salicylic acid
while inhibition was observed with 2,3-DHBA and 2,5-DHBA,
especially at higher concentrations (>750 µM; Fig. 2). Similar to CDK1 and CDK2, aspirin
and salicylic acid both failed to inhibit CDK4 and CDK6 enzyme
activity (Figs. 3 and 4). 2,3-DHBA and 2,5-DHBA inhibited CDK4
activity at 1,000 µM. 2,3-DHBA was able to inhibit CDK6 enzyme
activity beginning at 250 µM, while 2,5-DHBA inhibited CDK6 enzyme
activity at concentrations >750 µM. These results show 2,3-DHBA
and 2,5-DHBA were able to inhibit CDKs 1, 2, 4, and 6 activities to
different degrees. It is also important to note that the inhibition
observed with CDK1 by 2,3-DHBA and 2,5-DHBA, and CDK-6 by 2,3-DHBA
was significantly greater compared to inhibition of other CDKs by
these compounds.
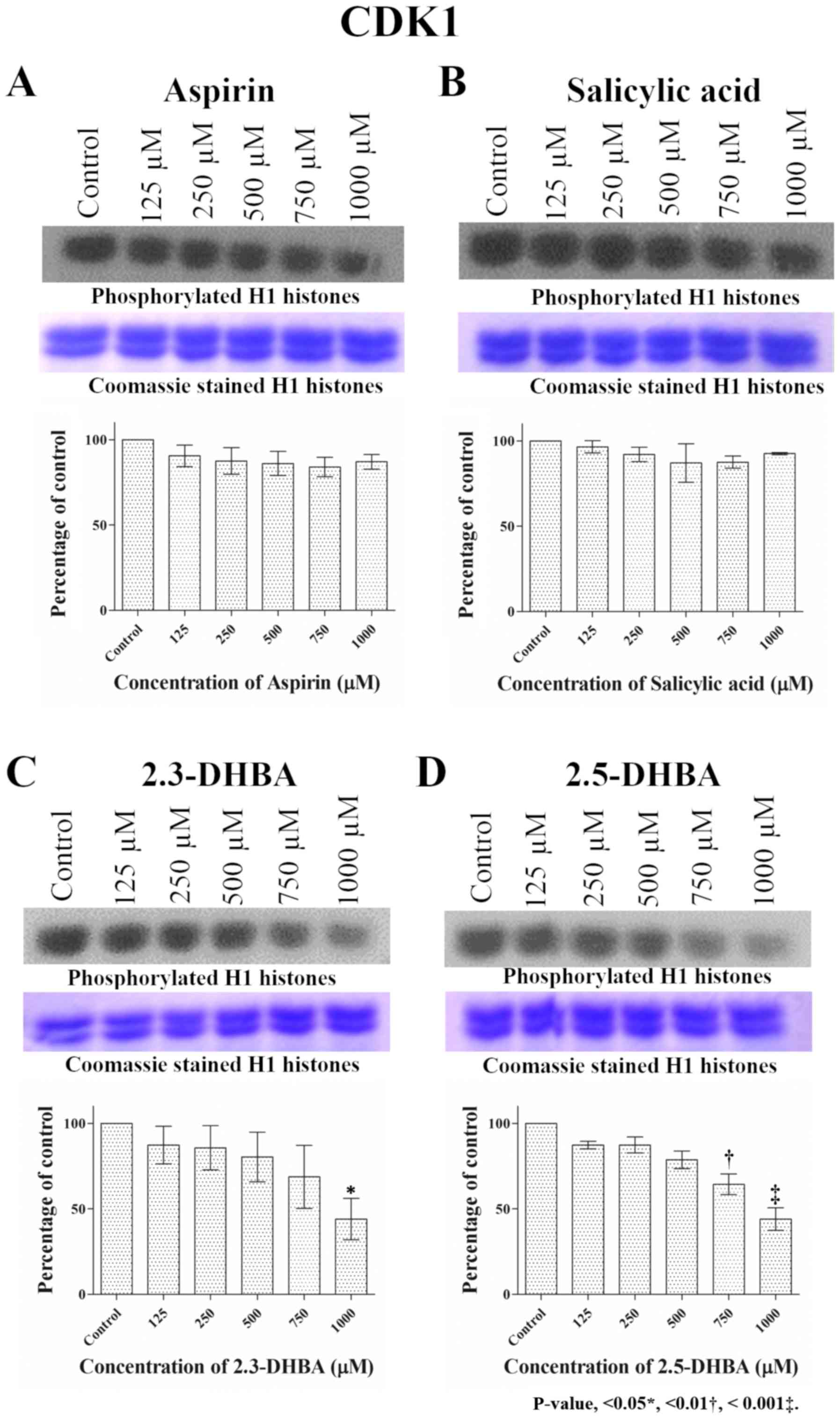 | Figure 1.Effect of different concentrations of
(A) aspirin, (B) salicylic acid, (C) 2,3-DHBA and (D) 2,5-DHBA on
CDK1 enzyme activity. H1 histones were used as the substrate for
CDK1. Upper panel shows phosphorylation of H1 Histones in the
presence of different concentrations of each compound. The middle
panel shows the coomassie blue stained proteins. Lower panel
represents the quantification of the bands in upper panel.
*P<0.05, **P<0.01, ***P<0.001 vs. Control. 2,3-DHBA,
2,3-dihydroxybenzoic acid; 2,5-DHBA, 2,5-dihydroxybenzoic acid. |
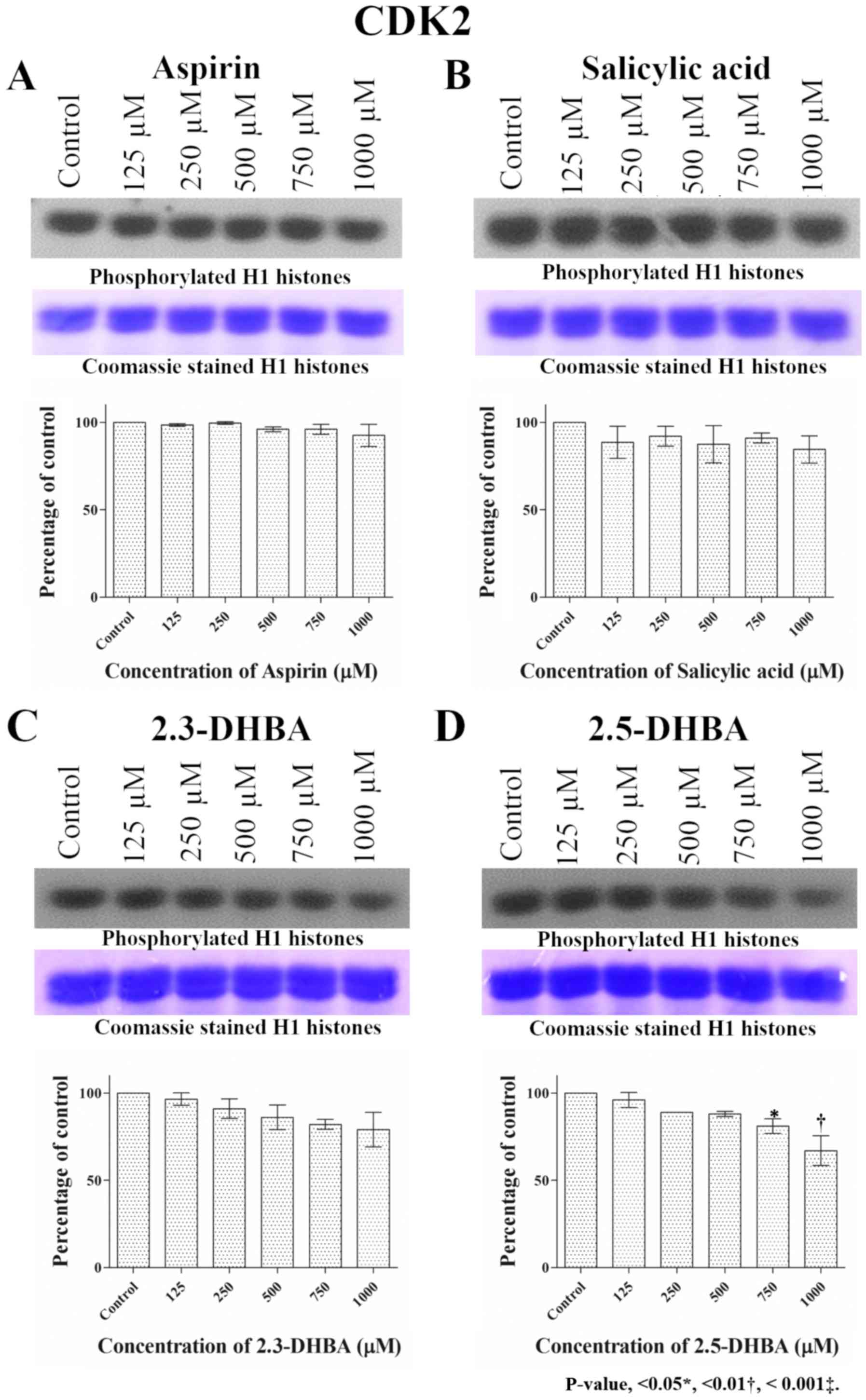 | Figure 2.Effect of different concentrations of
(A) aspirin, (B) salicylic acid, (C) 2,3-DHBA and (D) 2,5-DHBA on
CDK2 enzyme activity. H1 histones were used as the substrate for
CDK2. Upper panel shows phosphorylation of H1 Histones in the
presence of different concentrations of each compound. The middle
panel shows the coomassie blue stained proteins. Lower panel
represents the quantification of the bands in upper panel.
*P<0.05, **P<0.01 vs. Control.2,3-DHBA, 2,3-dihydroxybenzoic
acid; 2,5-DHBA, 2,5-dihydroxybenzoic acid. |
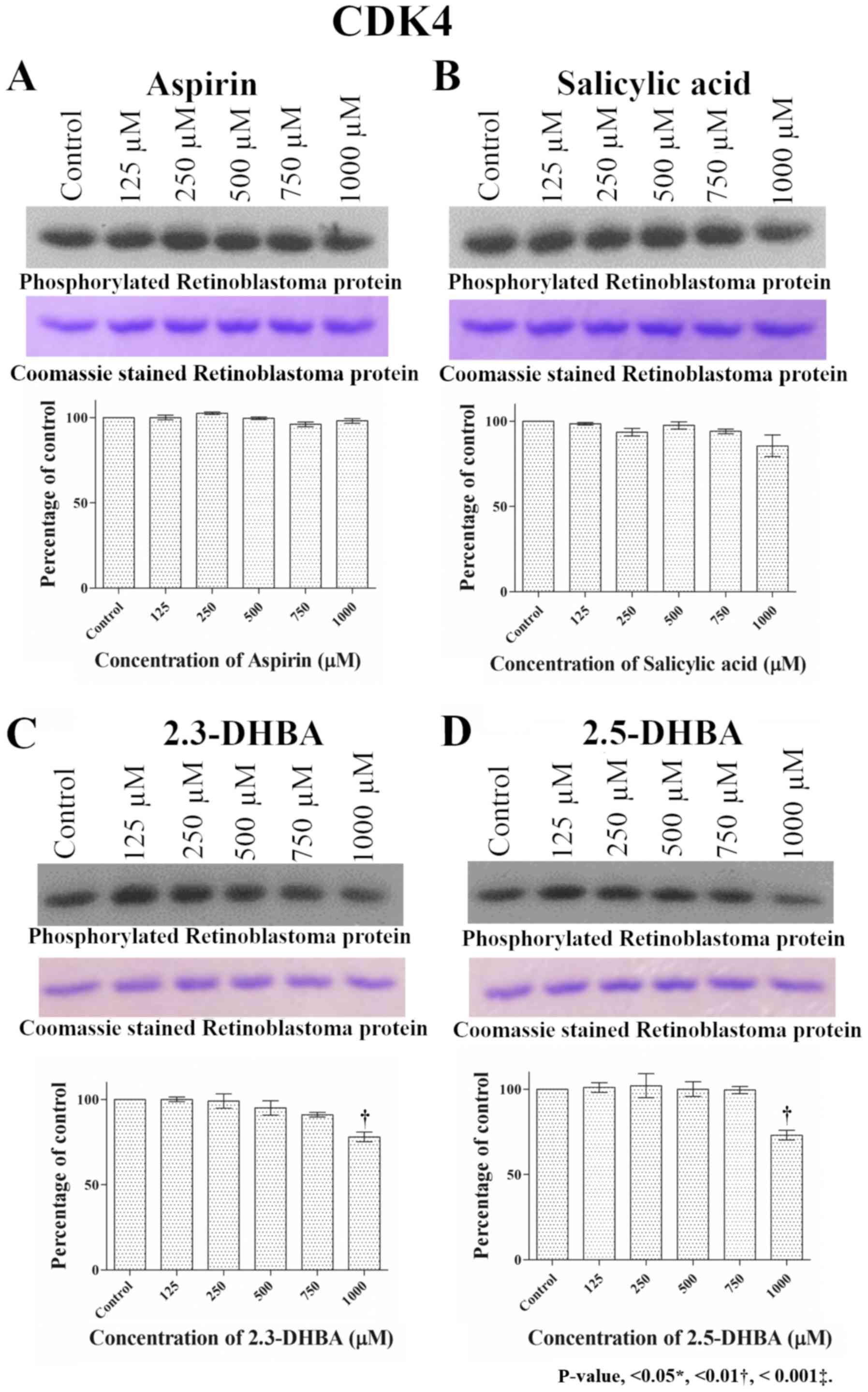 | Figure 3.Effect of different concentrations of
(A) aspirin, (B) salicylic acid, (C) 2,3-DHBA and (D) 2,5-DHBA on
CDK4 enzyme activity. Retinoblastoma protein was used as the
substrate for CDK4. Upper panel shows phosphorylation of
retinoblastoma protein in the presence of different concentrations
of each compound. The middle panel shows the coomassie blue stained
proteins. Lower panel represents the quantification of the bands in
upper panel. **P<0.01 vs. Control.2,3-DHBA, 2,3-dihydroxybenzoic
acid; 2,5-DHBA, 2,5-dihydroxybenzoic acid. |
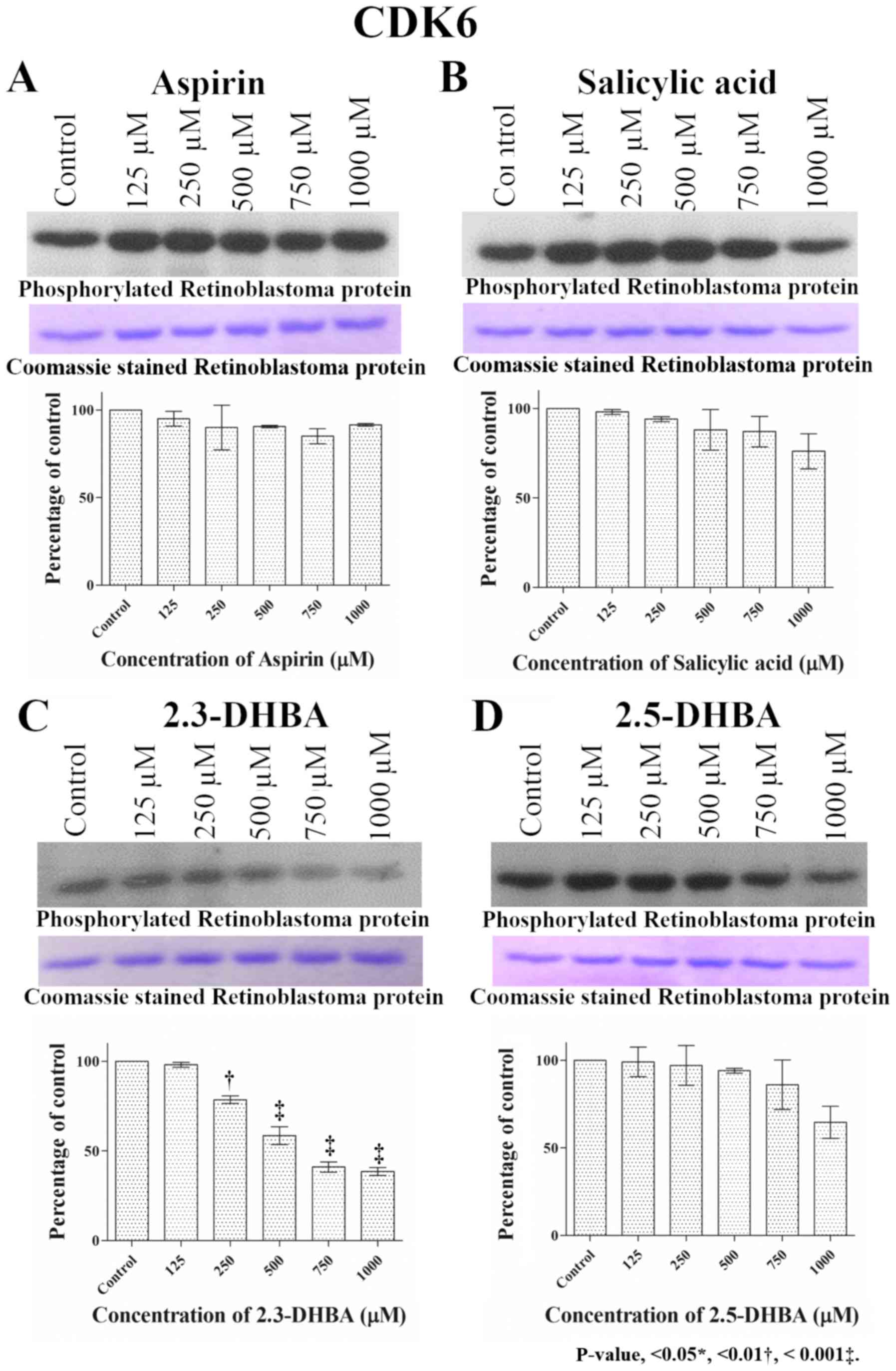 | Figure 4.Effect of different concentrations of
(A) aspirin, (B) salicylic acid, (C) 2,3-DHBA and (D) 2,5-DHBA on
CDK6 enzyme activity. Retinoblastoma protein was used as the
substrate for CDK6. Upper panel shows phosphorylation of
retinoblastoma protein in the presence of different concentrations
of each compound. The middle panel shows the coomassie blue stained
proteins. Lower panel represents the quantification of the bands in
upper panel. **P<0.01, ***P<0.001 vs. Control. 2,3-DHBA,
2,3-dihydroxybenzoic acid; 2,5-DHBA, 2,5-dihydroxybenzoic acid. |
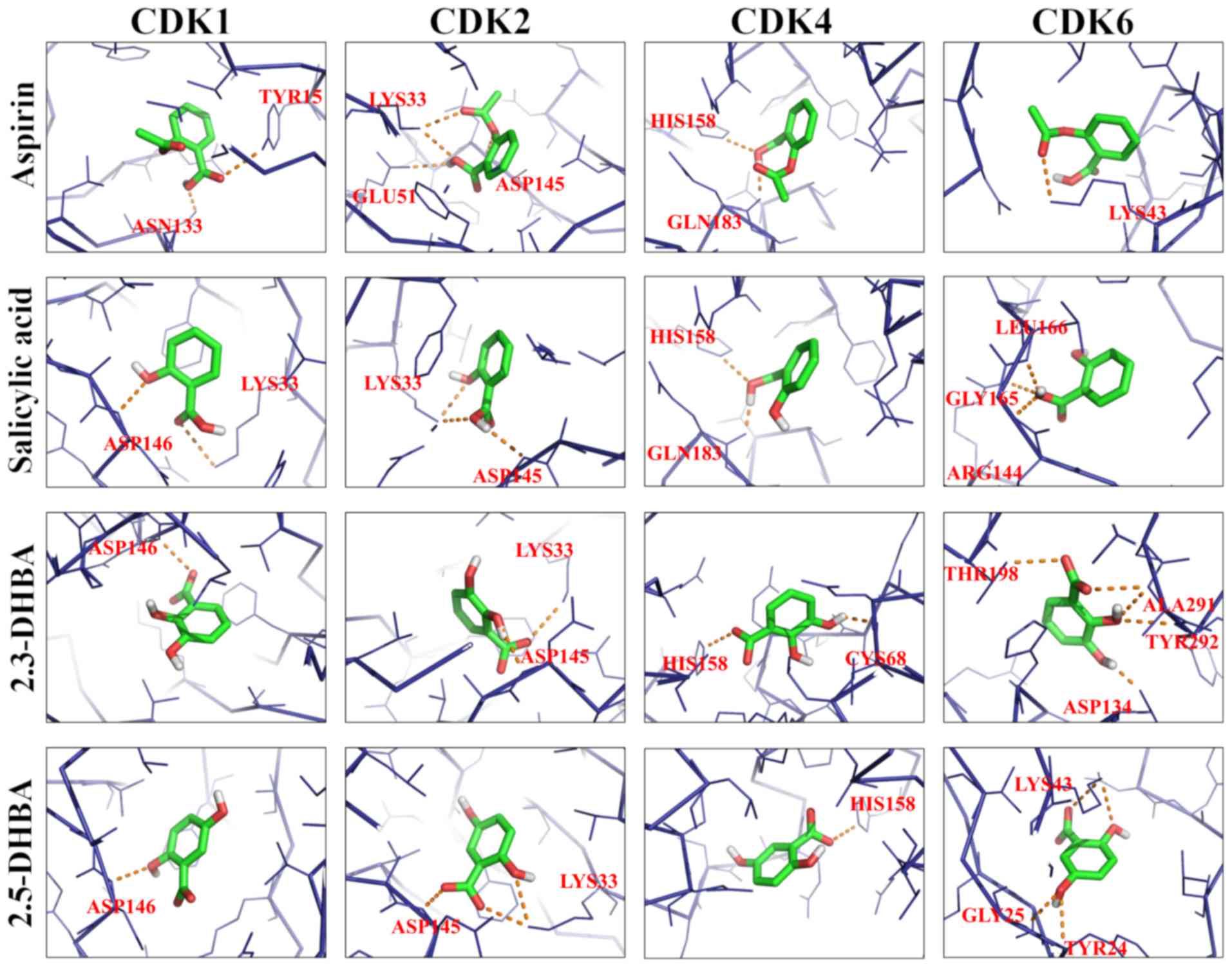
Docking studies reveal binding pockets
for aspirin, salicylic, 2,3-DHBA, and 2,5-DHBA
AutoDock Vina was used to predict the potential
interactions of aspirin, salicylic acid, 2,3-DHBA and 2,5-DHBA with
CDKs 1, 2, 4 and 6. The binding free energy and hydrogen bond
lengths were also determined. The results of the docking studies
are shown in Table I and Fig. 5 (line model). All the compounds
tested interacted with CDKs and utilized amino acids either
residing in the active site or at alternate binding pockets.
Docking studies revealed that 2,3-DHBA potentially interacts with
CDK1 through Asp146; with CDK2 it utilizes Lys33, and Asp145; with
CDK4, it interacts through Cys68 and His158 and with CDK6 it binds
to Asp134, Tyr292, Ala291 and Thr198. 2,5-DHBA interacts with CDK1
using Asp146; with CDK2 it utilizes Asp145 and Lys33; with CDK4
through His158; and with CDK6 it uses Lys43, Tyr24 and Gly25.
Salicylic acid and aspirin also interacted with all 4 CDKs
(Table I), although CDK inhibition
was not observed in vitro (Figs.
1–4). Notable interactions are
that between the drug molecules and Asp146 of CDK1, as this amino
acid is involved in CDK-ATP interactions (39). Although salicylic acid interacted
with CDK1 through Asp146 and Lys33 and CDK2 through Asp145 and
Lys33, it is not clear at this stage why it failed to cause
inhibition of the enzyme activity. This may be due to the formation
of an intramolecular hydrogen bond between its carboxyl and
hydroxyl groups which may hinder its interactions with CDKs.
 | Table I.Free energy binding values and
hydrogen bond lengths for the interaction of aspirin, salicylic
acid, 2,3-DHBA and 2,5-DHBA with CDK1, CDK2, CDK4 and CDK6. |
Table I.
Free energy binding values and
hydrogen bond lengths for the interaction of aspirin, salicylic
acid, 2,3-DHBA and 2,5-DHBA with CDK1, CDK2, CDK4 and CDK6.
| A, CDK1 |
|---|
|
|---|
| Ligand | Interacting amino
acids | Measurement,
A° | No of H bonds | Energy value,
kCal/mol |
|---|
| Aspirin | Asn133, Tyr15 | 2.5, 3.0 | 2 | −6.6 |
| Salicylic acid | Asp146, Lys33 | 2.8, 3.5 | 2 | −6.4 |
| 2,3-DHBA | Asp146 | 2.9 | 1 | −5.9 |
| 2,5-DHBA | Asp146 | 2.9 | 1 | −5.9 |
|
| B, CDK2 |
|
| Ligand | Interacting
amino acids | Measurement,
A° | No of H
bonds | Energy value,
kCal/mol |
|
| Aspirin | Asp145, Glu51,
Lys33(2) | 3.2, 2.6, 3.1,
3.0 | 4 | −6.2 |
| Salicylic acid | Asp145,
Lys33(2) | 3.3, 3.0, 3.3 | 3 | −5.7 |
| 2,3-DHBA | Lys33, Asp145 | 3.2, 3.2 | 2 | −5.6 |
| 2,5-DHBA | Asp145,
Lys33(2) | 3.2, 2.9, 2.9 | 3 | −5.9 |
|
| C, CDK4 |
|
| Ligand | Interacting
amino acids | Measurement,
A° | No of H
bonds | Energy value,
kCal/mol |
|
| Aspirin | His158, Gln183 | 2.9, 2.0 | 2 | −5.8 |
| Salicylic acid | His158, Gln183 | 2.8, 1.8 | 2 | −5.4 |
| 2,3-DHBA | His158, Cys68 | 3.1, 2.2 | 2 | −5.8 |
| 2,5-DHBA | His158 | 3.1 | 1 | −5.4 |
|
| D, CDK6 |
|
| Ligand | Interacting
amino acids | Measurement,
A° | No of H
bonds | Energy value,
kCal/mol |
|
| Aspirin | Lys43 | 2.9 | 1 | −5.4 |
| Salicylic acid | Gly165, Leu166,
Arg144 | 3.1, 2.8, 3.0 | 3 | −5.1 |
| 2,3-DHBA | Asp134, Tyr292,
Ala291(2), Thr198 | 2.0, 3.0, 3.2, 2.9,
2.9 | 5 | −5.4 |
| 2,5-DHBA | Lys43(2), Tyr24,
Gly25 | 3.2, 3.2, 3.2,
3.2 | 4 | −5.3 |
Effect of aspirin, salicylic acid,
2,3-DHBA and 2,5-DHBA on colony formation
Clonogenic assay was performed to determine the
effectiveness of aspirin, salicylic acid, 2,3-DHBA and 2,5-DHBA on
colony formation in HCT-116 and HT-29 colon cancer cells as well as
MDA-MB-231 breast cancer cells. HCT-116 and HT-29 cells were chosen
to study the effect of these compounds as these cell line have been
extensively used in studies pertaining to aspirin's biological
effects (40,41); MDA-MB-231 cells were chosen to
determine if this effect was mimicked in another cancer cell line.
We observed that aspirin and salicylic acid both failed to inhibit
colony formation in HCT-116 (Fig. 6A
and B), HT-29 (Fig. 7A and B)
and MDA-MB-231 cells (Fig. 8A and
B). 2,3-DHBA was effective in MDA-MB-231 cells at higher
concentrations (>500 µM) (Fig.
8C), while it was ineffective in HCT-116 (Fig. 6C) and HT-29 cells (Fig. 7C). Interestingly, 2,5-DHBA
exhibited a dose dependent effect on colony formation in all cell
lines examined (Figs. 6D, 7D, 8D).
In HCT-116 cells, decreased colony formation was observed with
2,5-DHBA at concentrations >250 µM, in HT-29 cells at
concentrations >200 µM, and in MDA-MB-231 cells, beginning at 50
µM. In contrast, two other salicylic acid derivatives 2,4-DHBA and
2,6-DHBA were unable to inhibit colony formation in MDA-MB-231
cells (Fig. S1). These results
clearly show that although the sensitivity varies among different
cell lines, both 2,3-DHBA and 2,5-DHBA exhibit the ability to
inhibit cancer cell proliferation in contrast to aspirin and
salicylic acid.
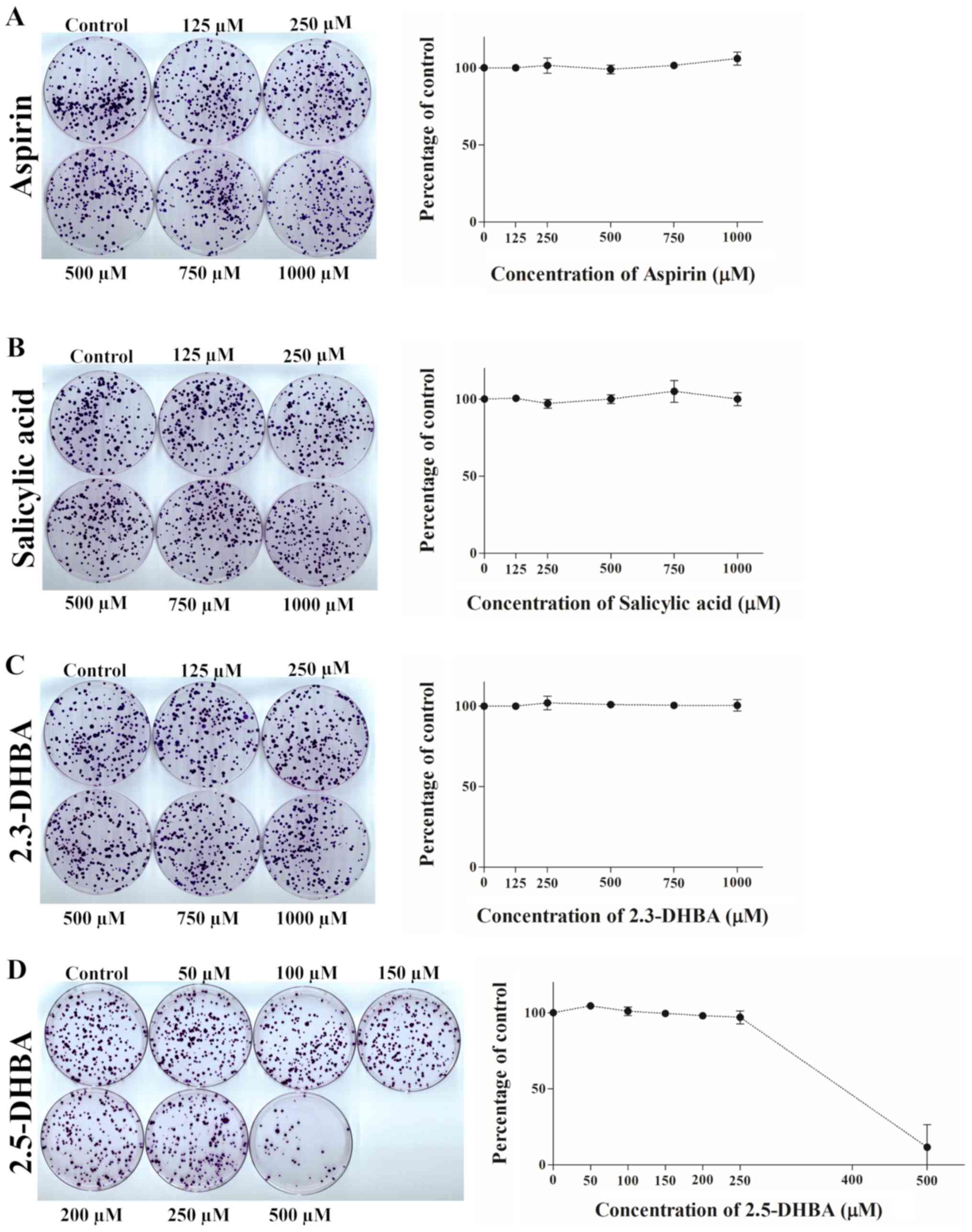 | Figure 6.Effect of (A) aspirin, (B) salicylic
acid, (C) 2,3-DHBA and (D) 2,5-DHBA on colony formation in HCT-116
cells. Quantification of the data is shown beside the crystal
violet stained image of the colonies. The graph is represented as
the mean ± standard deviation. 2,3-DHBA, 2,3-dihydroxybenzoic acid;
2,5-DHBA, 2,5-dihydroxybenzoic acid. |
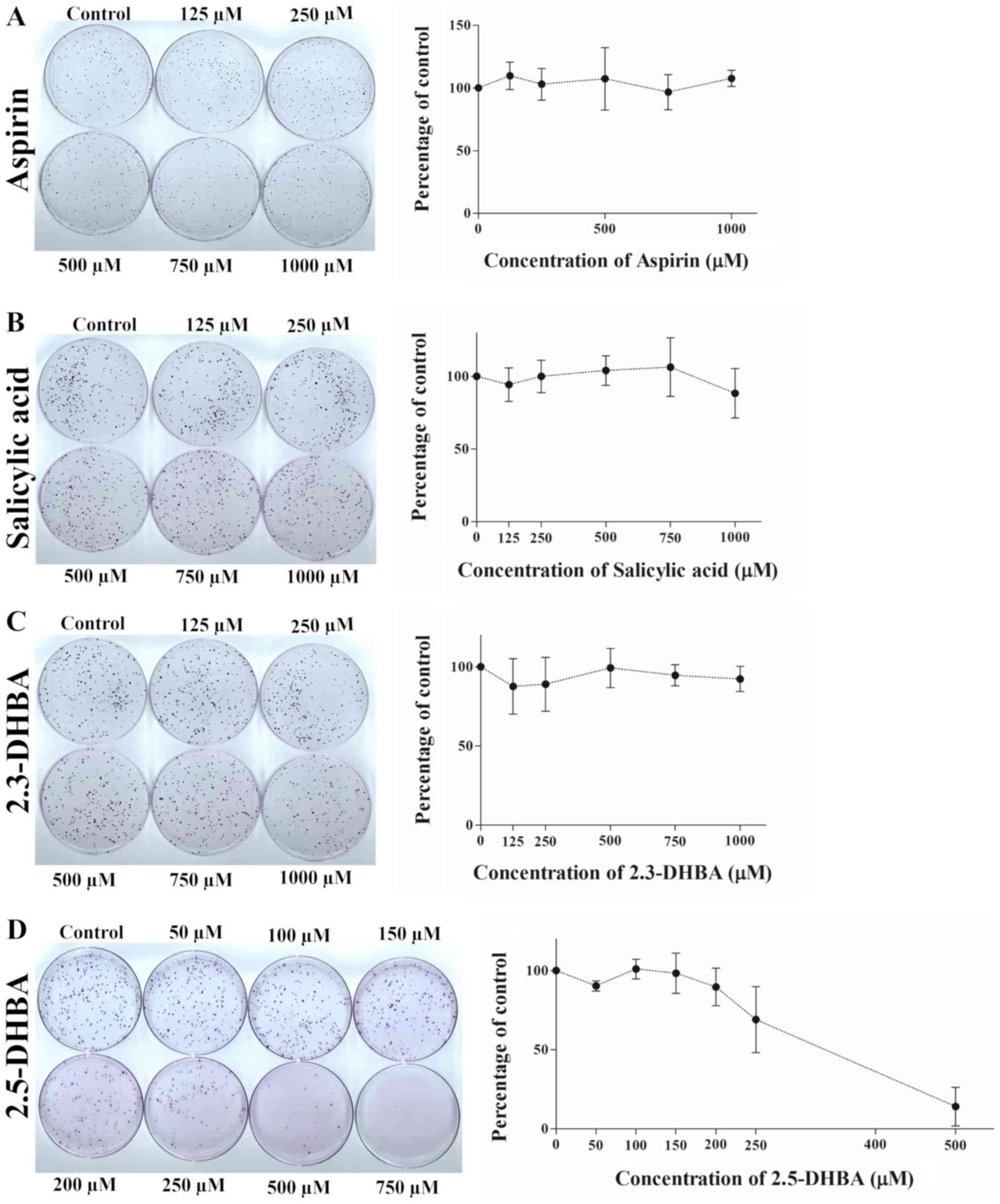 | Figure 7.Effect of (A) aspirin, (B) salicylic
acid, (C) 2,3-DHBA and (D) 2,5-DHBA on colony formation in HT-29
cells. Quantification of the data is shown beside the crystal
violet stained image of the colonies. The graph is represented as
mean ± standard deviation. 2,3-DHBA, 2,3-dihydroxybenzoic acid;
2,5-DHBA, 2,5-dihydroxybenzoic acid. |
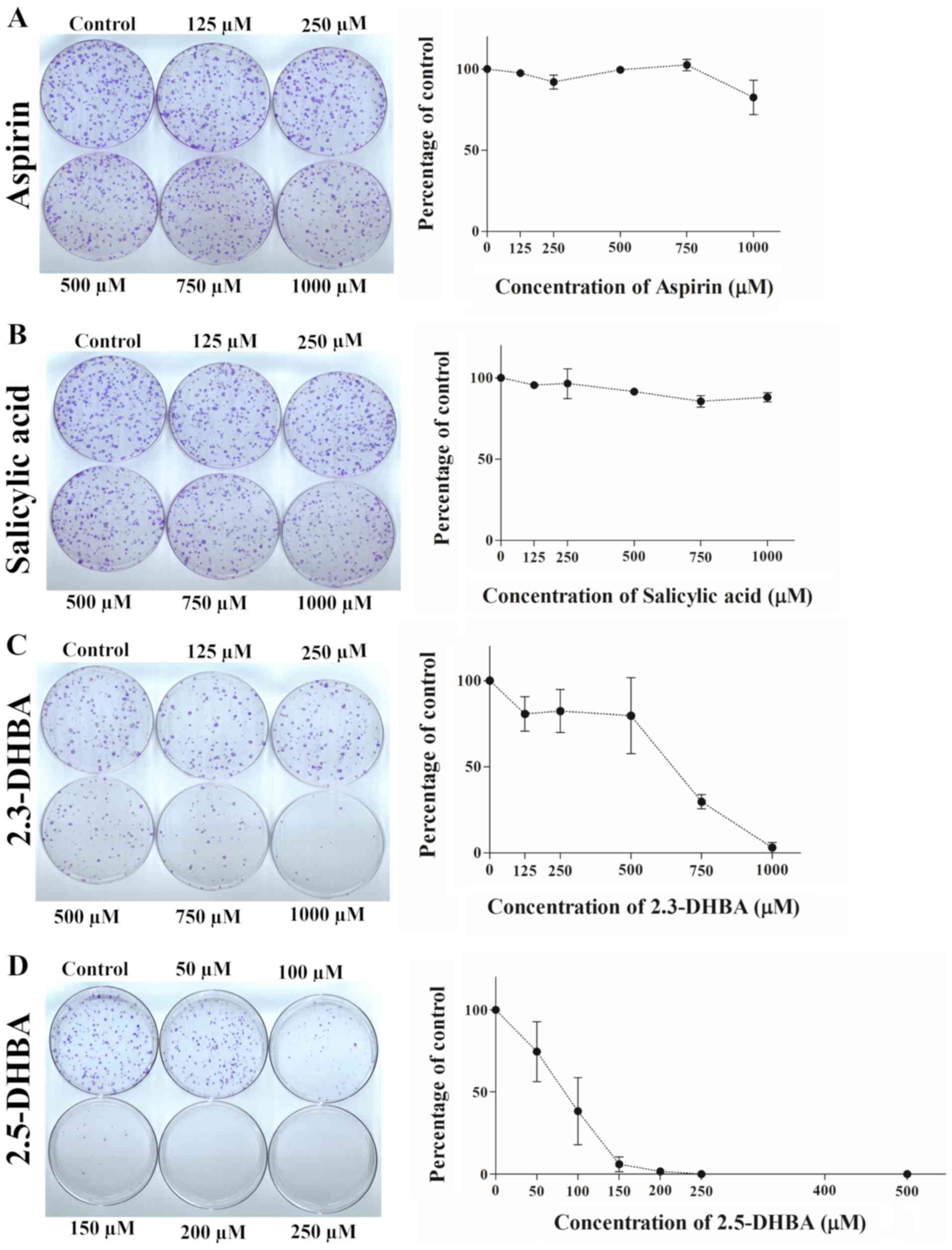 | Figure 8.Effect of (A) aspirin, (B) salicylic
acid, (C) 2,3-DHBA and (D) 2,5-DHBA on colony formation in
MDA-MB-231 cells. Quantification of the data is shown beside the
crystal violet stained image of the colonies. The graph is
represented as mean ± standard deviation. 2,3-DHBA,
2,3-dihydroxybenzoic acid; 2,5-DHBA, 2,5-dihydroxybenzoic acid. |
Interestingly, 2,3-DHBA and 2,5-DHBA were
ineffective in inhibiting cell proliferation in MDA-MB-231 cells
over a range of concentrations (125-1,000 µM) for 24–48 h (Fig. S2). We suggest that the lack of
inhibition in acute treatment observed may be related to the poor
uptake of these compounds.
Discussion
Despite the growing evidence for aspirin's ability
to decrease CRC, the mechanisms involved remain poorly defined.
Numerous hypotheses commencing from the COX inhibition in platelets
(9,11,12)
to increased expression of DNA repair proteins, transcription
factors and interconnected signaling pathways have been proposed
(42–45); however, a consensus has not been
reached regarding the target/pathway(s) primarily responsible for
its anti-cancer effects (46). In
this research paper, we tested the hypothesis that aspirin
metabolites 2,3-DHBA and 2,5-DHBA may mediate its chemopreventive
actions by inhibiting CDKs and affecting cell growth. We provide
evidence that 2,3-DHBA and 2,5-DHBA inhibit CDKs involved in
cell-cycle regulation (CDKs 1, 2, 4 and 6), although to different
degrees. Inhibition of CDK1 by 2,3-DHBA and 2,5-DHBA was observed
at 500 µM, and CDK-6 by 2,3-DHBA was observed starting at 250 µM,
however, inhibition of other CDKs by these compounds required much
higher concentrations (750 µM), suggesting that CDK1 and CDK6 are
better targets for 2,3-DHBA and/or 2,5-DHBA. In contrast, aspirin
and salicylic acid both failed to inhibit CDK-1, 2, 4 and 6 enzyme
activity. Through molecular docking studies we identified potential
sites of these interactions; most of the interacting amino acids
appear to be localized in the active site of these enzymes.
Importantly, we also demonstrated the ability of aspirin/salicylic
acid metabolites to inhibit clonal formation in cancer cell lines.
2,5-DHBA inhibited colony formation in a dose-dependent manner in
three different cancer cell lines (HCT-116, HT-29 and MDA-MB-231).
In contrast, 2,3-DHBA exhibited effective inhibition in MDA-MB-231
cells alone. These results suggest that 2,3-DHBA and 2,5-DHBA may
be involved in aspirin's chemopreventive actions. Our findings
raise two important questions: 1) What are the sources of the
aspirin metabolites 2,3-DHBA and 2,5-DHBA?; 2) Are there unique
scenarios that gives an anatomical advantage to 2,3-DHBA and
2,5-DHBA to act against CRC?.
Plain aspirin (non-enteric coated) is absorbed in
the stomach where the acidic environment protects it from
deacetylation and ionization, resulting in its faster absorption;
however, aspirin may also be absorbed in the upper small intestine
and enter into the hepatic portal vein (47–49).
In addition, lymphatic uptake of some aspirin and salicylic acid
has been reported (49). We
hypothesize that aspirin's preferential effect against CRC (over
other tissues) may be related to the generation of salicylic acid
that occurs both through hydrolysis and the action of gut esterases
(50) in the intestine or colon.
Intact aspirin's bioavailability is between 50–68% (51,52),
and it is estimated that nearly 29–39% of aspirin may be hydrolyzed
in the GI tract, particularly in the duodenum and ileum (52). Therefore, it can be argued that
epithelial cells of the intestine and colon are likely to be
exposed to substantially higher concentrations of salicylic acid
compared to other tissues in the body. With enteric coated aspirin,
the concentration of aspirin/salicylic acid exposed to the GI cells
may be even greater than non-enteric coated tablets as the
bioavailability of intact aspirin from enteric coated tablets is
significantly lower compared to plain aspirin due to slow release
and absorption in the intestine (47,53).
Aspirin in the systemic circulation is known to be
metabolized in the liver to 2,3-DHBA and 2,5-DHBA through the
action of cytochrome P450 (CYP450s) enzymes. However, it is
unlikely that these liver generated metabolites contribute to the
anti-cancer effect in colorectal tissues due to their low
concentrations in the plasma (29). We suggest that there are two routes
through which intestinal epithelial cells may get exposed to
2,3-DHBA and 2,5-DHBA. As intestinal epithelial cells also express
CYP450s similar to the isoforms expressed in the liver involved in
aspirin metabolism (54), one
would expect aspirin metabolites to be generated in these cells;
however, it is not clear at this stage to what extent these
metabolites are produced within the GI cells. Since exposure of
HCT-116, HT-29 and MDA-MB-231 cells to aspirin or salicylic acid
(up to 1 mM) failed to inhibit clonal formation, it is likely that
the contributions of cellular CYP450s to the formation of 2,3-DHBA
and 2,5-DHBA is negligible, although a small amount may still be
produced through this route. We have not determined the effect of
aspirin and salicylic acid on clonal formation at concentrations
greater than 1 mM, therefore we are not sure if higher
concentrations will have an effect on clonal formation. An
alternative possibility and an attractive hypothesis is that the
significant amount of aspirin/salicylic acid left unabsorbed
(32–50%) in the lumen of the intestine and colon maybe metabolized
by the intestinal microflora to generate hydroxyl derivatives of
salicylic acid, which may include both 2,3-DHBA and 2,5-DHBA.
Bacteria are capable of metabolizing drugs through hydroxylations
(55), and one report in 2016
showed that incubation of aspirin with human fecal suspensions
containing microbiota resulted in the degradation of aspirin to
salicylic acid and hydroxylated salicylic acids (30). The authors also showed that in rats
orally administered with aspirin and ampicillin, the
bioavailability of aspirin in the plasma was much higher compared
to rats administered with aspirin alone, suggesting that microbiota
contributes to the degradation of aspirin in the GI tract. An
estimation of the salicylic acid present in the lumen of human gut
has not been made, nor has the characterization of the hydroxyl
derivatives of salicylic acid produced in the lumen been reported
till date. The absence of experimental evidence on the potential
formation of 2,3-DHBA and 2,5-DHBA, beyond what is reported in
literature by Kim et al (30), is a limitation of this study.
Further work is also required to determine how these metabolites
are taken up by the normal, non-cancerous epithelial cells of the
intestine and colon and their effect on colony formation, which is
also a limitation of this study. Regarding the uptake mechanism,
one possibility is that 2,3-DHBA and 2,5-DHBA may be taken up by
the monocarboxylate transporter (MCT) SLC5A8 as it is implicated in
the transport of other derivatives of salicylic acid (36). The differential sensitivity of
HCT-116 and HT-29 vs. MDA-MB-231 could be attributed to the
differential expression of this transporter due to mutational/
methylation-driven inactivation of the SLC5A8 gene as previously
reported (56).
We observed that 2,5-DHBA was universally effective
in inhibiting colony formation as compared to 2,3-DHBA. Significant
inhibition of clonal formation was observed with 2,5-DHBA at 500 µM
in HCT-116 cells, at 250 µM in HT-29 cells and at 100 µM in
MDA-MB-231 cells, whereas 2,3-DHBA failed to inhibit colony
formation in HT-29 and HCT-116 cells (125-1,000 µM). However, it
inhibited the colony formation at ~500 µM in MDA-MB-231 cells. It
appears that 2,3-DHBA and 2,5-DHBA show differential inhibitory
effects in different cancer cell lines and it is unclear whether
this is due to the differences in uptake of these compounds by the
cell lines tested. Two other salicylic acid derivatives namely
2,4-DHBA and 2,6-DHBA failed to inhibit colony formation in
MDA-MB-231 cells (Fig. S1). This
suggests that among the different salicylic acid derivatives
tested, 2,5-DHBA is universally effective whereas 2,3-DHBA is more
selective as an anti-proliferative agent. This observation is
similar to our previous report in which we showed that another
salicylic acid derivative and flavonoid metabolite 2,4,6-THBA is a
potent inhibitor of colony formation in cells expressing functional
SLC5A8 (36).
In contrast to the inhibitory effect of 2,3-DHBA and
2,5-DHBA in clonal formation (21 days), both compounds failed to
show any appreciable inhibitory effect on cell number when cells
were treated for 24–48 h (acute treatment) at various
concentrations (125 to 1,000 µM; see Fig. S2). Similarly, in acute treatment
experiments, both compounds failed to show an effect on cell cycle
progression as measured by flow cytometry, and owing to the lack of
an effect, we have not included this data. This discrepancy in the
results between acute and chronic treatment may be related to the
time required for uptake and accumulation of these compounds to
exert an inhibitory effect. We suggest that acute exposure for
24–48 h may result in limited uptake of the drugs resulting in
milder effects which are difficult to discern in flow cytometry
analysis and cell counting, whereas chronic exposure for 21 days
may result in significant accumulation of the drugs sufficient to
cause inhibition of cell proliferation as measured by clonal
formation assays. As we have not performed flow cytometry analysis
on clonal cells following chronic exposure, it is not clear what
stages of the cell cycle are affected by these compounds, and this
is another limitation of the study.
Although the IC50 of the compounds tested
in this study are in micromolar concentrations for both CDK
inhibition and clonal formation, it could be argued that it is
physiologically relevant in view of their abundance in the
intestine due to the hydrolysis of aspirin to salicylic acid in the
gut (51,52). It is also important to note that
2,3-DHBA and 2,5-DHBA are abundantly available in fruits and
vegetables (57) and one study
demonstrated that the phenolic acid content generated from the diet
in the gut can rise to micromolar concentrations (58). CDKs and other potential cellular
targets probably have evolved to be less sensitive to these
compounds to avoid complete inhibition of cell cycle at lower
concentrations. The importance of 2,5-DHBA in cancer prevention was
also demonstrated in a recent study which showed that it is
effective in inhibiting colony formation and DNA synthesis in C6
glioma cells in vitro, and enhanced survival of Ehrlich
breast ascites carcinoma bearing mice (59). In addition, 2,5-DHBA has been
implicated in a previous report as a potential aspirin metabolite
formed through the action of tyrosinase enzyme in melanoma cells.
The study stated that p-quinone formation from 2,5-DHBA was
responsible for aspirin's anti-melanoma effects through ROS
formation (60). Thus, although
both the studies implicate anti-cancer effects of 2,5-DHBA, the
mechanisms described are starkly different; in melanocytes it
occurs through depletion of GSH and generation of ROS leading to
cell apoptosis (60), while in our
study (in HT-29, HCT-116, and MDA-MB-231 cells) it likely involves
inhibition of CDKs leading to suppression of cancer cell growth.
This suggest that 2,5-DHBA may contribute to anti-cancer effects
through distinct mechanisms in different cell types.
The demonstration of 2,3-DHBA and 2,5-DHBA to
inhibit cancer cell growth is a significant observation in view of
aspirin's reported ability to decrease CRC. An unanswered question
is: What is/are the target(s) for 2,3-DHBA and 2,5-DHBA in cells?
Our previously published data (31) and the present study show that
2,3-DHBA and 2,5-DHBA inhibited CDK1/CDK6 activity. However, at
this stage it is not known if these two observations are related,
or if the inhibition of proliferation by these compounds is due to
an alternative mechanism not involving CDKs. In this context, a
previous study reported that 2,5-DHBA can interfere with FGF
function by disrupting the receptor-growth factor signaling complex
in vitro and in cell culture studies (61). At this stage, we believe that
2,5-DHBA does not target COX-2 to mediate its chemo-preventive
effect in the GI epithelial cells as HCT-116 cells are reported to
lack COX-2 expression while HT-29 cells express inactive COX-2
(62). Since both these cell lines
were inhibited by 2,5-DHBA, it suggests that the aspirin
metabolites can inhibit cancer cell proliferation through a COX-2
independent mechanism. Therefore, greater efforts need to be placed
into these relatively unexplored areas to determine how 2,3-DHBA
and 2,5-DHBA exert their chemopreventive actions. Whether
microbiota generated salicylic acid metabolites becomes available
for CDK inhibition following their uptake into the GI cells is an
interesting area for future study. If proven, this will highlight
the contribution of the gut microflora to aspirin's chemoprevention
against CRC, through formation of 2,3-DHBA and 2,5-DHBA. A model
depicting how 2,3-DHBA and 2,5-DHBA generated in the GI lumen
through the microbial degradation of aspirin/salicylic acid,
leading to inhibition of CDKs and cell proliferation is shown in
Fig. 9. We suggest that the
ability of aspirin to inhibit tumor formation in the
intestinal/colonic mucosa may be a local effect via salicylic acid
metabolites generated in the gut acting on epithelial cells, and
may not require absorption into the blood.
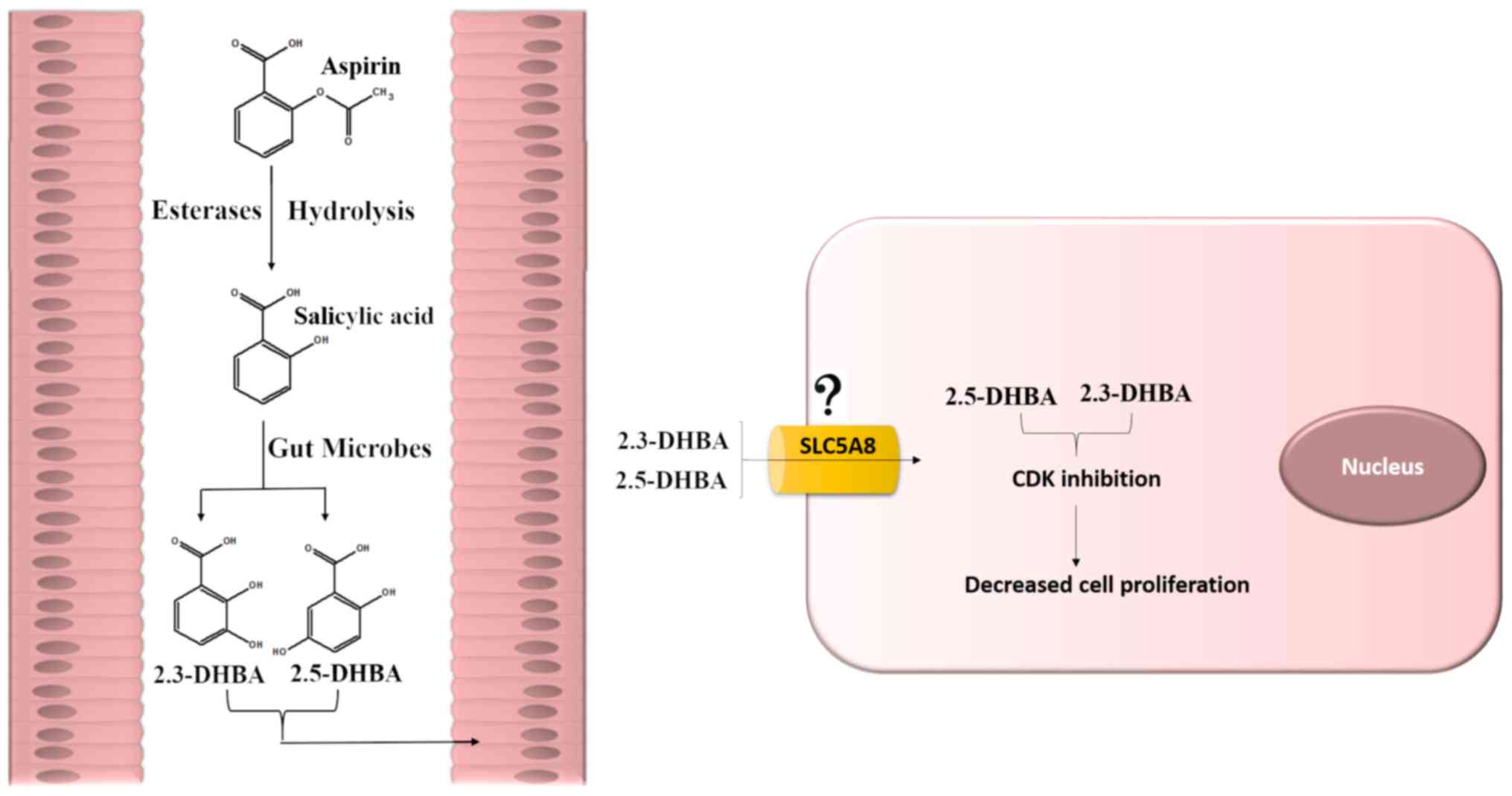 | Figure 9.A model depicting how aspirin
metabolites may exert its chemopreventive effects on colonic tissue
to protect against CRC. It was hypothesized that aspirin and
salicylic acid may impede events/pathways crucial to tumor
formation through its metabolites 2,3-DHBA and 2,5-DHBA. It was
suggested that 2,3-DHBA and 2,5-DHBA generated from salicylic acid
by the action of gut microflora is taken up by the colonic
epithelial cells. The uptake of these metabolites may occur through
SLC5A8, a monocarboxylic acid transporter. Accumulation of these
metabolites within the epithelial cells may result in CDK
inhibition. This may cause reduced cell proliferation, allowing an
opportunity for DNA repair and immune surveillance, thereby
preventing tumorigenesis. Alternatively, it is also possible that
2,3-DHBA and 2,5-DHBA may have additional targets contributing to
cancer prevention. 2,3-DHBA, 2,3-dihydroxybenzoic acid; 2,5-DHBA,
2,5-dihydroxybenzoic acid. |
Nearly 35 years have passed since the initial report
of aspirin's ability to decrease incidences of CRC (63), however, debates still continue on
its mode of action. Although the platelet hypothesis is widely
discussed in literature, it has its own short-comings and appears
to work indirectly through a set of sequential steps involving two
COX isoforms (COX-1 and COX-2) (9–14).
COX-1 is the only form of COX in platelets and is constitutively
expressed in all tissues, whereas COX-2 is inducible and expressed
under inflammatory conditions. COX-2 levels are also elevated in
many cancers (48,64–67).
The half-life of intact aspirin is about 15–20 min (68) in the systemic circulation and this
period appears to be enough to cause inhibition of COX-1 in
platelets (1). As chronic
inflammation is linked to cancer, it is suggested that aspirin may
mediate its effect through inhibition of these COX enzymes.
However, aspirin is more effective in the inhibition COX-1
(IC50 1.67 µM) than COX-2 (IC50 278 µM)
(69). Since low dose aspirin
(75–160 mg) is as effective as higher doses (6), the possibility of aspirin directly
targeting COX-2 as a potential mechanism to decrease cancer is
thought to be unlikely because lower doses administered per day is
insufficient to cause COX-2 inhibition. It is suggested that
aspirin's effects may involve sequential inhibition of COX-1 and
COX-2 (9,10,12).
This theory states that inhibition of COX-1 in platelets leads to
prevention of platelet aggregation which subsequently prevents the
release of lipid mediators (e.g., PGE-2; TXA2), cytokines and
growth factors (e.g., PDGF) from the alpha-granules. These events
could inhibit the induction of COX-2 in regions of GI mucosal
lesions, preventing tumorigenesis.
If inhibition of COX-1 in platelets in the blood
following its absorption is the primary mechanism as proposed in
the platelet hypothesis (9–11),
one would expect aspirin to decrease the incidences of cancer to
the same extent in all tissues in the body, unless one assumes that
GI mucosal tissues are more prone to lesions which leads to
platelet aggregation, creating an environment suitable for
eliciting an inflammatory response. It is intriguing that aspirin
that is only present for less than 20 min. in the circulation,
while capable of preventing platelet activation and aggregation,
could have such a profound effect against CRC development. While
inhibition of COX-1 in platelets may play a role (e.g., in the
inhibition of metastasis), it is likely that other pathways may
play a more significant role in aspirin's cancer preventive
actions.
Although the ‘platelet hypothesis’ is an attractive
theory, we suggest that alternative mechanisms as proposed in this
study involving locally generated salicylic acid metabolites by the
gut microflora from aspirin as potential contributors to
chemoprevention should be considered. As ~50% of orally
administered aspirin is left unabsorbed in the GI lumen (51,52),
one would expect the concentration of aspirin from an 81 mg tablet
to be in the range of 0.3 to 1.4 mM in the gut assuming that the
total GI fluid volume under fed and fasting conditions are ~750 and
~160 ml respectively (70).
Presence of microflora in the GI lumen may thus be able to generate
2,3-DHBA and/or 2,5-DHBA from aspirin/salicylic acid, sufficient to
cause inhibition of cell proliferation, allowing aspirin to act
locally on colorectal tissues, thus adding merit to our hypothesis.
In our opinion, locally generated metabolites will provide a more
direct effect on colorectal tissues against CRC as compared to the
indirect mechanism involving sequential steps proposed in platelet
hypothesis through COX-1 and COX-2 inhibition. In vivo
studies utilizing germ-free mice should shed light on the role of
aspirin/salicylic metabolites and the microflora in aspirin's
chemo-preventive properties.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Hemachand Tummala
(Department of Pharmaceutical Sciences, South Dakota State
University) for the helpful discussions and Ms. Vijaya Gaddipati
(volunteer researcher; Department of Pharmaceutical Sciences, South
Dakota State University) for assisting in cell culture studies.
Funding
The present study was funded by The National
Institutes of Health (grant no. 5R03CA133061-02) and
Research/Scholarship Support Fund 2018 from The Office of Research,
South Dakota State University to GJB.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
GJB and RS conceptualized the study. CKV, RD and TL
contributed to the experimental work. DRK and RS conducted the
formal analysis (molecular docking). GJB and RS performed the
experimental investigation and analysis. RS and GJB wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
2,3-DHBA
|
2,3-dihydroxybenzoic acid
|
|
2,5-DHBA
|
2,5-dihydroxybenzoic acid
|
References
|
1
|
Vane JR and Botting RM: The mechanism of
action of aspirin. Thromb Res. 110:255–258. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vane JR: Inhibition of prostaglandin
synthesis as a mechanism of action for aspirin-like drugs. Nat New
Biol. 231:232–235. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Roth GJ and Majerus PW: The mechanism of
the effect of aspirin on human platelets. I. Acetylation of a
particulate fraction protein. J Clin Invest. 56:624–632. 1975.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hennekens CH: Update on aspirin in the
treatment and prevention of cardiovascular disease. Am J Manag
Care. 8 (22 Suppl):S691–S700. 2002.PubMed/NCBI
|
|
5
|
Rothwell PM, Price JF, Fowkes FG,
Zanchetti A, Roncaglioni MC, Tognoni G, Lee R, Belch JF, Wilson M,
Mehta Z and Meade TW: Short-term effects of daily aspirin on cancer
incidence, mortality, and non-vascular death: Analysis of the time
course of risks and benefits in 51 randomised controlled trials.
Lancet. 379:1602–1612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rothwell PM, Wilson M, Elwin CE, Norrving
B, Algra A, Warlow CP and Meade TW: Long-term effect of aspirin on
colorectal cancer incidence and mortality: 20-year follow-up of
five randomised trials. Lancet. 376:1741–1750. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rothwell PM, Wilson M, Price JF, Belch JF,
Meade TW and Mehta Z: Effect of daily aspirin on risk of cancer
metastasis: A study of incident cancers during randomised
controlled trials. Lancet. 379:1591–1601. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bibbins-Domingo K; U.S. Preventive
Services Task Force, : Aspirin use for the primary prevention of
cardiovascular disease and colorectal cancer: U.S. Preventive
services task force recommendation statement. Ann Intern Med.
164:836–845. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thun MJ, Jacobs EJ and Patrono C: The role
of aspirin in cancer prevention. Nat Rev Clin Oncol. 9:259–267.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Patrono C: The Multifaceted clinical
readouts of platelet inhibition by low-dose aspirin. J Am Coll
Cardiol. 66:74–85. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Patrignani P and Patrono C: Aspirin,
platelet inhibition and cancer prevention. Platelets. 29:779–785.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Patrignani P and Patrono C: Aspirin and
cancer. J Am Coll Cardiol. 68:967–976. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dovizio M, Alberti S, Guillem-Llobat P and
Patrignani P: Role of platelets in inflammation and cancer: Novel
therapeutic strategies. Basic Clin Pharmacol Toxicol. 114:118–127.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chan AT, Arber N, Burn J, Chia WK, Elwood
P, Hull MA, Logan RF, Rothwell PM, Schrör K and Baron JA: Aspirin
in the chemoprevention of colorectal neoplasia: An overview. Cancer
Prev Res (Phila). 5:164–178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kopp E and Ghosh S: Inhibition of NF-kappa
B by sodium salicylate and aspirin. Science. 265:956–959. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dovizio M, Bruno A, Tacconelli S and
Patrignani P: Mode of action of aspirin as a chemopreventive agent.
Recent Results Cancer Res. 191:39–65. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Din FV, Valanciute A, Houde VP, Zibrova D,
Green KA, Sakamoto K, Alessi DR and Dunlop MG: Aspirin inhibits
mTOR signaling, activates AMP-activated protein kinase, and induces
autophagy in colorectal cancer cells. Gastroenterology.
142:1504–1515.e3. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hawley SA, Fullerton MD, Ross FA,
Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green
KA, Mustard KJ, et al: The ancient drug salicylate directly
activates AMP-activated protein kinase. Science. 336:918–922. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gala MK and Chan AT: Molecular pathways:
Aspirin and Wnt signaling-a molecularly targeted approach to cancer
prevention and treatment. Clin Cancer Res. 21:1543–1548. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ai G, Dachineni R, Muley P, Tummala H and
Bhat GJ: Aspirin and salicylic acid decrease c-Myc expression in
cancer cells: A potential role in chemoprevention. Tumour Biol.
37:1727–1738. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Goel A, Chang DK, Ricciardiello L, Gasche
C and Boland CR: A novel mechanism for aspirin-mediated growth
inhibition of human colon cancer cells. Clin Cancer Res. 9:383–390.
2003.PubMed/NCBI
|
|
22
|
Pietrocola F, Castoldi F, Markaki M,
Lachkar S, Chen G, Enot DP, Durand S, Bossut N, Tong M, Malik SA,
et al: Aspirin recapitulates features of caloric restriction. Cell
Rep. 22:2395–2407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ai G, Dachineni R, Kumar DR, Marimuthu S,
Alfonso LF and Bhat GJ: Aspirin acetylates wild type and mutant p53
in colon cancer cells: Identification of aspirin acetylated sites
on recombinant p53. Tumour Biol. 37:6007–6016. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ai G, Dachineni R, Kumar DR, Alfonso LF,
Marimuthu S and Bhat GJ: Aspirin inhibits glucose-6-phosphate
dehydrogenase activity in HCT 116 cells through acetylation:
Identification of aspirin-acetylated sites. Mol Med Rep.
14:1726–1732. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rigas B and Tsioulias GJ: The evolving
role of nonsteroidal anti-inflammatory drugs in colon cancer
prevention: A cause for optimism. J Pharmacol Exp Ther. 353:2–8.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bosetti C, Rosato V, Gallus S, Cuzick J
and La Vecchia C: Aspirin and cancer risk: A quantitative review to
2011. Ann Oncol. 23:1403–1415. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Harris RE, Beebe-Donk J, Doss H and Burr
Doss D: Aspirin, ibuprofen, and other non-steroidal
anti-inflammatory drugs in cancer prevention: A critical review of
non-selective COX-2 blockade (review). Oncol Rep. 13:559–583.
2005.PubMed/NCBI
|
|
28
|
Cao Y, Nishihara R, Wu K, Wang M, Ogino S,
Willett WC, Spiegelman D, Fuchs CS, Giovannucci EL and Chan AT:
Population-wide impact of long-term use of aspirin and the risk for
cancer. JAMA Oncol. 2:762–769. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bojić M, Sedgeman CA, Nagy LD and
Guengerich FP: Aromatic hydroxylation of salicylic acid and aspirin
by human cytochromes P450. Eur J Pharm Sci. 73:49–56. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim IS, Yoo DH, Jung IH, Lim S, Jeong JJ,
Kim KA, Bae ON, Yoo HH and Kim DH: Reduced metabolic activity of
gut microbiota by antibiotics can potentiate the antithrombotic
effect of aspirin. Biochem Pharmacol. 122:72–79. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dachineni R, Kumar DR, Callegari E,
Dachineni R, Kumar DR, Callegari E, Kesharwani SS, Sankaranarayanan
R, Seefeldt T, Tummala H and Bhat GJ: Salicylic acid metabolites
and derivatives inhibit CDK activity: Novel insights into aspirin's
chemopreventive effects against colorectal cancer. Int J Oncol.
51:1661–1673. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
DK2 kinase assay, . https://www.signalchem.com/shared_product_sheets/N211-4.pdf
|
|
33
|
CDK1 Kinase assay, . https://www.signalchem.com/shared_product_sheets/F625-2.pdf
|
|
34
|
CDK4 Kinase assay, . https://www.signalchem.com/shared_product_sheets/R219-1.pdf
|
|
35
|
CDK6 Kinase assay, . https://www.signalchem.com/shared_product_sheets/D062-1.pdf
|
|
36
|
Sankaranarayanan R, Valiveti CK, Kumar DR,
Van Slambrouck S, Kesharwani SS, Seefeldt T, Scaria J, Tummala H
and Bhat GJ: The flavonoid metabolite 2,4,6-trihydroxybenzoic acid
is a CDK inhibitor and an anti-proliferative agent: A potential
role in cancer prevention. Cancers (Basel). 11(pii): E4272019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dachineni R, Ai G, Kumar DR, Sadhu SS,
Tummala H and Bhat GJ: Cyclin A2 and CDK2 as novel targets of
aspirin and salicylic acid: A potential role in cancer prevention.
Mol Cancer Res. 14:241–252. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Van Der Spoel D, Lindahl E, Hess B,
Groenhof G, Mark AE and Berendsen HJ: GROMACS: Fast, flexible, and
free. J Comput Chem. 26:1701–1718. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu SY, McNae I, Kontopidis G, McClue SJ,
McInnes C, Stewart KJ, Wang S, Zheleva DI, Marriage H, Lane DP, et
al: Discovery of a novel family of CDK inhibitors with the program
LIDAEUS: Structural basis for ligand-induced disordering of the
activation loop. Structure. 11:399–410. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Din FV, Dunlop MG and Stark LA: Evidence
for colorectal cancer cell specificity of aspirin effects on NF
kappa B signalling and apoptosis. Br J Cancer. 91:381–388. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Qiao L, Hanif R, Sphicas E, Shiff SJ and
Rigas B: Effect of aspirin on induction of apoptosis in HT-29 human
colon adenocarcinoma cells. Biochem Pharmacol. 55:53–64. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dibra HK, Brown JE, Hooley P and Nicholl
ID: Aspirin and alterations in DNA repair proteins in the SW480
colorectal cancer cell line. Oncol Rep. 24:37–46. 2010.PubMed/NCBI
|
|
43
|
Chen J and Stark LA: Aspirin prevention of
colorectal cancer: Focus on NF-κB signalling and the nucleolus.
Biomedicines. 5(pii): E432017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mitrugno A, Sylman JL, Ngo AT, Pang J,
Sears RC, Williams CD and McCarty OJ: Aspirin therapy reduces the
ability of platelets to promote colon and pancreatic cancer cell
proliferation: Implications for the oncoprotein c-MYC. Am J Physiol
Cell Physiol. 312:C176–C189. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ma J, Cai Z, Wei H, Liu X, Zhao Q and
Zhang T: The anti-tumor effect of aspirin: What we know and what we
expect. Biomed Pharmacother. 95:656–661. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bruno A, Dovizio M, Tacconelli S, Contursi
A, Ballerini P and Patrignani P: Antithrombotic agents and cancer.
Cancers (Basel). 10(pii): E2532018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cox D, Maree AO, Dooley M, Conroy R, Byrne
MF and Fitzgerald DJ: Effect of enteric coating on antiplatelet
activity of low-dose aspirin in healthy volunteers. Stroke.
37:2153–2158. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dovizio M, Maier TJ, Alberti S, Di
Francesco L, Marcantoni E, Münch G, John CM, Suess B, Sgambato A,
Steinhilber D and Patrignani P: Pharmacological inhibition of
platelet-tumor cell cross-talk prevents platelet-induced
overexpression of cyclooxygenase-2 in HT29 human colon carcinoma
cells. Mol Pharmacol. 84:25–40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lichtenberger LM, Fang D, Bick RJ,
Poindexter BJ, Phan T, Bergeron AL, Pradhan S, Dial EJ and Vijayan
KV: Unlocking aspirin's chemopreventive activity: Role of
irreversibly inhibiting platelet cyclooxygenase-1. Cancer Prev Res
(Phila). 10:142–152. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Inoue M, Morikawa M, Tsuboi M, Ito Y and
Sugiura M: Comparative study of human intestinal and hepatic
esterases as related to enzymatic properties and hydrolizing
activity for ester-type drugs. Jpn J Pharmacol. 30:529–535. 1980.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pedersen AK and FitzGerald GA:
Dose-related kinetics of aspirin. Presystemic acetylation of
platelet cyclooxygenase. N Engl J Med. 311:1206–1211. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Rowland M, Riegelman S, Harris PA and
Sholkoff SD: Absorption kinetics of aspirin in man following oral
administration of an aqueous solution. J Pharm Sci. 61:379–385.
1972. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Haastrup PF, Gronlykke T and Jarbol DE:
Enteric coating can lead to reduced antiplatelet effect of low-dose
acetylsalicylic acid. Basic Clin Pharmacol Toxicol. 116:212–215.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gervot L, Carriere V and Costet P: CYP3A5
is the major cytochrome P450 3A expressed in human colon and
colonic cell lines. Environ Toxicol Pharmacol. 2:381–388. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Swanson HI: Drug metabolism by the host
and gut microbiota: A partnership or rivalry? Drug Metab Dispos.
43:1499–1504. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ganapathy V, Thangaraju M, Gopal E, Martin
PM, Itagaki S, Miyauchi S and Prasad PD: Sodium-coupled
monocarboxylate transporters in normal tissues and in cancer. AAPS
J. 10:193–199. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Juurlink BH, Azouz HJ, Aldalati AM,
AlTinawi BM and Ganguly P: Hydroxybenzoic acid isomers and the
cardiovascular system. Nutr J. 13:632014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jenner AM, Rafter J and Halliwell B: Human
fecal water content of phenolics: The extent of colonic exposure to
aromatic compounds. Free Radic Biol Med. 38:763–772. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Altinoz MA, Elmaci I, Cengiz S,
Emekli-Alturfan E and Ozpinar A: From epidemiology to treatment:
Aspirin's prevention of brain and breast-cancer and
cardioprotection may associate with its metabolite gentisic acid.
Chem Biol Interact. 291:29–39. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Vad NM, Yount G and Moridani MY:
Biochemical mechanism of acetylsalicylic acid (Aspirin) selective
toxicity toward melanoma cell lines. Melanoma Res. 18:386–399.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Fernández IS, Cuevas P, Angulo J,
López-Navajas P, Canales-Mayordomo A, González-Corrochano R, Lozano
RM, Valverde S, Jiménez-Barbero J, Romero A and Giménez-Gallego G:
Gentisic acid, a compound associated with plant defense and a
metabolite of aspirin, heads a new class of in vivo fibroblast
growth factor inhibitors. J Biol Chem. 285:11714–11729. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hsi LC, Baek SJ and Eling TE: Lack of
cyclooxygenase-2 activity in HT-29 human colorectal carcinoma
cells. Exp Cell Res. 256:563–570. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kune GA, Kune S and Watson LF: Colorectal
cancer risk, chronic illnesses, operations, and medications: Case
control results from the melbourne colorectal cancer study. Cancer
Res. 48:4399–4404. 1988.PubMed/NCBI
|
|
64
|
Eberhart CE, Coffey RJ, Radhika A,
Giardiello FM, Ferrenbach S and DuBois RN: Up-regulation of
cyclooxygenase 2 gene expression in human colorectal adenomas and
adenocarcinomas. Gastroenterology. 107:1183–1188. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sano H, Kawahito Y, Wilder RL, Hashiramoto
A, Mukai S, Asai K, Kimura S, Kato H, Kondo M and Hla T: Expression
of cyclooxygenase-1 and −2 in human colorectal cancer. Cancer Res.
55:3785–3789. 1995.PubMed/NCBI
|
|
66
|
Gupta RA and Dubois RN: Colorectal cancer
prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev
Cancer. 1:11–21. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kargman SL, O'Neill GP, Vickers PJ, Evans
JF, Mancini JA and Jothy S: Expression of prostaglandin G/H
synthase-1 and −2 protein in human colon cancer. Cancer Res.
55:2556–2559. 1995.PubMed/NCBI
|
|
68
|
Costello PB and Green FA: Aspirin survival
in human blood modulated by the concentration of erythrocytes.
Arthritis Rheum. 25:550–555. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Vane JR, Bakhle YS and Botting RM:
Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 38:97–120.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Schiller C, Fröhlich CP, Giessmann T,
Siegmund W, Mönnikes H, Hosten N and Weitschies W: Intestinal fluid
volumes and transit of dosage forms as assessed by magnetic
resonance imaging. Aliment Pharmacol Ther. 22:971–979. 2005.
View Article : Google Scholar : PubMed/NCBI
|