Introduction
Timely and successful myocardial reperfusion with
either primary percutaneous coronary intervention or thrombolytic
therapy has resulted in a significant decrease in infarct area size
and mortality rates of acute myocardial infarction (AMI) (1). However, ischemic heart disease (IHD)
following AMI is one of the principal causes of death and
disability worldwide (2).
MicroRNAs (miRNAs/miRs) are a class of small non-coding RNAs~20
ribonucleotides in length that negatively regulate target genes by
binding to sequences in the 3′-untranslated regions of target
messenger RNAs (3). Currently, a
growing body of evidence has indicated that miRNAs may play an
important role in the processes that lead to the pathophysiological
consequences of IHD by altering key signaling elements and
regulating proliferation, differentiation and survival in mRNA
levels (4–7).
miR-210 has been demonstrated to be markedly
upregulated in response to hypoxia in a broad spectrum of cell
types (8), including
cardiomyocytes. A murine model study revealed that miR-210
overexpression in the heart using minicircle vectors enhanced
cardiac function, decreased infarct size, inhibited apoptosis and
induced neovascularization following AMI (9). Other studies have revealed that
apoptosis-inducing factor mitochondrion-associated 3 (AIFM3) and
caspase 8 associated protein 2 (Casp8ap2), known as proapoptotic
proteins, were inhibited by miR-210 overexpression (9,10).
miR-210 has also been demonstrated as being involved in several
other cellular functions, from stem cell survival (9,11)
and reactive oxygen species (ROS) generation (10) to angiogenesis (12). Thus, miR-210 is suggested to be a
potential therapeutic target for treating IHD.
Cardiac shock wave therapy (SWT) is a promising,
noninvasive and safe modality that has been demonstrated to
alleviate myocardial ischemia and improve cardiac function in
patients with IHD (13,14). The improvement of myocardial
perfusion and exercise capacity has been observed in numerous
clinical trials (15,16). In vitro studies have
indicated that cardiac SWT decreased hypoxia-induced apoptosis in
H9c2 cells by activating the PI3K-Akt pathway (17). A recent report revealed that
cardiac SWT protected cardiomyocytes from apoptosis by attenuating
cytochrome c release from the mitochondria in an in vivo rat
AMI model (18). However, few
studies have focused on miRNAs in regard to their protective
effects during cardiac SWT. Taken together, an evaluation of the
influence of cardiac SWT on miR-210 following myocardial ischemic
injury would be of use.
The present study used an in vitro model of
AMI in order to investigate whether cardiac SWT could protect
cardiomyocytes against hypoxia through modulating miR-210 and the
underlying molecular mechanisms.
Materials and methods
Reagents
Dulbecco's Modified Eagle's medium (DMEM), RPMI-1640
medium and protease inhibitor cocktails were purchased from
Sigma-Aldrich; Merck KGaA. Trypsin-EDTA, PBS,
penicillin/streptomycin and fetal bovine serum (FBS) were from
Thermo Fisher Scientific, Inc. Antibodies (Abs) directed against
GAPDH, Bcl-2, Bax, p38 mitogen-activated protein kinase (MAPK),
phosphorylated (p)-p38MAPK, Akt, p-Akt, horseradish peroxidase
(HRP)-coupled anti-rabbit IgG secondary Ab and lysis buffer were
purchased from Cell Signaling Technology, Inc. Protein
concentration was determined by bicinchoninic acid (BCA) protein
assay kit from Pierce; Thermo Fisher Scientific, Inc. Immobilon
Western HRP Substrate was purchased from Merck KGaA. Fluorescent
assays for apoptosis was from Beijing Solarbio Science &
Technology Co., Ltd. The Cell Titer 96® AQueous One
Solution Cell Proliferation Assay was obtained from Promega
Corporation. miR-210 mimics, miR-210 inhibitors and negative
controls (NC) of miRNA were all designed and synthesized by Sangon
Biotech Co., Ltd. The sequences of miR-210 inhibitor negative
controls and mimics negative controls were as follows (5′ to 3′):
miR-210 inhibitor negative controls, CAGUACUUUUGUGUAGUACAA; miR-210
mimics negative controls sense, UUCUCCGAACGUGUCACGUTT; and miR-210
mimics negative controls antisense, ACGUGACACGUUCGGAGAATT.
TRIzol® and Lipofectamine® RNAiMAX reagent
were obtained from Thermo Fisher Scientific, Inc. MicroRNA reverse
transcription kit was from New England BioLabs, Inc. SYBR Green PCR
Master Mix was purchased from Takara Biotechnology Co., Ltd. A
lipid peroxidation malondialdehyde (MDA) assay kit was purchased
from Beyotime Institute of Biotechnology (cat. no. S0131).
HL-1 cell culture
HL-1 cells were provided by Dr William Claycomb
(Louisiana State University Health Science Center), an immortalized
cell line derived from mouse atrial cardiac myocytes, were cultured
in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 100
µg/ml streptomycin. Cells were maintained at 37°C in a humidified
chamber with an atmosphere of 95% air and 5% CO2.
Hypoxia treatment
When the cells reached a confluence of 60–70%, HL-1
cells were cultured in FBS-free media for 24 h before all
experiments. To mimic ischemic injury in vivo, cells were
exposed to ischemia-mimetic solution as previously described
(19). Subsequently, the cells
were incubated in a 3-gas hypoxic chamber (HERAcell VIOS 160i,
Thermo Fisher Scientific, Inc.) equilibrated with 94%
N2, 5% CO2 and 1% O2 for 5 h.
SWT
After 5 h of hypoxia, cells were subjected to SWT as
previously described (20). In
brief, the HL-1 cells were digested with trypsin after hypoxia,
transferred to centrifuge tubes (1 ml, 5×106 cells/ml)
and treated with a shock wave device (Duolith VET; Storz Medical
AG). Both the ultrasonic probe and the tubes were submerged in a
water bath at 37°C. Then, 200 pulses of shock waves with an energy
of 0.05 mJ/mm2 were administered to the cells. After
SWT, cells were placed in standard medium under normoxic conditions
for 12 h before harvest.
MTS assay
Cell viability was measured by Cell Titer 96 AQueous
One Solution Cell Proliferation Assay. In brief, HL-1 cells were
obtained after hypoxia and SWT treatment and re-suspended to a
density of 5×106 cells/ml. Then, cells were seeded in
24-well plates in quadruplicate. MTS reagent (45 µl) was added to
each well of the 24-well plates with 225 µl of culture medium.
After 4 h, the absorbance was detected at 490 nm using a microplate
reader.
Flow cytometry
Cell apoptosis was analyzed using flow cytometry
after fluorescein isothiocyanate-conjugated Annexin V (FITC-Annexin
V; Beijing Solarbio Science & Technology Co., Ltd.) and
propidium iodide (PI) staining. The staining procedure was
performed according to the instructions. HL-1 cells in early
logarithmic growth phase were seeded in 6-well plates, harvested by
centrifugation at 800 g for 5 min at room temperature, and then
washed twice with PBS. Cells were resuspended in 100 µl of 1X
binding buffer to a density of 1×106 cells/ml, 5 µl of
Annexin V-FITC was added, and the cells were incubated in the dark
at room temperature for 10 min. Then, 5 µl of PI was added and the
cells were incubated in the dark at room temperature for another 5
min. Finally, 500 µl of 1X binding buffer was added to the mixture,
which was then loaded onto a flow cytometer (FACSCalibur; BD
Biosciences) for the apoptosis analysis using FlowJo 7.6.1 (BD
Biosciences). Cells that were considered viable were both Annexin V
and PI negative, while cells that were in early apoptosis were
Annexin V positive and PI negative, and necrotic cells were both
Annexin V and PI positive.
Lipid peroxidation
MDA is a natural byproduct of lipid peroxidation and
used as a reliable marker to measure the level of oxidative stress.
Free MDA reacts with thiobarbituric acid (TBA) and generates an
MDA-TBA adduct, which can be quantified colorimetrically (optical
density 532 nm). MDA levels were measured using the TBA method
according to the manufacturer's protocol.
Measuring intracellular ROS
levels
ROS generation was measured with dihydroethidium
(DHE; Invitrogen; Thermo Fisher Scientific, Inc.) staining and
observed under a fluorescent microscope (Olympus BX51; Olympus
Corporation; magnification, ×50). Briefly, When the HL-1 cells
reached the appropriate confluence (70–80%) in 24-well plates, the
cells were incubated with DHE (8 µm) for 30 min at 37°C in the
dark, and then washed with PBS. Following the manufacturer's
protocols, DHE was alternately excited at a wavelength of 535 nm
and emitted at a wavelength of 512 nm. The fluorescence was
measured and expressed as a percentage of the integrated optical
density in the control.
Western blot analysis
HL-1 cells were lysed in ice-cold lysis buffer (Cell
Signaling Technology, Inc.) supplemented with phosphatase and
protease inhibitors. Cell lysates were incubated on ice for 5 min
and centrifuged at 13,000 g for 15 min (4°C). Protein
concentrations of the supernatants were determined using a Pierce™
BCA Protein Assay Kit. All samples were boiled for 10 min at 100°C.
Soluble proteins (20 µg/lane) were electrophoresed on 12% SDS
polyacrylamide gels and transferred to polyvinylidene fluoride
membranes that were then blocked with TBS with 0.1% Tween-20 buffer
containing 5% non-fat milk for 2 h at room temperature. Membranes
were incubated overnight at 4°C with specific primary Abs (1:1,000)
against GAPDH (cat. no. 5174), Bcl-2 (cat. no. 2772), p38 MAPK
(cat. no. 8690), p-p38 MAPK (cat. no. 4511), Akt (cat. no. 4691)
and p-Akt (cat. no. 4060), and then incubated with secondary Ab
(1:5,000; ct. no. 7074) for 2 h at room temperature. The
immunoreaction was visualized with Immobilon Western HRP substrate.
Densitometry was performed using ImageJ software Java 1.8.0
(National Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNAs from cardiomyocytes were isolated by
TRIzol® reagent. The concentration of RNA was detected
using a NanoDrop™ 2,000 Spectrophotometer (NanoDrop Technologies;
Thermo Fisher Scientific, Inc.). Using a microRNA reverse
transcription kit (New England BioLabs, Inc.), 800 ng of total RNA
was reverse transcribed. The RNA specific primers and SYBR Green
were used for RT-qPCR to examine the relative quantification of
RNAs with a Bio-Rad iCycler RT-PCR Detection System (Bio-Rad
Laboratories, Inc.). The thermocycling conditions were: 95°C for 10
min, 95°C for 15 sec and 60°C for 60 sec. Then, steps 2–3 were
repeated for 40 cycles. U6 and GAPDH were chosen as internal
controls. Each reaction was performed in triplicate, and analysis
was performed by the 2−∆∆Cq method (21). The primers are shown in Table I.
 | Table I.Primers for RT-qPCR. |
Table I.
Primers for RT-qPCR.
| Gene | Primer sequence
(5′-3′) |
|---|
| U6 | Forward:
GCGCGTCGTGAAGCGTTC |
|
| Universal:
GTGCAGGGTCCGAGGT |
|
| Stem loop:
GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAAAATATG |
| miR-210 | Forward:
GTGCGTGTGACAGCGGC |
|
| Universal:
GTGCAGGGTCCGAGGT |
|
| Stem loop:
GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTCAGCC |
| GAPDH | Forward:
TGGCCTTCCGTGTTCCTACC |
|
| Reverse:
TGTAGGCCATGAGGTCCACCAC |
| Casp8ap2 | Forward:
ACAGTGAAGCAAGGACGGAG |
|
| Reverse:
TCGAGTGTGTGGCCTTTCTC |
| AIFM3 | Forward:
CTGGCCTAGTGTGTGCAGAA |
|
| Reverse:
CCGGTCATAGGGCAGATGTC |
Transient transfection
HL-1 cells (70% confluent) were transfected with
miR-210 mimics (20 µM) or inhibitors (20 µM) or miRNA negative
controls using Lipofectamine-RNAiMAX reagent according to the
manufacturer's protocols. After 24 h of transfection, cells were
treated with hypoxia and SWT.
Monodansylcadaverine (MDC)
staining
The MDC kit was obtained from Nanjing KeyGen Biotech
Co., Ltd. The cells were washed twice with 300 µl 1X washing
buffer. Then, 100 µl working solution was added to each well. The
cells were incubated at room temperature for 20 min in the dark.
The cells were washed four times with 1X washing buffer to remove
redundant dye. Finally, cells were visualized using a fluorescent
microscope (magnification, ×50) with an excitation wavelength of
355 nm and an emission filter of 512 nm.
Statistical analysis
Data are presented as the mean ± SEM. Student's
t-test and one-way ANOVA with Bonferroni correction were performed
to compare two conditions and multiple groups, respectively. All
statistical calculations were performed by GraphPad Prism 5
software (GraphPad Software, Inc.) and P<0.05 was considered to
indicate a statistically significant difference.
Results
Hypoxia induces cell injury in HL-1
cardiomyocytes
HL-1 cardiomyocytes have previously been applied in
hypoxia-induced studies, and have been demonstrated as a cell line
for cardiomyocyte study (22).
Therefore, the present study created an in vitro model of
myocardial ischemia using HL-1 cells. When using the MTS assay,
cell viability was significantly decreased by 29.6±1.6% after 5 h
of exposure to hypoxia, followed by 12 h of reoxygenation when
compared with the control, which was considered to be a moderate
injury (Fig. 1A). In order to
further investigate hypoxia-induced injury in cardiomyocytes, an
Annexin V/PI staining assay was used to detect cardiomyocyte
apoptosis. Hypoxia significantly increased the apoptotic rate
compared with normoxic cells (Fig.
1B). Furthermore, western blot analysis verified that hypoxia
induced a decrease in the Bcl-2/Bax ratio, indicating an increase
in cell apoptosis (Fig. 2).
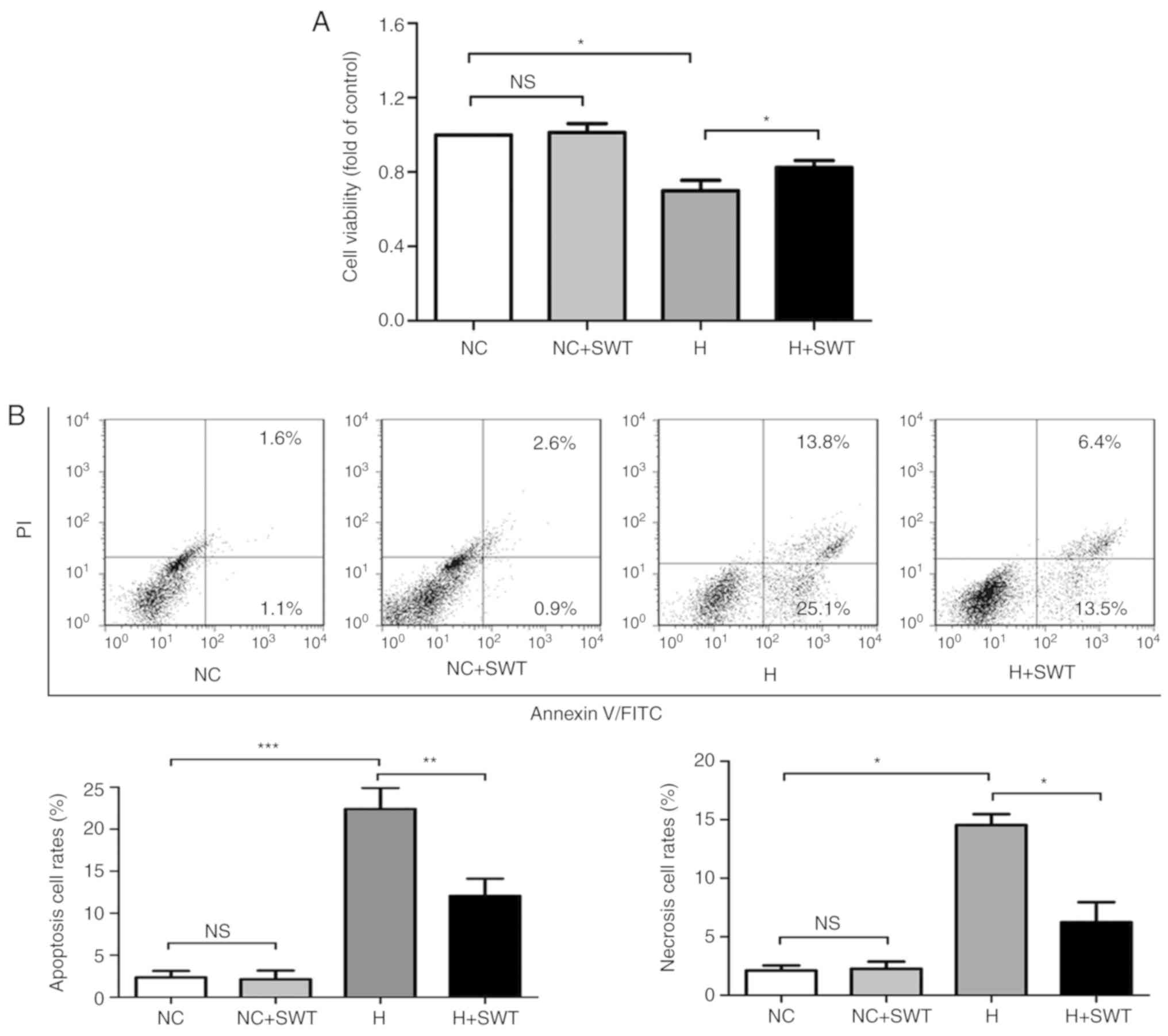 | Figure 1.Hypoxia induces cell injury in HL-1
cells, which can be reversed by SWT. (A) Cell viability was
determined by MTS assay (n=4). (B) Annexin V/PI double staining was
used to assess the apoptotic rates of HL-1 cells in the NC, NC +
SWT, hypoxia and hypoxia + SWT groups (n=4). *P<0.05,
**P<0.01, ***P<0.001. SWT, shock wave therapy; ns, not
significant; NC, negative control; H, hypoxia; PI, propidium
iodide. |
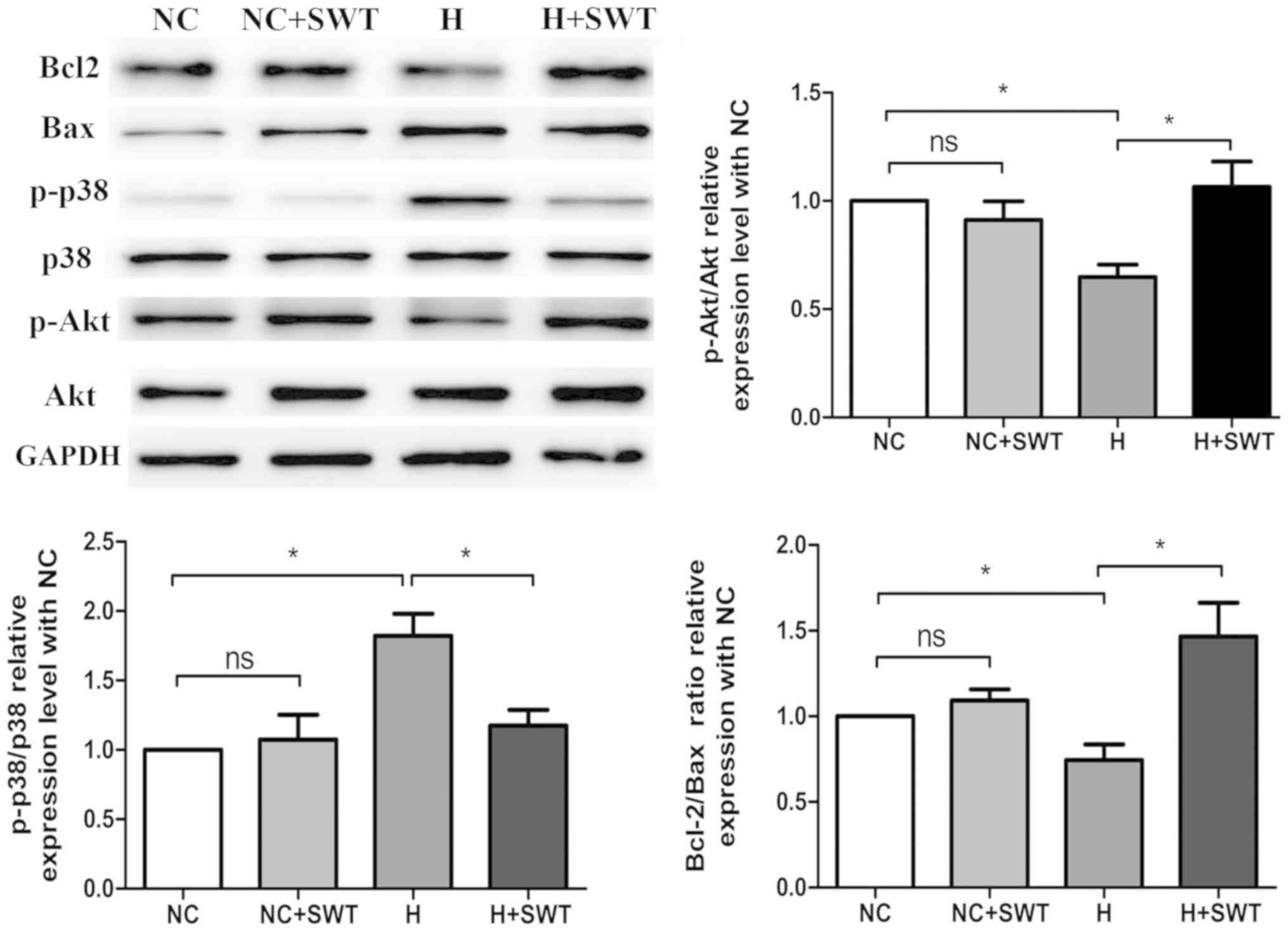 | Figure 2.Western blotting and average data for
Bcl-2, Bax, Akt, p-Akt, p38MAPK and p-p38MAPK levels in HL-1 cells
exposed to hypoxia and SWT (n=4). *P<0.05. MAPK,
mitogen-activated protein kinase; SWT, shock wave therapy; ns, not
significant; NC, negative control; H, hypoxia; MAPK, p,
phosphorylated. |
SWT alleviates hypoxia-induced
oxidative stress and apoptosis in HL-1 cells
In order to investigate the effect of SWT on hypoxic
injury, SWT was applied to HL-1 cells during the first 10 min of
reperfusion. SWT led to increased cell viability (Fig. 1A), a significant decrease in
apoptosis as manifested by reduced apoptotic cells (Fig. 1B) and an elevated Bcl-2/Bax ratio
specifically in hypoxic cells (Fig.
2), which was demonstrated by the morphological changes of HL1
cells and the decreased number of dead cells after SWT (Fig. S1). Accumulating evidence has
suggested that excessive ROS generation during ischemia could
stimulate apoptosis (23).
Therefore, the present study determined whether SWT inhibited ROS
production in the hypoxic model. Using DHE staining, the present
study revealed increased ROS production following hypoxia
treatment, and ROS levels were decreased by SWT (Fig. 3A). Excessive production of ROS
results in lipid peroxidation that generates oxidized by-products,
such as MDA (24). Thus, the
present study detected MDA levels using the TBA method. As
presented in Fig. 3B, the hypoxic
challenge caused a markedly increased MDA level, which was
downregulated by SWT (NC group vs. HR group vs. HR + SWT group,
1.0780±0.1229 nmol/mg protein vs. 3.5000±0.1600 nmol/mg protein vs.
2.8000±0.2300 nmol/mg protein, respectively). Furthermore, in order
to determine the molecular mechanisms underlying the protective
effects of SWT on hypoxic injury, cell survival-associated
proteins, including p38MAPK and Akt, were measured. Western blot
analysis revealed that cells exposed to hypoxia exhibited
significantly decreased expression of p-Akt and significantly
increased expression of p-p38MAPK (Fig. 2). In addition, it was revealed that
SWT significantly suppressed hypoxia-induced p38MAPK
phosphorylation and increased p-Akt in HL-1 cells. Overall, these
data suggested that SWT could reverse oxidative stress and
apoptosis in HL-1 cells exposed to hypoxia.
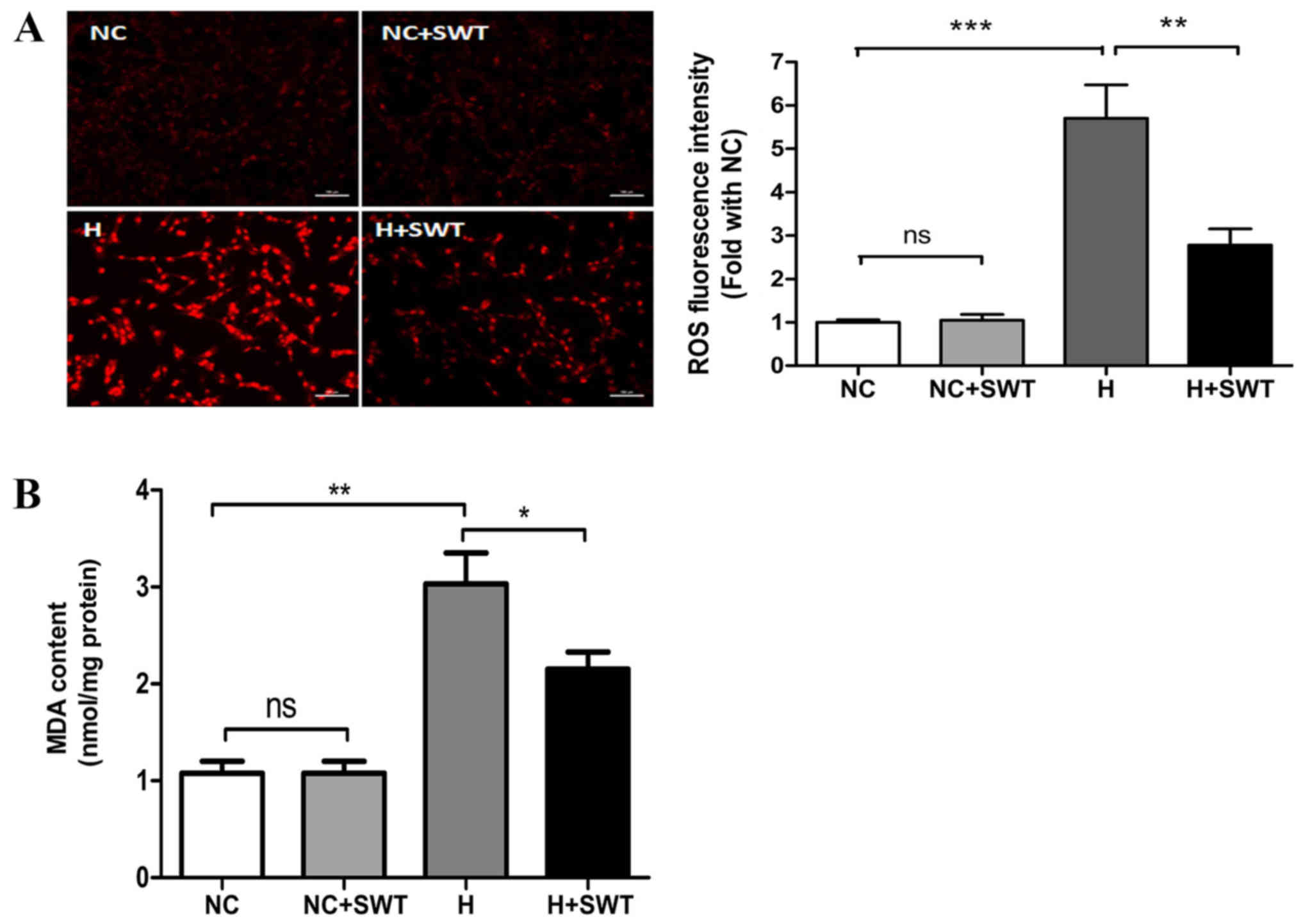 | Figure 3.SWT alleviates hypoxia-induced
oxidative stress in HL-1 cells. (A) ROS production, as measured
using dihydroethidium staining, in the NC, NC + SWT, hypoxia and
hypoxia + SWT groups (n=3; scale bar, 100 µm). (B) MDA levels were
assessed using the thiobarbituric acid method in HL-1 cells exposed
to hypoxia and SWT (n=4). *P<0.05, **P<0.01, ***P<0.001.
SWT, shock wave therapy; ROS, reactive oxygen species; NC, negative
control; ns, not significant; H, hypoxia; MDA, malondialdehyde. |
SWT increased miR-210 level in
cardiomyocytes exposed to hypoxia
Previous reports have demonstrated that miR-210 is
one of several hypoxia-induced miRNAs that may play an important
role in cell survival (25). In
order to investigate whether hypoxia regulated miR-210 in HL-1
cells, the present study used RT-qPCR to determine miR-210 levels
in HL-1 cells following hypoxia treatment. As presented in Fig. 4A, hypoxia led to a substantial
increase in miR-210 expression. Notably, the administration of SWT
to the hypoxic cells resulted in a significant increase in miR-210
expression in comparison with the hypoxia group.
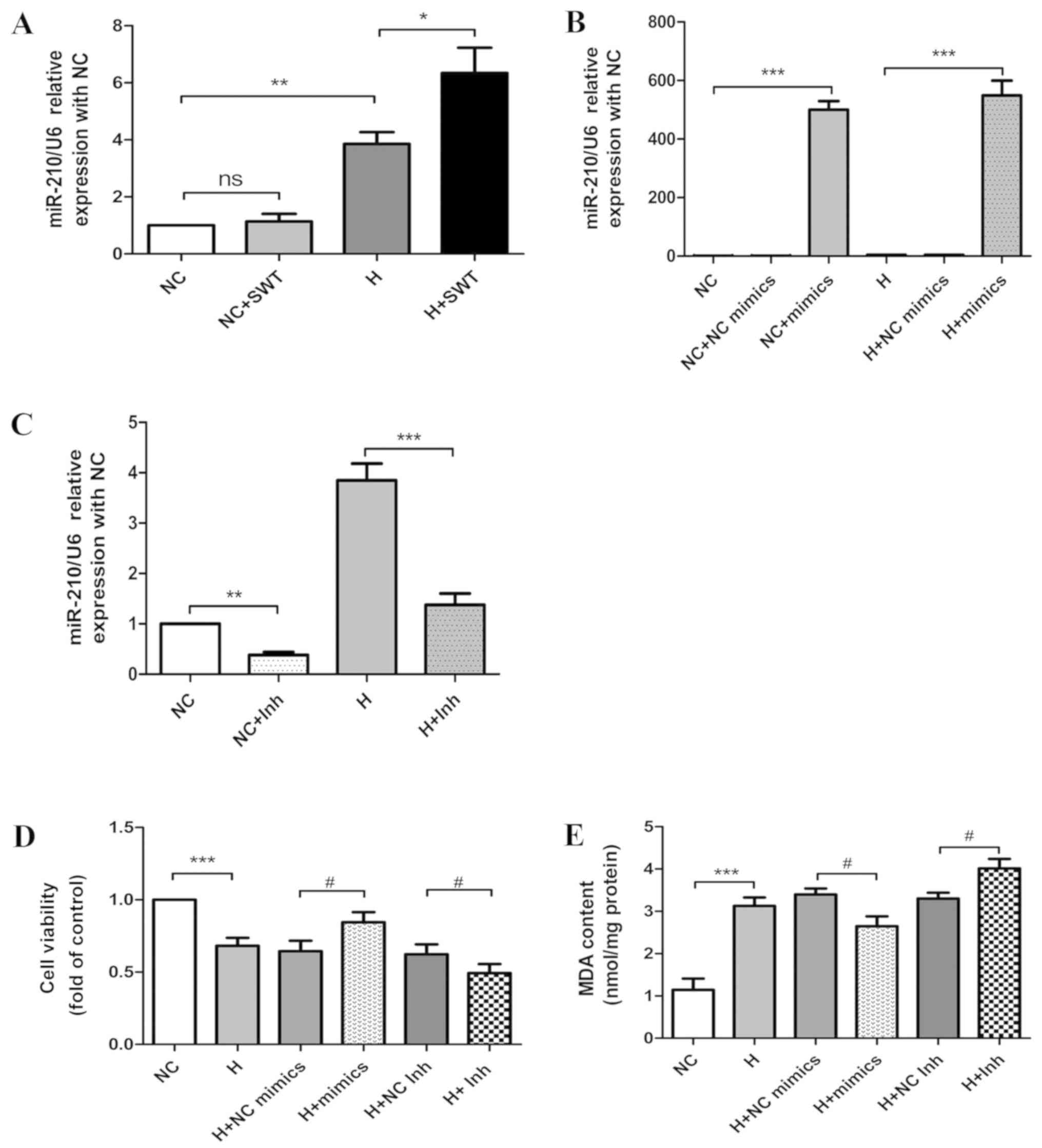 | Figure 4.miR-210 protects cardiomyocytes
against hypoxia and SWT enhances miR-210 expression in HL-1 cells
exposed to hypoxia. (A) miR-210 expression in HL-1 cells exposed to
hypoxia as demonstrated by RT-qPCR in the NC, NC + SWT, hypoxia,
and hypoxia + SWT groups (n=4). (B) miR-210 expression in HL-1
cells exposed to normoxic and hypoxic conditions using RT-qPCR
following the transfection of miR-210 mimics. Data are expressed as
the relative fold change in comparison with the negative control
(n=3). (C) miR-210 expression in HL-1 cells exposed to normoxic and
hypoxic conditions as determined by RT-qPCR following the
transfection of miR-210 inhibitors. Data are expressed as the
relative fold change in comparison with the negative control (n=3).
(D) Cell viability was assessed following the transfection of
miR-210 mimics or miR-210 inhibitors into HL-1 cardiomyocytes
compared with negative controls (n=4). (E) MDA levels were detected
following the transfection of miR-210 mimics or miR-210 inhibitors
into HL-1 cardiomyocytes (n=4). *P<0.05, **P<0.01,
***P<0.001; #P<0.05. miR, microRNA; RT-qPCR,
reverse transcription-quantitative PCR; NC, negative control; SWT,
shock wave therapy; ns, not significant; H, hypoxia; Inh,
inhibitors; MDA, malondialdehyde. |
miR-210 attenuates hypoxia-induced
oxidative stress and apoptosis in cardiomyocytes
In order to investigate the effect of miR-210 on
cardiomyocytes following hypoxia, miR-210 mimics and inhibitors
were transfected in order to alter the expression of miR-210 in
HL-1 cells. RT-qPCR analysis revealed that miR-210 expression in
the mimics group was significantly increased compared with HL-1
cells transfected with the negative control in normoxic and hypoxic
cells (Fig. 4B); in contrast,
inhibitors transfection decreased miR-210 expression (Fig. 4C). Using an MTS assay, the data
from the present study revealed that increased expression of
miR-210 significantly increased cell viability, while inhibition of
miR-210 decreased cell viability when compared with the respective
controls (Fig. 4D). Consistently,
oxidative stress was evaluated by MDA levels. As presented in
Fig. 4E, overexpression of miR-210
led to a significant decrease in MDA level induced by hypoxia,
while miR-210 inhibition resulted in the opposite effect. In order
to determine whether miR-210 plays a role in regulating
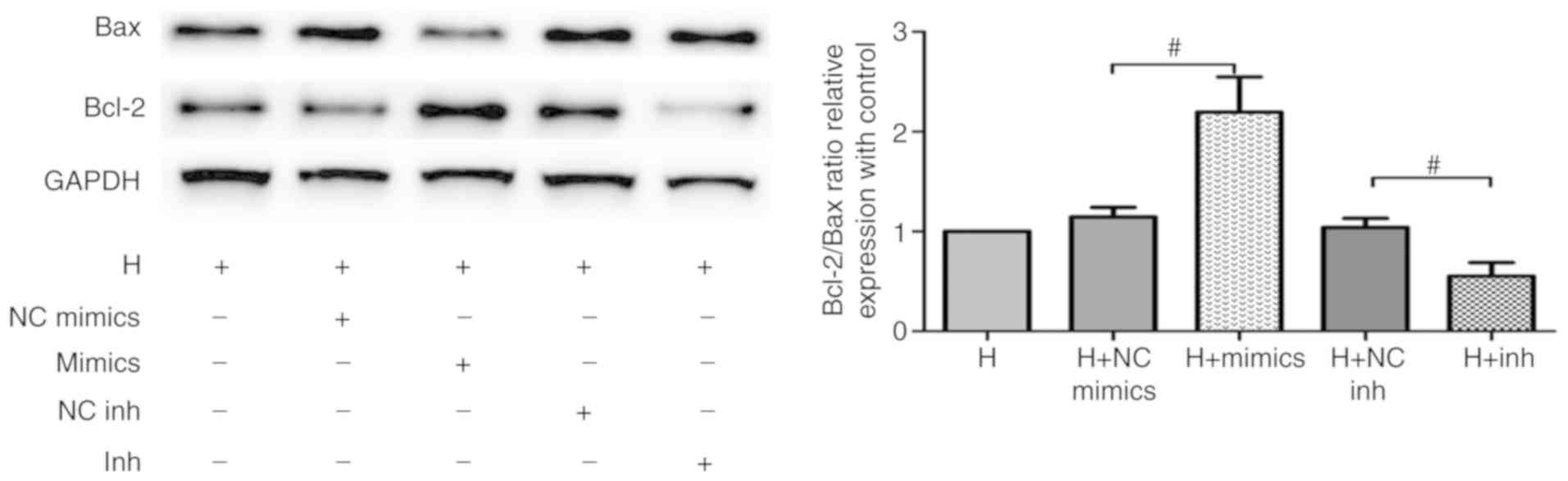
hypoxia-induced apoptosis in cardiomyocytes, Bax and Bcl-2
expression levels were evaluated via western blot analysis. As
presented in Fig. 5, the Bcl-2/Bax
ratio was significantly increased following transfection with
miR-210 mimics when compared with controls, while miR-210
inhibitors decreased this level. Overall, these data demonstrated
that miR-210 protected HL-1 cells against hypoxia-induced injury by
inhibiting apoptosis and oxidative stress.
miR-210 inhibition blocks the
protective effect of SWT
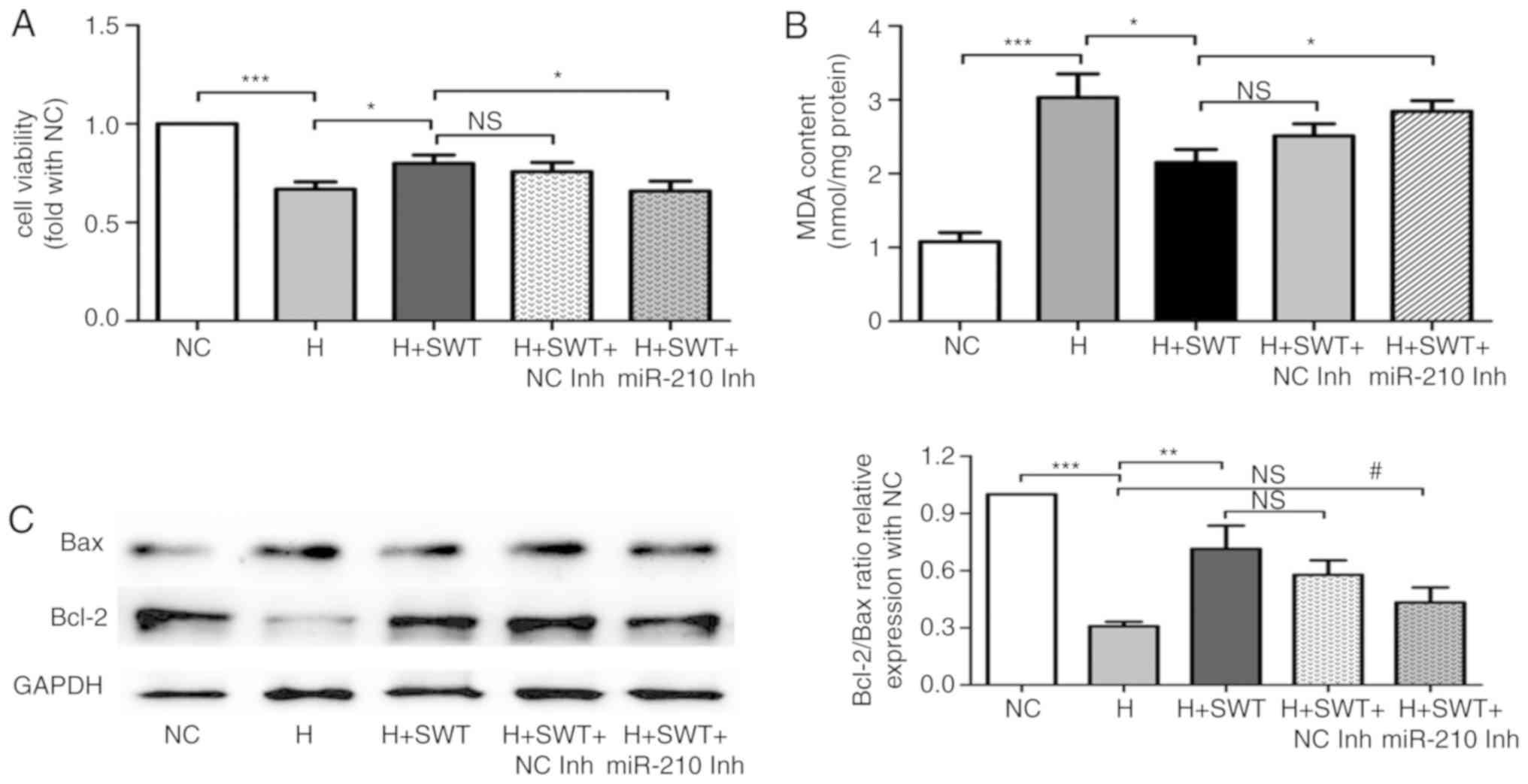
In order to verify whether SWT protects
cardiomyocytes from hypoxia injury by regulating miR-210, HL-1
cells were transfected with miR-210 inhibitors or the negative
controls prior to hypoxia treatment. The data in Fig. 6A and B demonstrated that the
increased cell viability and decreased MDA levels regulated by SWT
were reversed by miR-210 inhibition. Furthermore, the effects of
SWT on apoptosis-associated proteins including Bax and Bcl-2 were
attenuated by miR-210 inhibition (Fig.
6C). Collectively, miR-210 inhibition affected cytoprotection
of SWT in hypoxia-induced injury.
SWT protects HL-1 cells from
hypoxia-induced apoptosis by regulating mRNA levels of Casp8ap2 and
AIFM3
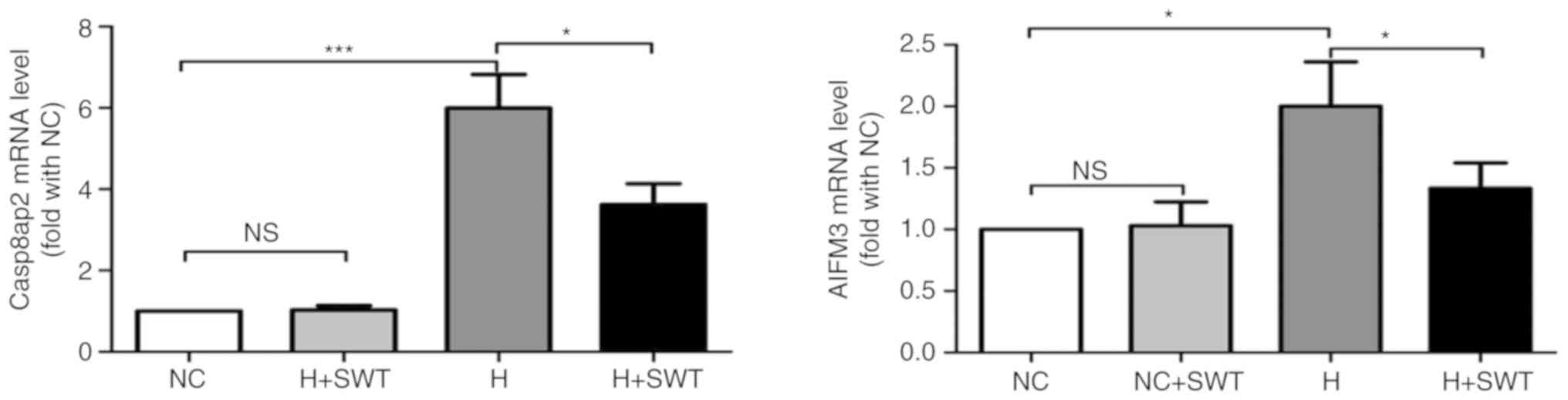
Previous studies have reported that miR-210 exerts
its cardioprotective action against hypoxia-induced apoptosis by
regulating Casp8ap2 and AIFM3 (9,10).
In the present study, RT-qPCR analysis demonstrated that miR-210
overexpression decreased the mRNA expression levels of AIFM3 and
Casp8ap2, and miR-210 inhibitors increased the mRNA expression
levels of AIFM3 and Casp8ap2 (Fig.
S2). In order to further identify the underlying molecular
mechanisms responsible for the cytoprotective effects of SWT on
hypoxia-induced apoptosis in HL-1 cells, RT-qPCR was performed to
detect the mRNA levels of Casp8ap2 and AIFM3. Fig. 7 demonstrates that SWT led to a
significant decrease in Casp8ap2 and AIFM3 mRNA levels,
which were upregulated by hypoxia.
Discussion
Using a well-established murine cardiac hypoxia
model ex vivo, the present study reported that miR-210 was
significantly increased following hypoxia exposure.
Hypoxia-triggered myocardial injury could be alleviated by
overexpression of miR-210. SWT may protect cardiomyocytes from
hypoxia-induced injury by upregulating miR-210.
Ischemic injury to the myocardium results from
events leading to a decrease in or interruption to coronary blood
flow, including IHD. Myocardial ischemia leads to a typical pattern
of metabolic and ultrastructural alterations that induce
irreversible injury, causing different forms of cardiomyocyte
death, referred to as apoptosis, necrosis and necroptosis (26,27).
Apoptosis, known as type I programmed cell death, is a highly
regulated and energy-dependent process that primarily contributes
to cell death in AMI, and could be initiated via the death receptor
and mitochondrial pathways (28,29).
The mitochondrial pathway is activated by a wide range of
non-receptor mediated stimuli, including hypoxia and oxidative
stress (30). Apoptosis presents
with a decrease in Bcl-2 protein levels and an increase in Bax
protein levels (28). Consistent
with previous studies, the data from the present study demonstrated
that hypoxia induced myocardial injury and apoptosis, as evidenced
by decreased cell viability, increased ROS generation, decreased
Bcl-2/Bax ratio and increased apoptotic rates.
Cardiac SWT is a novel effective therapy for IHD.
Both in vivo and in vitro studies demonstrated that
SWT could protect cardiomyocytes from hypoxia-induced apoptosis
(17,18). Similarly, the results from the
present study demonstrated that SWT exhibited antiapoptotic
activities. Upon hypoxia exposure, ROS originates from mitochondria
and activates proapoptotic p38MAPK thereafter (31). In the present study, SWT attenuated
p38MAPK activity, which was markedly increased in hypoxic
cardiomyocytes.
It is well known that numerous genes are abnormally
expressed in IHD, which are responsible for cardiomyocyte survival
and cardiac remodeling following acute and chronic ischemia
(32,33). Previous studies have demonstrated
that miR-210, as one of several hypoxia-induced miRNAs, plays a
critical role in cell survival and possesses antiapoptotic
properties (34–37). Supporting evidence suggested that
miR-210 expression was ubiquitously and robustly increased in
hypoxic cells (34), which is in
line with the results of the present study. miR-210 overexpression
decreased hypoxia-induced injury, and miR-210 inhibition aggravated
hypoxic injury as evidenced by decreased cell viability and
increased apoptosis. Mutharasan et al (10) demonstrated that miR-210
counteracted oxidative stress by decreasing mitochondrial ROS
production and mitochondrial biogenesis in cardiomyocytes exposed
to hypoxia. However, contradictory results have been reported that
suggest increased expression of miR-210 in hypoxia stress activates
ROS production, suppresses mitochondrial function and elevates
glycolysis, leading to increased ATP levels (38). These diverging results may be
attributed to cell-specific effects and varied oxygen
concentrations. The data from the present study demonstrated that
miR-210 overexpression decreased lipid peroxidation, which could be
reversed by miR-210 inhibition. Therefore, both loss- and gain-of
function experiments revealed that miR-210 was a positive regulator
of cytoprotective action against hypoxia-induced injury. SWT
significantly increased miR-210 expression following hypoxic insult
and exerted cytoprotection, which could be prevented by miR-210
supression. To the best of our knowledge, the present study
revealed for the first time that SWT prevented hypoxic injury in
cardiomyocytes through regulating miRNA expression.
Both Casp8ap2 and AIFM3 have been confirmed
as miR-210 targets in the heart, indicating the role of miR-210 in
cell apoptosis (10,11,39).
Casp8ap2 is a component of the death-signaling complex that
is involved in Fas-mediated apoptosis (40), and AIFM3 plays an important role in
initiating the caspase-independent pathway of apoptosis (41). The present study demonstrated that
SWT negatively regulated Casp8ap2 and AIFM3 in
cardiomyocytes exposed to hypoxia with an increase in miR-210
expression, which may contribute to its anti-apoptotic effect.
In summary, the present study demonstrated that
cardiac SWT could protect cardiomyocytes from hypoxia-induced
apoptosis and oxidative stress by modulating miR-210. SWT may act
as an effective therapeutic strategy for the prevention of
hypoxia-induced injury in patients with IHD.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from The
Capital Health Project (grant no. Z131100004013032) and The Beijing
Hospital Clinical Research 121 Project (grant no. 121-2016004).
Availability of data and material
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QH and TS conceived and designed the experiments;
QQ, XXY and QW performed the experiments; NJ, QQ and TS analyzed
the data and wrote the paper.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ibanez B, Heusch G, Ovize M and Van de
Werf F: Evolving therapies for myocardial ischemia/reperfusion
injury. J Am Coll Cardiol. 65:1454–1471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shepard D, VanderZanden A, Moran A,
Naghavi M, Murray C and Roth G: Ischemic heart disease worldwide,
1990 to 2013: Estimates from the global burden of disease study
2013. Circulation. Circ Cardiovasc Qual Outcomes. 8:455–456. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ha M and Kim VN: Regulation of microRNA
biogenesis. Nat Rev Mol Cell Biol. 15:5092014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tang Y, Zheng J, Sun Y, Wu Z, Liu Z and
Huang G: MicroRNA-1 regulates cardiomyocyte apoptosis by targeting
Bcl-2. Int Heart J. 50:377–387. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sayed D, He M, Hong C, Gao S, Rane S, Yang
Z and Abdellatif M: MicroRNA-21 is a downstream effector of AKT
that mediates its antiapoptotic effects via suppression of Fas
ligand. J Biol Chem. 285:20281–20290. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ren XP, Wu J, Wang X, Sartor MA, Jones K,
Qian J, Nicolaou P, Pritchard TJ and Fan GC: MicroRNA-320 is
involved in the regulation of cardiac ischemia/reperfusion injury
by targeting heat-shock protein 20. Circulation. 119:2357–2366.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang JX, Zhang XJ, Li Q, Wang K, Wang Y,
Jiao JQ, Feng C, Teng S, Zhou LY, Gong Y, et al: MicroRNA-103/107
regulate programmed necrosis and myocardial ischemia/reperfusion
injury through targeting FADD. Circ Res. 117:352–363. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kulshreshtha R, Ferracin M, Wojcik SE,
Garzon R, Alder H, Agosto-Perez FJ, Davuluri R, Liu CG, Croce CM,
Negrini M, et al: A microRNA signature of hypoxia. Mol Cell Biol.
27:1859–1867. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim HW, Haider HK, Jiang S and Ashraf M:
Ischemic preconditioning augments survival of stem cells via
miR-210 expression by targeting caspase-8-associated protein 2. J
Biol Chem. 284:33161–33168. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mutharasan RK, Nagpal V, Ichikawa Y and
Ardehali H: microRNA-210 is upregulated in hypoxic cardiomyocytes
through Akt- and p53-dependent pathways and exerts cytoprotective
effects. Am J Physiol Heart Circ Physiol. 301:H1519–H1530. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim HW, Jiang S, Ashraf M and Haider KH:
Stem cell-based delivery of Hypoxamir-210 to the infarcted heart:
Implications on stem cell survival and preservation of infarcted
heart function. J Mol Med (Berl). 90:997–1010. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fasanaro P, D'Alessandra Y, Di Stefano V,
Melchionna R, Romani S, Pompilio G, Capogrossi MC and Martelli F:
MicroRNA-210 modulates endothelial cell response to hypoxia and
inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol
Chem. 283:15878–15883. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nishida T, Shimokawa H, Oi K, Tatewaki H,
Uwatoku T, Abe K, Matsumoto Y, Kajihara N, Eto M, Matsuda T, et al:
Extracorporeal cardiac shock wave therapy markedly ameliorates
ischemia-induced myocardial dysfunction in pigs in vivo.
Circulation. 110:3055–3061. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fukumoto Y, Ito A, Uwatoku T, Matoba T,
Kishi T, Tanaka H, Takeshita A, Sunagawa K and Shimokawa H:
Extracorporeal cardiac shock wave therapy ameliorates myocardial
ischemia in patients with severe coronary artery disease. Coron
Artery Dis. 17:63–70. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cassar A, Prasad M, Rodriguez-Porcel M,
Reeder GS, Karia D, DeMaria AN and Lerman A: Safety and efficacy of
extracorporeal shock wave myocardial revascularization therapy for
refractory angina pectoris. Mayo Clin Proc. 89:346–354. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Khattab AA, Brodersen B,
Schuermann-Kuchenbrandt D, Beurich H, Tölg R, Geist V, Schäfer T
and Richardt G: Extracorporeal cardiac shock wave therapy: First
experience in the everyday practice for treatment of chronic
refractory angina pectoris. Int J Cardiol. 121:84–85. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu W, Shen T, Liu B, Wang S, Li J, Dai D,
Cai J and He Q: Cardiac shock wave therapy attenuates H9c2 myoblast
apoptosis by activating the AKT signal pathway. Cell Physiol
Biochem. 33:1293–1303. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Y, Shen T, Liu B, Dai D, Cai J, Zhao
C, Du L, Jia N and He Q: Cardiac shock wave therapy attenuates
cardiomyocyte apoptosis after acute myocardial infarction in rats.
Cell Physiol Biochem. 49:1734–1746. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hamacher-Brady A, Brady NR and Gottlieb
RA: Enhancing macroautophagy protects against ischemia/reperfusion
injury in cardiac myocytes. J Biol Chem. 281:29776–29787. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Du L, Shen T, Liu B, Zhang Y, Zhao C, Jia
N, Wang Q and He Q: Shock wave therapy promotes cardiomyocyte
autophagy and survival during hypoxia. Cell Physiol Biochem.
42:673–684. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. 25:402–408. 2001.PubMed/NCBI
|
|
22
|
White SM, Constantin PE and Claycomb WC:
Cardiac physiology at the cellular level: Use of cultured HL-1
cardiomyocytes for studies of cardiac muscle cell structure and
function. Am J Physiol Heart Circ Physiol. 286:H823–H829. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Levraut J, Iwase H, Shao ZH, Hoek TLV and
Schumacker PT: Cell death during ischemia: Relationship to
mitochondrial depolarization and ROS generation. Am J Physiol Heart
Circ Physiol. 284:H549–H558. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pryor WA and Stanley JP: Suggested
mechanism for the production of malonaldehyde during the
autoxidation of polyunsaturated fatty acids. Nonenzymic production
of prostaglandin endoperoxides during autoxidation. J Org Chem.
40:3615–3617. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chan SY and Loscalzo J: MicroRNA-210: A
unique and pleiotropic hypoxamir. Cell Cycle. 9:1072–1083. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kang Peter M, Haunstetter A, Aoki H,
Usheva A and Izumo S: Morphological and molecular characterization
of adult cardiomyocyte apoptosis during hypoxia and reoxygenation.
Circ Res. 87:118–125. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Buja LM: Myocardial ischemia and
reperfusion injury. Cardiovasc Pathol. 14:170–175. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Misao J, Hayakawa Y, Ohno M, Kato S,
Fujiwara T and Fujiwara H: Expression of bcl-2 protein, an
inhibitor of apoptosis, and Bax, an accelerator of apoptosis, in
ventricular myocytes of human hearts with myocardial infarction.
Circulation. 94:1506–1512. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Olivetti G, Quaini F, Sala R, Lagrasta C,
Corradi D, Bonacina E, Gambert SR, Cigola E and Anversa P: Acute
myocardial infarction in humans is associated with activation of
programmed myocyte cell death in the surviving portion of the
heart. J Mol Cell Cardiol. 28:2005–2016. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang BC, Zander DS and Mehta JL:
Hypoxia-reoxygenation-induced apoptosis in cultured adult rat
myocytes and the protective effect of platelets and transforming
growth factor-beta(1). J Pharmacol Exp Ther. 291:733–738.
1999.PubMed/NCBI
|
|
31
|
See F, Thomas W, Way K, Tzanidis A, Kompa
A, Lewis D, Itescu S and Krum H: p38 mitogen-activated protein
kinase inhibition improves cardiac function and attenuates left
ventricular remodeling following myocardial infarction in the rat.
J Am Coll Cardiol. 44:1679–1689. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lima J, Batty JA, Sinclair H and Kunadian
V: MicroRNAs in ischemic heart disease: From pathophysiology to
potential clinical applications. Cardiol Rev. 25:117–125. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boon RA and Dimmeler S: MicroRNAs in
myocardial infarction. Nat Rev Cardiol. 12:135–142. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hu S, Huang M, Li Z, Jia F, Ghosh Z,
Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, et al:
MicroRNA-210 as a novel therapy for treatment of ischemic heart
disease. Circulation. 122 (11 Suppl):S124–S131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Arif M, Pandey R, Alam P, Jiang S,
Sadayappan S, Paul A and Ahmed RPH: MicroRNA-210-mediated
proliferation, survival, and angiogenesis promote cardiac repair
post myocardial infarction in rodents. J Mol Med (Berl).
95:1369–1385. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li T, Song X, Zhang J, Zhao L, Shi Y, Li
Z, Liu J, Liu N, Yan Y, Xiao Y, et al: Protection of human
umbilical vein endothelial cells against oxidative stress by
MicroRNA-210. Oxid Med Cell Longev. 2017:35656132017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Diao H, Liu B, Shi Y, Song C, Guo Z, Liu
N, Song X, Lu Y, Lin X and Li Z: MicroRNA-210 alleviates oxidative
stress-associated cardiomyocyte apoptosis by regulating BNIP3.
Biosci Biotechnol Biochem. 81:1712–1720. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen Z, Li Y, Zhang H, Huang P and Luthra
R: Hypoxia-regulated microRNA-210 modulates mitochondrial function
and decreases ISCU and COX10 expression. Oncogene. 29:4362–4368.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim HW, Mallick F, Durrani S, Ashraf M,
Jiang S and Haider KH: Concomitant activation of miR-107/PDCD10 and
hypoxamir-210/Casp8ap2 and their role in cytoprotection during
ischemic preconditioning of stem cells. Antioxid Redox Signal.
17:1053–1065. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Imai Y, Kimura T, Murakami A, Yajima N,
Sakamaki K and Yonehara S: The CED-4-homologous protein FLASH is
involved in Fas-mediated activation of caspase-8 during apoptosis.
Nature. 398:777–785. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xie Q, Lin T, Zhang Y, Zheng J and Bonanno
JA: Molecular cloning and characterization of a human AIF-like gene
with ability to induce apoptosis. J Biol Chem. 280:19673–19681.
2005. View Article : Google Scholar : PubMed/NCBI
|